
Frasier Syndrome: A 15-Year-Old Phenotypically Female Adolescent Presenting with Delayed Puberty and Nephropathy

Abstract
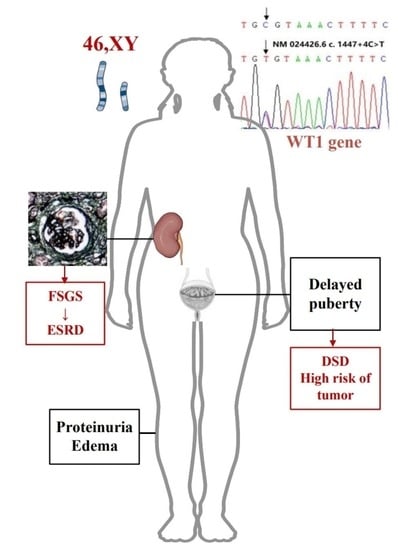
:
1. Introduction
2. Case Presentation
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohsaka, T.; Tagawa, M.; Koinuma, S.; Yamada, M. Frasier syndrome. Nihon Rinsho 2006, 2006 (Suppl. 2), 499–504. [Google Scholar]
- Gessler, M.; Konig, A.; Moore, J.; Qualman, S.; Arden, K.; Cavenee, W.; Bruns, G. Homozygous inactivation of WT1 in a Wilms’ tumor associated with the WAGR syndrome. Genes Chromosomes Cancer 1993, 7, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Patek, C.E.; Little, M.H.; Fleming, S.; Miles, C.; Charlieu, J.P.; Clarke, A.R.; Miyagawa, K.; Christie, S.; Doig, J.; Harrison, D.J.; et al. A zinc finger truncation of murine WT1 results in the characteristic urogenital abnormalities of Denys-Drash syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 2931–2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasier, S.D.; Bashore, R.A.; Mosier, H.D. Gonadoblastoma Associated with Pure Gonadal Dysgenesis in Monozygous Twins. J. Pediatr. 1964, 64, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y.; Yamamura, T.; Nagano, C.; Horinouchi, T.; Sakakibara, N.; Ishiko, S.; Aoto, Y.; Rossanti, R.; Okada, E.; Tanaka, E.; et al. Systematic Review of Genotype-Phenotype Correlations in Frasier Syndrome. Kidney Int. Rep. 2021, 6, 2585–2593. [Google Scholar] [CrossRef]
- Li, J.; Zhao, D.; Ding, J.; Xiao, H.; Guan, N.; Fan, Q.; Zhang, H. WT1 mutation and podocyte molecular expression in a Chinese Frasier syndrome patient. Pediatr. Nephrol. 2007, 22, 2133–2136. [Google Scholar] [CrossRef]
- Yu, Z.; Yang, Y.; Feng, D. Discordant phenotypes in monozygotic twins with identical de novo WT1 mutation. Clin. Kidney J. 2012, 5, 221–222. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Feng, D.; Huang, J.; Nie, X.; Yu, Z. A child with isolated nephrotic syndrome and WT1 mutation presenting as a 46, XY phenotypic male. Eur. J. Pediatr. 2013, 172, 127–129. [Google Scholar] [CrossRef]
- Benoit, G.; Machuca, E.; Antignac, C. Hereditary nephrotic syndrome: A systematic approach for genetic testing and a review of associated podocyte gene mutations. Pediatr. Nephrol. 2010, 25, 1621–1632. [Google Scholar] [CrossRef] [Green Version]
- Barbaux, S.; Niaudet, P.; Gubler, M.C.; Grunfeld, J.P.; Jaubert, F.; Kuttenn, F.; Fekete, C.N.; Souleyreau-Therville, N.; Thibaud, E.; Fellous, M.; et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat. Genet. 1997, 17, 467–470. [Google Scholar] [CrossRef]
- Berta, P.; Morin, D.; Poulat, F.; Taviaux, S.; Lobaccaro, J.M.; Sultan, C.; Dumas, R. Molecular analysis of the sex-determining region from the Y chromosome in two patients with Frasier syndrome. Horm. Res. 1992, 37, 103–106. [Google Scholar] [CrossRef]
- Blanchet, P.; Daloze, P.; Lesage, R.; Papas, S.; Van Campenhout, J. XY gonadal dysgenesis with gonadoblastoma discovered after kidney transplantation. Am. J. Obstet. Gynecol. 1977, 129, 221–222. [Google Scholar] [CrossRef]
- Hashimoto, K.; Horibe, Y.U.; Ezaki, J.; Kanno, T.; Takahashi, N.; Akizawa, Y.; Matsui, H.; Yamamoto, T.; Shibata, N. Laparoscopically Removed Streak Gonad Revealed Gonadoblastoma in Frasier Syndrome. Anticancer Res. 2017, 37, 3975–3979. [Google Scholar]
- Schumacher, V.; Gueler, B.; Looijenga, L.H.J.; Becker, J.U.; Amann, K.; Engers, R.; Dotsch, J.; Stoop, H.; Schulz, W.; Royer-Pokora, B. Characteristics of testicular dysgenesis syndrome and decreased expression of SRY and SOX9 in Frasier syndrome. Mol. Reprod. Dev. 2008, 75, 1484–1494. [Google Scholar] [CrossRef]
- Anderson, E.; Aldridge, M.; Turner, R.; Harraway, J.; McManus, S.; Stewart, A.; Borzi, P.; Trnka, P.; Burke, J.; Coman, D. WT1 complete gonadal dysgenesis with membranoproliferative glomerulonephritis: Case series and literature review. Pediatr. Nephrol. 2022, 37, 2369–2374. [Google Scholar] [CrossRef]
- Denamur, E.; Bocquet, N.; Mougenot, B.; Da Silva, F.; Martinat, L.; Loirat, C.; Elion, J.; Bensman, A.; Ronco, P.M. Mother-to-child transmitted WT1 splice-site mutation is responsible for distinct glomerular diseases. J. Am. Soc. Nephrol. 1999, 10, 2219–2223. [Google Scholar] [CrossRef]
- Ezaki, J.; Hashimoto, K.; Asano, T.; Kanda, S.; Akioka, Y.; Hattori, M.; Yamamoto, T.; Shibata, N. Gonadal tumor in Frasier syndrome: A review and classification. Cancer Prev. Res. 2015, 8, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Guaragna, M.S.; Lutaif, A.C.; Bittencourt, V.B.; Piveta, C.S.; Soardi, F.C.; Castro, L.C.; Belangero, V.M.; Maciel-Guerra, A.T.; Guerra-Junior, G.; Mello, M.P. Frasier syndrome: Four new cases with unusual presentations. Arq. Bras. Endocrinol. Metabol. 2012, 56, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Zugor, V.; Zenker, M.; Schrott, K.M.; Schott, G.E. Frasier syndrome: A rare syndrome with WT1 gene mutation in pediatric urology. Aktuelle Urol. 2006, 37, 64–66. [Google Scholar] [CrossRef]
- Howard, S.R. The Genetic Basis of Delayed Puberty. Front. Endocrinol. 2019, 10, 423. [Google Scholar] [CrossRef] [Green Version]
- Sultan, C.; Gaspari, L.; Maimoun, L.; Kalfa, N.; Paris, F. Disorders of puberty. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 48, 62–89. [Google Scholar] [CrossRef] [PubMed]
- Trotman, G.E. Delayed puberty in the female patient. Curr. Opin. Obstet. Gynecol. 2016, 28, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.R.; Dunkel, L. Delayed Puberty-Phenotypic Diversity, Molecular Genetic Mechanisms, and Recent Discoveries. Endocