
Caudal Regression Syndrome—A Narrative Review: An Orthopedic Point of View
Abstract
:1. Introduction
2. Material and Methods
- Recognized key publications in the history of CRS studies;
- Studies fully describing the etiology/epidemiology/morphology of the spinal defect;
- Studies of interest from the orthopedic point of view (both authors are orthopedic surgeons);
- The latest treatment solutions.
3. Etiology and Genetic Background
4. Classifications History and Present
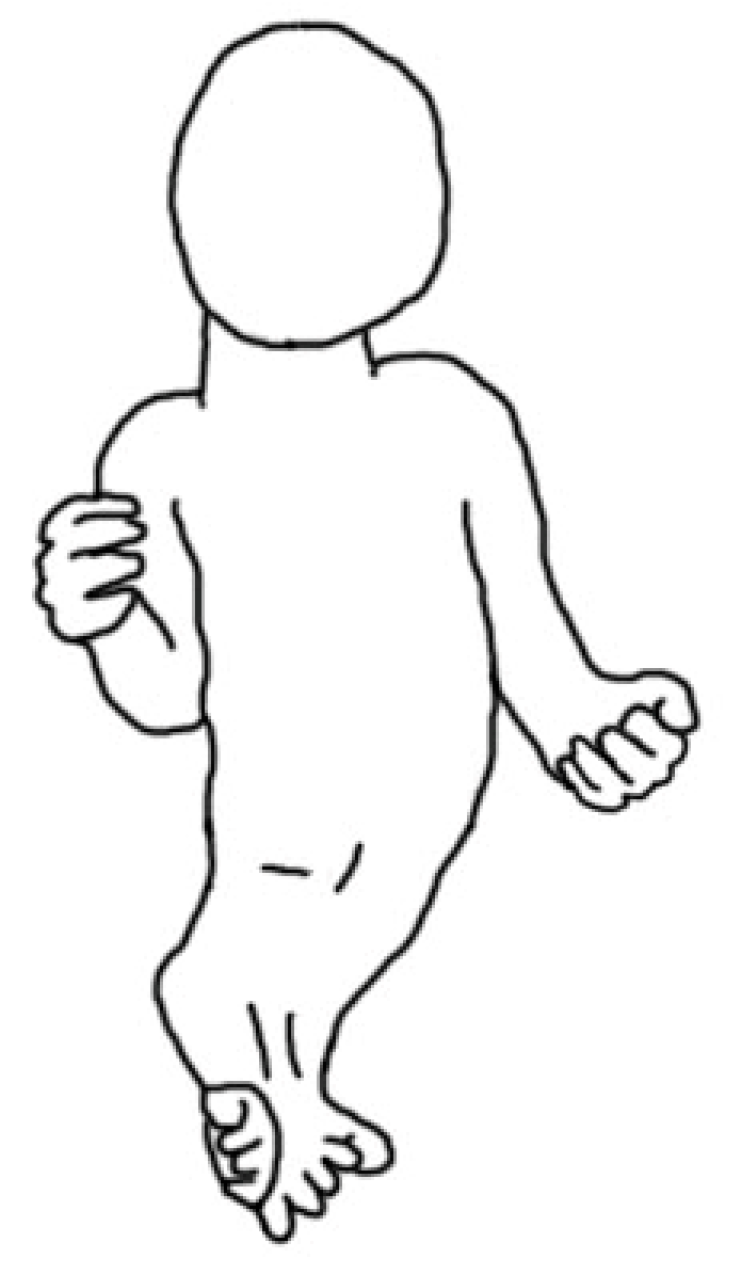
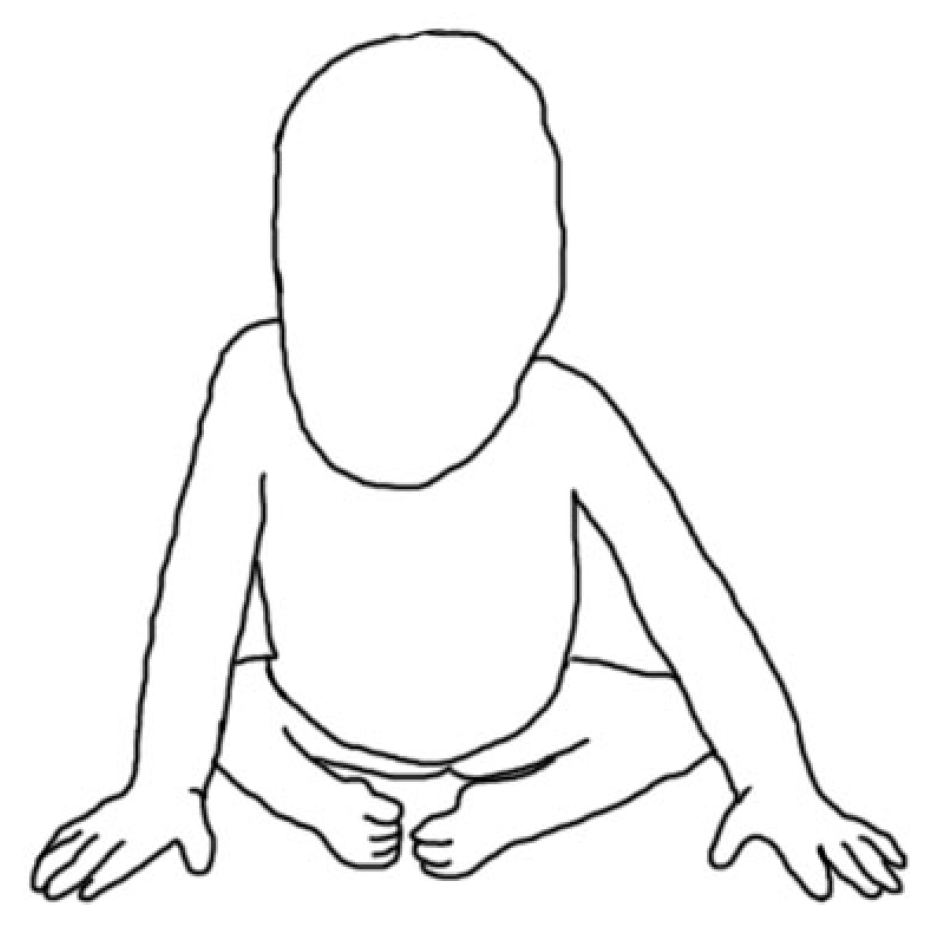
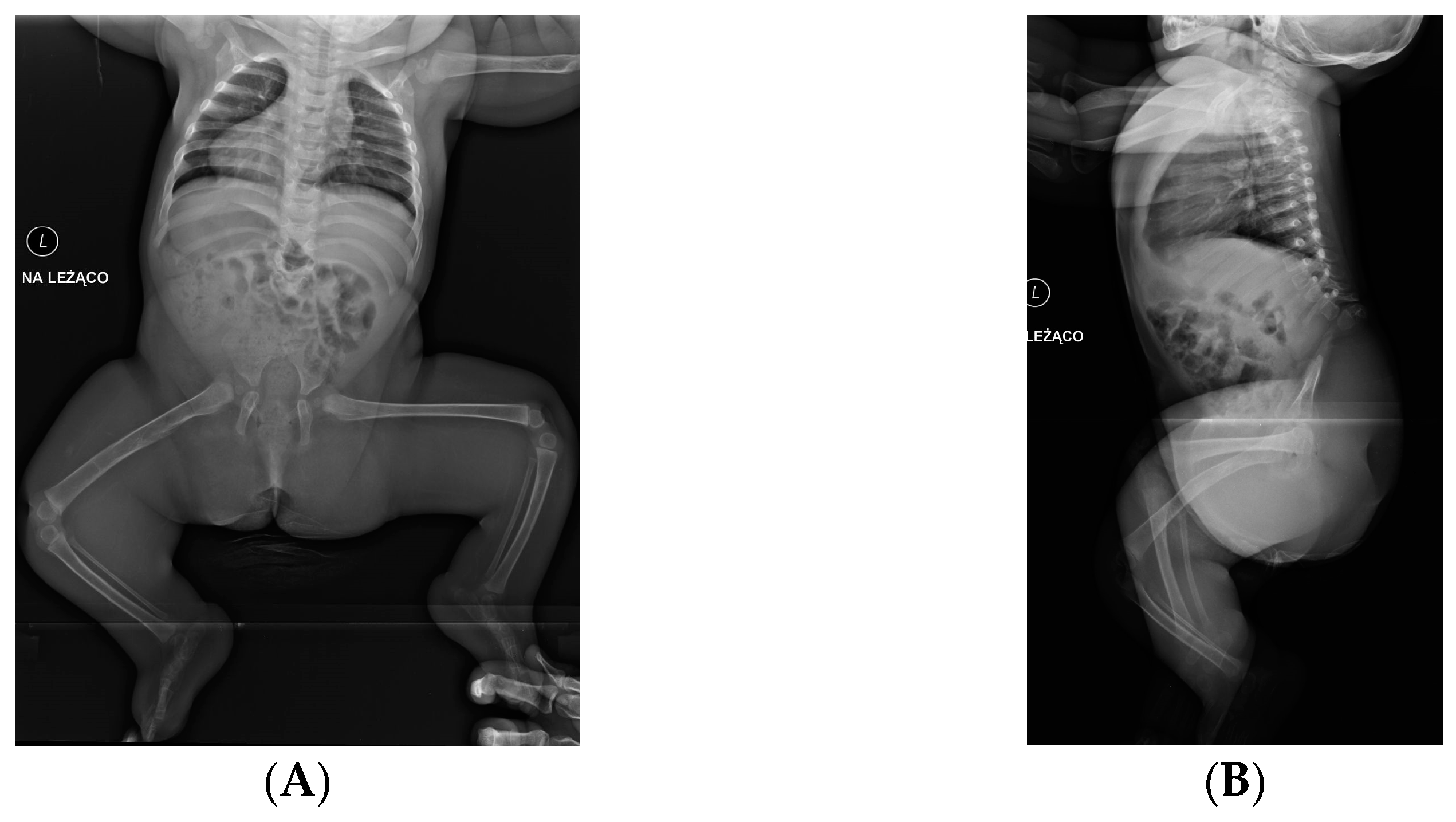
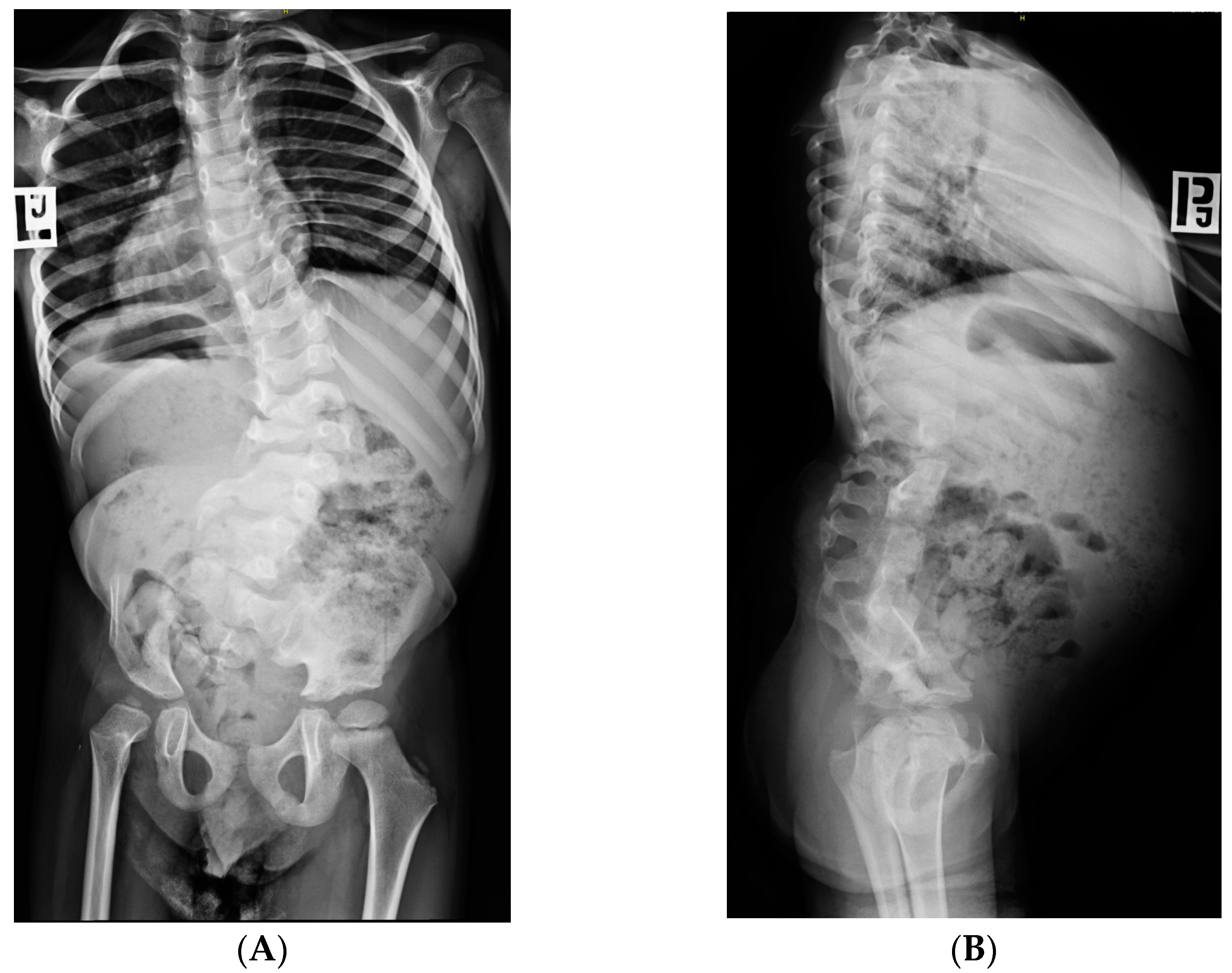
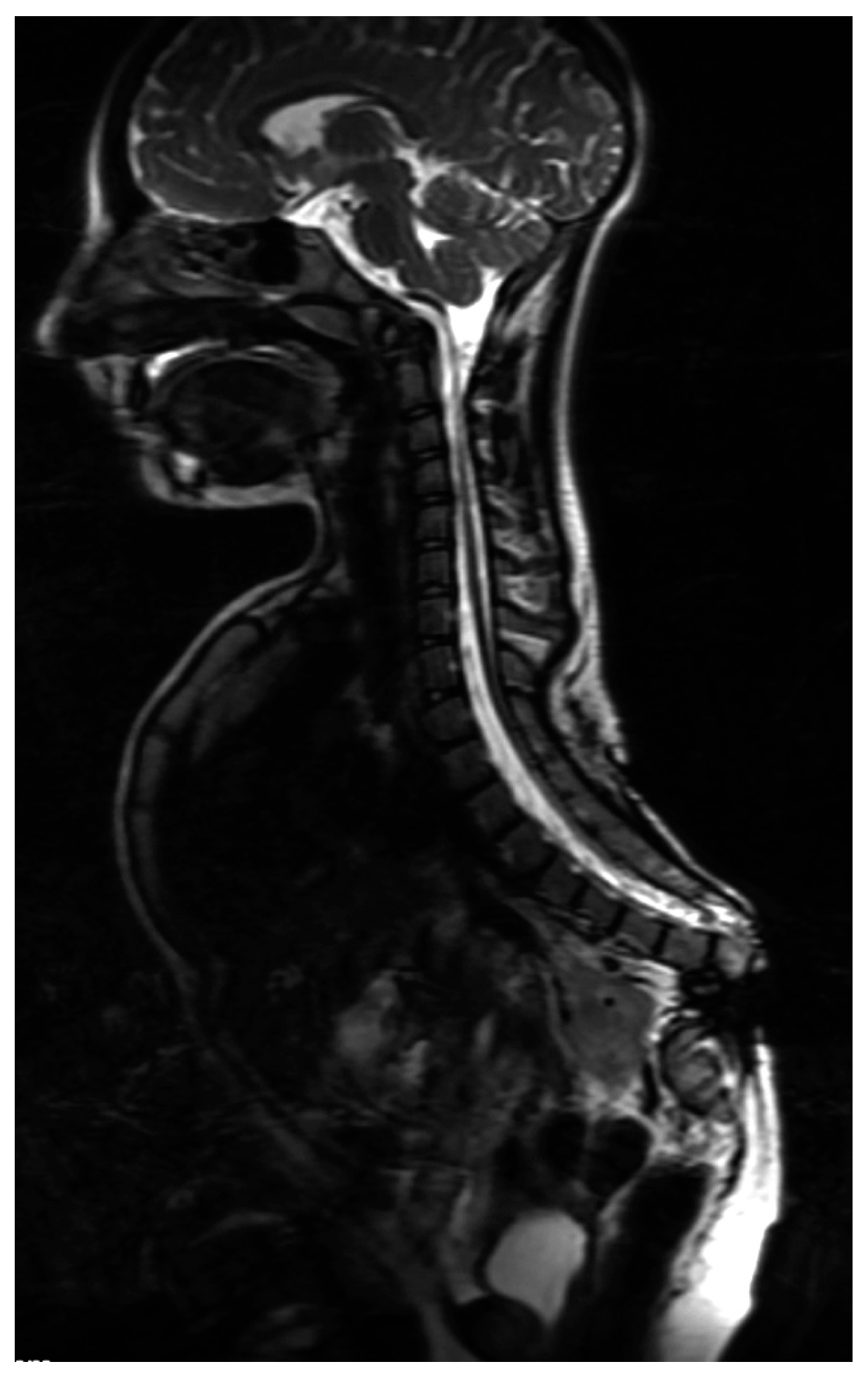
5. Morphology Symptoms
6. Urogenital and Gastrointestinal Problems
7. Treatment
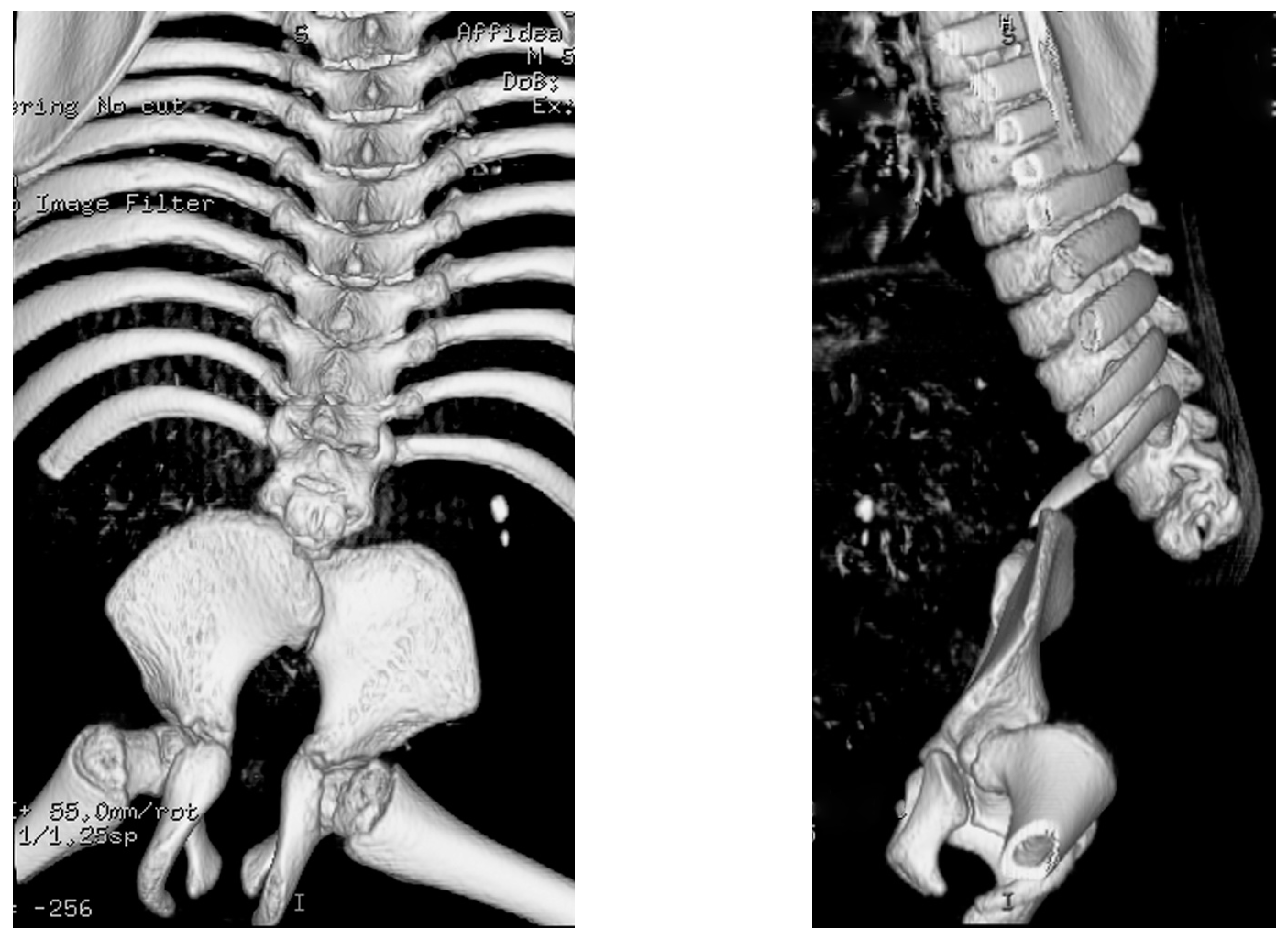
7.1. Spine
7.2. Lower Limbs
7.2.1. Knee
7.2.2. Hip
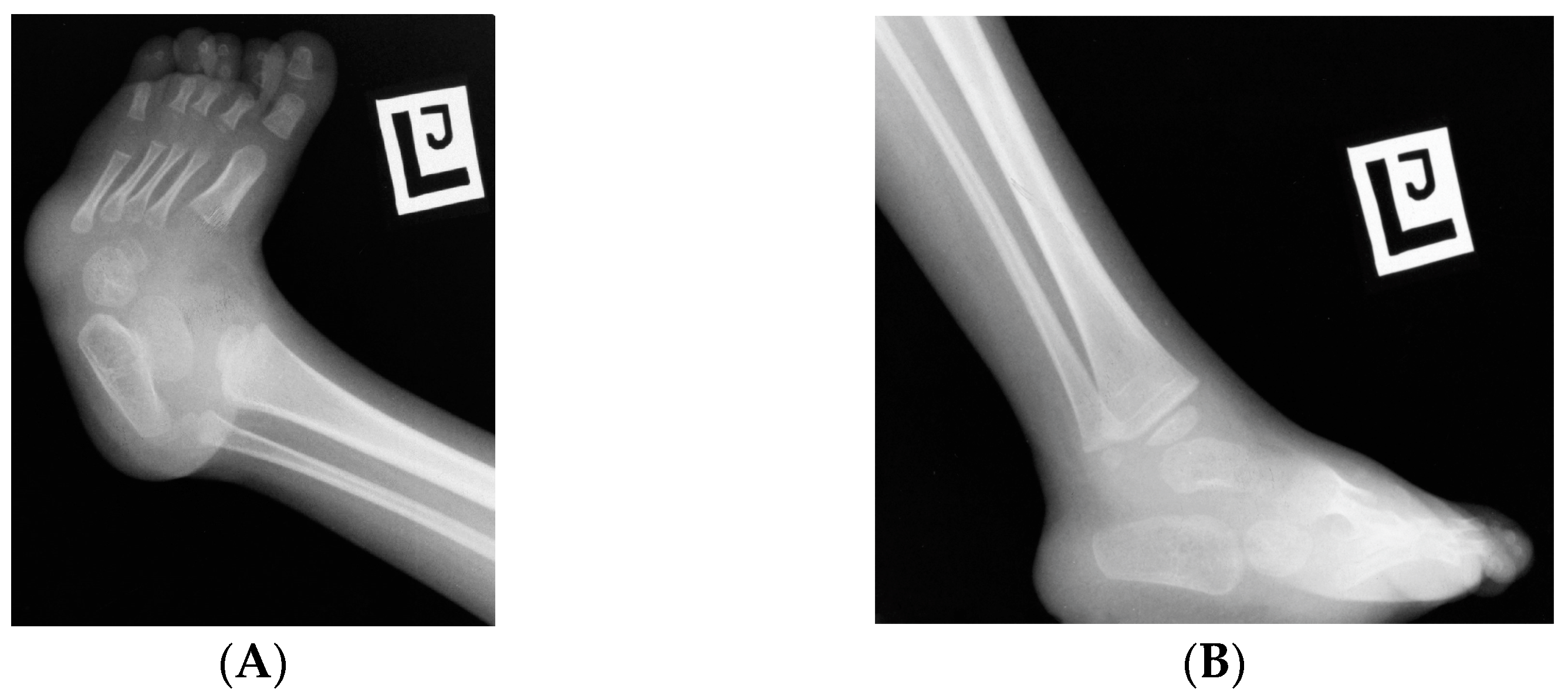
7.2.3. Foot
8. Discussion
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Szumera, E.; Jasiewicz, B.; Potaczek, T. Atypical caudal regression syndrome with agenesis of lumbar spine and presence of sacrum–case report and literature review. J. Spinal Cord Med. 2017, 41, 496–500. [Google Scholar] [CrossRef]
- Chan, B.W.; Chan, K.-S.; Koide, T.; Yeung, S.-M.; Leung, M.B.; Copp, A.J.; Loeken, M.R.; Shiroishi, T.; Shum, A.S. Maternal Diabetes Increases the Risk of Caudal Regression Caused by Retinoic Acid. Diabetes 2002, 51, 2811–2816. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Totonelli, G.; Iacobelli, B.D.; Longo, D.; Caldaro, T.; Blasetti, G.; Bevilacqua, F.; Santato, F.; Lucignani, G.; Sollini, M.L.; et al. Continence management in children with severe caudal regression syndrome: Role of multidisciplinary team and long-term follow-up. Pediatr. Surg. Int. 2022, 38, 1461–1472. [Google Scholar] [CrossRef]
- Duhamel, B. From the Mermaid to Anal Imperforation: The Syndrome of Caudal Regression. Arch. Dis. Child. 1961, 36, 152–155. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, S.; Pradhan, G.; Gautam, V.K.; Ratan, S.K.; Singh, A. Scoliotic deformity and asymptomatic cervical syrinx in a 9 year old with caudal regression syndrome. J. Pediatr. Neurosci. 2012, 7, 191–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Singh, R.D.; Sharma, A. Caudal regression syndrome—Case report and review of literature. Pediatr. Surg. Int. 2005, 21, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Pang, D. Sacral Agenesis and Caudal Spinal Cord Malformations. Neurosurgery 1993, 32, 755–779. [Google Scholar] [CrossRef]
- Pauli, R.M. Lower mesodermal defects: A common cause of fetal and early neonatal death. Am. J. Med. Genet. 1994, 50, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Tandon, A.; Singh, A.K.; Manchanda, S.; Jain, S.; Meena, N. Caudal Regression Syndrome. J. Diagn. Med. Sonogr. 2016, 33, 130–133. [Google Scholar] [CrossRef]
- Qudsieh, H.; Aborajooh, E.; Daradkeh, A. Caudal regression syndrome: Postnatal radiological diagnosis with literature review of 83 cases. Radiol. Case Rep. 2022, 17, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Wilmshurst, J.M.; Kelly, R.; Borzyskowski, M. Presentation and outcome of sacral agenesis: 20 years’ experience. Dev. Med. Child Neurol. 1999, 41, 806–812. [Google Scholar] [CrossRef]
- Lee, J.Y.; Shim, Y.; Wang, K.-C. Caudal Agenesis: Understanding the Base of the Wide Clinical Spectrum. J. Korean Neurosurg. Soc. 2021, 64, 380–385. [Google Scholar] [CrossRef]
- Stevens, S.J.C.; Stumpel, C.T.R.M.; Diderich, K.E.M.; van Slegtenhorst, M.A.; Abbott, M.; Manning, C.; Balciuniene, J.; Pyle, L.C.; Leonard, J.; Murrell, J.R.; et al. The broader phenotypic spectrum of congenital caudal abnormalities associated with mutations in the caudal type homeobox 2 gene. Clin. Genet. 2021, 101, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.; Scullen, T.A.; Iwanaga, J.; Loukas, M.; Bui, C.; Dumont, A.S.; Tubbs, R.S. Caudal Regression Syndrome—A Review Focusing on Genetic Associations. World Neurosurg. 2020, 138, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.P.; Aterman, K. The Syndrome of Caudal Dysplasia: A Review, Including Etiologic Considerations and Evidence of Heterogeneity. Pediatr. Pathol. 1984, 2, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Al Kaissi, A.; Klaushofer, K.; Grill, F. Caudal regression syndrome and popliteal webbing in connection with maternal diabetes mellitus: A case report and literature review. Cases J. 2008, 1, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Banta, J.V.; Nichols, O. Sacral agenesis. J. Bone Jt. Surg. Am. 1969, 51, 693–703. [Google Scholar] [CrossRef]
- Passarge, E.; Lenz, W. Syndrome of Caudal Regression in Infants of Diabetic Mothers: Observations of Further Cases. Pediatrics 1966, 37, 672–675. [Google Scholar] [CrossRef]
- Nievelstein, R.A.; Valk, J.; Smit, L.M.; Vermeij-Keers, C. MR of the caudal regression syndrome: Embryologic implications. Am. J. Neuroradiol. 1994, 15, 1021–1029. [Google Scholar] [PubMed]
- Boer, L.L.; Morava, E.; Klein, W.M.; Schepens-Franke, A.N.; Oostra, R.J. Sirenomelia: A Multi-systemic Polytopic Field Defect with Ongoing Controversies. Birth Defects Res. 2017, 109, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Orioli, I.M.; Amar, E.; Arteaga-Vazquez, J.; Bakker, M.K.; Bianca, S.; Botto, L.D.; Clementi, M.; Correa, A.; Csaky-Szunyogh, M.; Leoncini, E.; et al. Sirenomelia: An epidemiologic study in a large dataset from the International Clearinghouse of Birth Defects Surveillance and Research, and literature review. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porsch, R.M.; Merello, E.; De Marco, P.; Cheng, G.; Rodriguez, L.; So, M.; Sham, P.C.; Tam, P.K.; Capra, V.; Cherny, S.S.; et al. Sacral agenesis: A pilot whole exome sequencing and copy number study. BMC Med. Genet. 2016, 17, 98. [Google Scholar] [CrossRef] [Green Version]
- Allan, D.; Houle, M.; Bouchard, N.; Meyer, B.I.; Gruss, P.; Lohnes, D. RARgamma and Cdx1 interactions in vertebral patterning. Dev. Biol. 2001, 240, 46–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Tang, X.B.; Wang, L.L.; Bai, Y.Z.; Qiu, G.R.; Yuan, Z.W.; Wang, W.L. Mutations and Down-Regulation of CDX1 in Children with Anorectal Malformations. Int. J. Med. Sci. 2013, 10, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro-Domínguez, P.; Bass, J.; Hurteau-Miller, J. Currarino syndrome in a Fetus, Infant, Child, and Adolescent: Spectrum of Clinical Presentations and Imaging Findings. Can. Assoc. Radiol. J. 2016, 68, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Keppler-Noreuil, K.M. OEIS complex (omphalocele-exstrophy-imperforate anus-spinal defects): A review of 14 cases. Am. J. Med. Genet. 2001, 99, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D. VACTERL/VATER Association. Orphanet J. Rare Dis. 2011, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Belloni, E.; Martuciello, G.; Verderio, D.; Ponti, E.; Seri, M.; Jasonni, V.; Torre, M.; Ferrari, M.; Tsui, L.-C.; Scherer, S.W. Involvement of the HLXB9 homeoboxgene in Currarino syndrome. Am. J. Hum. Genet. 2000, 66, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.A.; Wang, Y.; Strachan, T.; Burn, J.; Lindsay, S. Autosomal dominant sacral agenesis: Currarino syndrome. J. Med. Genet. 2000, 37, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.J.; Ruiz-Perez, V.; Wang, Y.; Hagan, D.-M.; Scherer, S.; Lynch, S.A.; Lindsay, S.; Custard, E.; Belloni, E.; Wilson, D.I.; et al. A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat. Genet. 1998, 20, 358–361. [Google Scholar] [CrossRef]
- Merello, E.; De Marco, P.; Mascelli, S.; Raso, A.; Calevo, M.G.; Torre, M.; Cama, A.; Lerone, M.; Martucciello, G.; Capra, V. HLXB9 homeobox gene and caudal regression syndrome. Birth Defects Res. Part A Clin. Mol. Teratol. 2006, 76, 205–209. [Google Scholar] [CrossRef]
- Lee, S.-J.; Perera, L.; Coulter, S.J.; Mohrenweiser, H.W.; Jetten, A.; Goldstein, J.A. The discovery of new coding alleles of human CYP26A1 that are potentially defective in the metabolism of all-trans retinoic acid and their assessment in a recombinant cDNA expression system. Pharm. Genom. 2007, 17, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Tse, H.K.; Leung, M.B.; Woolf, A.S.; Menke, A.L.; Hastie, N.D.; Gosling, J.A.; Pang, C.-P.; Shum, A.S. Implication of Wt1 in the Pathogenesis of Nephrogenic Failure in a Mouse Model of Retinoic Acid-Induced Caudal Regression Syndrome. Am. J. Pathol. 2005, 166, 1295–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatkupt, S.; Speer, M.C.; Ding, Y.; Thomas, M.; Stenroos, E.S.; Dermody, J.J.; Koenigsberger, M.R.; Ott, J.; Johnson, W.G. Linkage analysis of a candidate locus (HLA) in autosomal dominant sacral defect with anterior meningocele. Am. J. Med. Genet. 1994, 52, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Achilleos, A.; Huffman, N.T.; Marcinkiewicyz, E.; Seidah, N.G.; Chen, Q.; Dallas, S.L.; Trainor, P.A.; Gorski, J.P. MBTPS1/SKI-1/S1P proprotein convertase is required for ECM signaling and axial elongation during somitogenesis and vertebral development. Hum. Mol. Genet. 2015, 24, 2884–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liška, F.; Peterková, R.; Peterka, M.; Landa, V.; Zídek, V.; Mlejnek, P.; Šilhavý, J.; Šimáková, M.; Křen, V.; Starker, C.G.; et al. Targeting of the Plzf Gene in the Rat by Transcription Activator-Like Effector Nuclease Results in Caudal Regression Syndrome in Spontaneously Hypertensive Rats. PLoS ONE 2016, 11, e0164206. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Kohlhase, J.; Bohm, D.; Schweiger, B.; Hoffmann, D.; Heitmann, M.; Horsthemke, B.; Wieczorek, D. Biallelic loss of function of the promyelocytic leukaemia zinc finger (PLZF) gene causes severe skeletal defects and genital hypoplasia. J. Med. Genet. 2008, 45, 731–737. [Google Scholar] [CrossRef]
- Pitsava, G.; Feldkamp, M.L.; Pankratz, N.; Lane, J.; Kay, D.M.; Conway, K.M.; Hobbs, C.; Shaw, G.M.; Reefhuis, J.; Jenkins, M.M.; et al. Exome sequencing identifies variants in infants with sacral agenesis. Birth Defects Res. 2022, 114, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Economides, A.; Yanagita, M.; Graf, D.; Yamada, G. New horizons at the caudal embryos: Coordinated urogenital/reproductive organ formation by growth factor signaling. Curr. Opin. Genet. Dev. 2009, 19, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Kampmeier, O.F. On sireniform monsters, with a considera-tion of the causation and predominance of the male sex amongthem. Anat. Rec. 1927, 34, 365. [Google Scholar] [CrossRef]
- Seidahmed, M.Z.; Abdelbasis, O.B.; Albussein, K.A.; Miqdad Am Khalid, M.I.; Salih, M.A. Sirenomelia and severe caudal regression syndrome. Saudi Med. J. 2014, 35, S36–S38. [Google Scholar]
- Valenzano, M.; Paoletti, R.; Rossi, A.; Farinini, D.; Garlaschi, G.; Fulcheri, E. Sirenomelia. Pathological features, antenatal ultrasonographic clues, and a review of current embryogenic theories. Hum. Reprod. Updat. 1999, 5, 82–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhuaire, M.; Jestin, A.; Boulagnon, C.; Loock, M.; Doco-Fenzy, M.; Gaillard, D.; Diebold, M.-D.; Avisse, C.; Labrousse, M. Sirenomelia: A new type, Showing VACTERL Association with thomas syndrome and a review of literature. Birth Defects Res. Part A Clin. Mol. Teratol. 2013, 97, 123–132. [Google Scholar] [CrossRef]
- Kjaer, K.W.; Keeling, J.W.; Opitz, J.M.; Gilbert-Barness, E.; Hartling, U.; Hansen, B.F.; Kjaer, I. Sirenomelia sequence according to the distance between the first sacral vertebra and the ilia. Am. J. Med. Genet. Part A 2003, 120, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, T.S. Sacral agenesis. J. Bone Jt. Surg. Am. 1978, 60, 373–383. [Google Scholar] [CrossRef]
- Stanley, J.; Owen, R.; Koff, S. Congenital sacral anomalies. J. Bone Jt. Surg. 1979, 61, 401–409. [Google Scholar] [CrossRef]
- Guille, J.T.; Benevides, R.; DeAlba, C.C.; Siriram, V.; Kumar, S.J. Lumbosacral Agenesis: A New Classification Correlating Spinal Deformity and Ambulatory Potential. J. Bone Jt. Surg. 2002, 84, 32–38. [Google Scholar] [CrossRef]
- Shojaee, A.; Ronnasian, F.; Behnam, M.; Salehi, M. Sirenomelia: Two case reports. J. Med. Case Rep. 2021, 15, 217. [Google Scholar] [CrossRef]
- Sparnon, A.L.; Ahmed, S. Urological anomalies in the caudal regression syndrome. ANZ J. Surg. 1984, 54, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Shah, M.A.; Babu, D.M. Symptomatic lower urinary tract dysfunction in sacral agenesis: Potentially high risk? Indian J. Urol. 2018, 34, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Emami-Naeini, P.; Rahbar, Z.; Nejat, F.; Kajbafzadeh, A.; El Khashab, M. Neurological Presentations, Imaging, and Associated Anomalies in 50 Patients with Sacral Agenesis. Neurosurgery 2010, 67, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Dewberry, L.; Peña, A.; Mirsky, D.; Ketzer, J.; Bischoff, A. Sacral agenesis and fecal incontinence: How to increase the index of suspicion. Pediatr. Surg. Int. 2018, 35, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Kylat, R.I.; Bader, M. Caudal Regression Syndrome. Children 2020, 7, 211. [Google Scholar] [CrossRef] [PubMed]
- Akhaddar, A. Caudal Regression Syndrome (Spinal Thoraco-lumbo-sacro-coccygeal Agenesis). World Neurosurg. 2020, 142, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Balioğlu, M.B.; Akman, Y.E.; Ucpunar, H.; Albayrak, A.; Kargın, D.; Atıcı, Y.; Büyük, A.F. Sacral agenesis: Evaluation of accompanying pathologies in 38 cases, with analysis of long-term outcomes. Child’s Nerv. Syst. 2016, 32, 1693–1702. [Google Scholar] [CrossRef]
- Devesa, J.; Alonso, A.; López, N.; García, J.; Puell, C.I.; Pablos, T.; Devesa, P. Growth Hormone (GH) and Rehabilitation Promoted Distal Innervation in a Child Affected by Caudal Regression Syndrome. Int. J. Mol. Sci. 2017, 18, 230. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Guo, H.; He, S.; Hui, H.; Hao, D. Sacral agenesis combined with spinopelvic dissociation: A case report and literature review. Medicine 2018, 97, e12162. [Google Scholar] [CrossRef]
- Phillips, W.A.; Cooperman, D.R.; Lindquist, T.C.; Sullivan, R.C.; Millar, E.A. Orthopaedic management of lumbosacral agenesis. Long-term follow-up. J. Bone Jt. Surg. 1982, 64, 1282–1294. [Google Scholar] [CrossRef]
- Canaz, H.; Alatas, I.; Canaz, G.; Gumussuyu, G.; Cacan, M.A.; Saracoglu, A.; Ucar, B.Y. Surgical treatment of patients with myelomeningocele-related spine deformities: Study of 26 cases. Child’s Nerv. Syst. 2018, 34, 1367–1374. [Google Scholar] [CrossRef]
- Perry, J.; Bonnett, C.A.; Hoffer, M.M. Vertebral Pelvic Fusions in the Rehabilitation of Patients with Sacral Agenesis. J. Bone Jt. Surg. 1970, 52, 288–294. [Google Scholar] [CrossRef]
- Vissarionov, S.; Schroder, J.E.; Kokushin, D.; Murashko, V.; Belianchikov, S.; Kaplan, L. Surgical Correction of Spinopelvic Instability in Children with Caudal Regression Syndrome. Glob. Spine J. 2018, 9, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Yazici, M.; Akel, I.; Demirkiran, H.G. Lumbopelvic fusion with a new fixation technique in lumbosacral agenesis: Three cases. J. Child. Orthop. 2011, 5, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Shu, S.; Jing, W.; Gu, Q.; Liu, Z.; Sun, X.; Wang, B.; Qiu, Y.; Zhu, Z.; Bao, H. Sacral Agenesis: A Neglected Deformity That Increases the Incidence of Postoperative Coronal Imbalance in Congenital Lumbosacral Deformities. Glob. Spine J. 2020, 12, 916–921. [Google Scholar] [CrossRef]
- Griffet, J.; Leroux, J.; El Hayek, T. Lumbopelvic stabilization with external fixator in a patient with lumbosacral agenesis. Eur. Spine J. 2010, 20, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Ferland, C.E.; Sardar, Z.M.; Abduljabbar, F.; Arlet, V.; Ouellet, J.A. Bilateral vascularized rib grafts to promote spinopelvic fixation in patients with sacral agenesis and spinopelvic dissociation: A new surgical technique. Spine J. 2015, 15, 2583–2592. [Google Scholar] [CrossRef] [PubMed]
- Dumont, C.E.; Damsin, J.P.; Forin, V.; Carlioz, H. Lumbosacral agenesis. Three cases of reconstruction using Cotrel-Dubousset or L-rod instrumentation. Spine 1993, 18, 1229–1235. [Google Scholar] [CrossRef]
- Rieger, M.A.; Hall, J.E.; Dalury, D.F. Spinal Fusion in a Patient with Lumbosacral Agenesis. Spine 1990, 15, 1382–1384. [Google Scholar] [CrossRef]
- Winter, R.B. Congenital absence of the lumbar spine and sacrum: One-stage reconstruction with subsequent two-stage spine lengthening. J. Pediatr. Orthop. 1991, 11, 666–670. [Google Scholar] [CrossRef]
- Zhang, T.; Bao, H.; Shu, S.; Liu, Z.; Sun, X.; Wang, B.; Qiu, Y.; Zhu, Z. Different distal fixation anchors in lumbosacral spinal deformities associated with sacral agenesis: Which one is better? J. Neurosurg. Spine 2021, 34, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.S.; Bumpass, D.B.; McCullough, F.L.; McCarthy, R.E. Expansion Thoracoplasty as a Life-Saving Procedure in an Adolescent with Severe Spinal Deformity and Sacral Agenesis. Spine Deform. 2018, 7, 171–175. [Google Scholar] [CrossRef]
- Krol, R.; Wasilewski, K. Leczenie zniekształceń towarzyszacych hypoplazji i aplazji kości krzyzowej. Chir Narz. Ruchu Ortop. Pol. 1992, 57, 204–208. (In Polish) [Google Scholar]
- Guidera, K.J.; Raney, E.; Ogden, J.A.; Highhouse, M.; Habal, M. Caudal regression: A review of seven cases, including the mermaid syndrome. J. Pediatr. Orthop. 1991, 11, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.J.H.; Crosswell, S.; Brown, R. Congenital talipes equinovarus and congenital vertical talus secondary to sacral agenesis. BMJ Case Rep. 2017, 2017, bcr2017219786. [Google Scholar] [CrossRef]
- Jasiewicz, B. Przepuklina oponowo-rdzeniowa oraz ukryte wady rdzenia kręgowego. Zniekształcenia neurogenne stóp. In Stopa Dziecięca w Praktyce Ortopedycznej; Napiontek Marek, Ed.; Wydawnictwo Lekarskie PZWL: Warszawa, Poland, 2021; pp. 236–244. ISBN 978-83-20-06213-7. [Google Scholar]
- Segal, L.S.; Czoch, W.; Hennrikus, W.L.; Shrader, M.W.; Kanev, P.M. The spectrum of musculoskeletal problems in lipomyelomeningocele. J. Child. Orthop. 2013, 7, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamichi, K. Caudal Regression Syndrome with Pressure Ulcers of the Foot: A Case Report. J. Clin. Diagn. Res. 2017, 11, PD19–PD20. [Google Scholar] [CrossRef]
- Matar, H.E.; Beirne, P.; Garg, N.K. Effectiveness of the Ponseti method for treating clubfoot associated with myelomeningocele: 3–9 years follow-up. J. Pediatr. Orthop. B 2017, 26, 133–136. [Google Scholar] [CrossRef]
- Adra, A.; Cordero, D.; Mejides, A.; Yasin, S.; Salman, F.; O’Sullivan, M.J. Caudal regression syndrome: Etiopathogenesis, prenatal diagnosis, and perinatal management. Obstet. Gynecol. Surv. 1994, 49, 508–516. [Google Scholar] [CrossRef]
- Mehdi, S.M.; Baig, U.; Zia, M.H.; Iftikhar, N. Caudal Regression Syndrome—A rare congenital disorder: A case report. J. Pak. Med. Assoc. 2022, 71, 2847–2849. [Google Scholar] [CrossRef]
- Van Buskirk, C.S.; Ritterbusch, J.F. Natural History of Distal Spinal Agenesis. J. Pediatr. Orthop. B 1997, 6, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Bicakci, I.; Turgut, S.T.; Turgut, B.; Icagasioglu, A.; Egilmez, Z.; Yumusakhuylu, Y. A case of caudal regression syndrome: Walking or sitting? Pan Afr. Med. J. 2014, 18, 92. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PubMed | Google Scholar | |
|---|---|---|
| “caudal regression syndrome” | 395 | 3390 |
| “sacral agenesis” | 415 | 6000 |
| “sirenomelia” | 2588 | 3050 |
| “caudal regression syndrome” AND “treatment” | 118 | 2850 |
| “caudal regression syndrome” AND “surgery” | 96 | 1700 |
| “sacral agenesis” AND “treatment” | 197 | 4200 |
| “sacral agenesis” AND “surgery” | 184 | 4100 |
| “caudal regression syndrome” AND “spine” | 176 | 1650 |
| “sacral agenesis” AND “spine” | 239 | 2870 |
| “caudal agenesis” | 2857 | 23,400 |
| “caudal agenesis” AND “treatment” | 1030 | 17,500 |
| Authors and Title | Year of Publication |
|---|---|
| Balioğlu MB et al.: Sacral agenesis: evaluation of accompanying pathologies in 38 cases, with analysis of long-term outcomes [55] | 2016 |
| Ferland CE et al.: Bilateral vascularized rib grafts to promote spinopelvic fixation in patients with sacral agenesis and spinopelvic dissociation: a new surgical technique [65] | 2015 |
| Griffet J et al.: Lumbopelvic stabilization with external fixator in a patient with lumbosacral agenesis [64] | 2011 |
| Mathews CS et al.: Expansion Thoracoplasty as a Life-Saving Procedure in an Adolescent With Severe Spinal Deformity and Sacral Agenesis [70] | 2019 |
| Szumera E et al.: Atypical caudal regression syndrome with agenesis of lumbar spine and presence of sacrum—case report and literature review [1] | 2018 |
| Vissarionov S et al.: Surgical Correction of Spinopelvic Instability in Children With Caudal Regression Syndrome [61] | 2019 |
| Yazici M et al.: Lumbopelvic fusion with a new fixation technique in lumbosacral agenesis: three cases [62] | 2011 |
| Zhang H et al.: Sacral agenesis combined with spinopelvic dissociation: A case report and literature review [57] | 2018 |
| Zhang T et al.: Different distal fixation anchors in lumbosacral spinal deformities associated with sacral agenesis: which one is better? [69] | 2021 |
| Zhang T et al.: Sacral Agenesis: A neglected deformity that increases the incidence of postoperative coronal imbalance in congenital lumbosacral deformities [63] | 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasiewicz, B.; Kacki, W. Caudal Regression Syndrome—A Narrative Review: An Orthopedic Point of View. Children 2023, 10, 589. https://doi.org/10.3390/children10030589
Jasiewicz B, Kacki W. Caudal Regression Syndrome—A Narrative Review: An Orthopedic Point of View. Children. 2023; 10(3):589. https://doi.org/10.3390/children10030589
Chicago/Turabian StyleJasiewicz, Barbara, and Wojciech Kacki. 2023. "Caudal Regression Syndrome—A Narrative Review: An Orthopedic Point of View" Children 10, no. 3: 589. https://doi.org/10.3390/children10030589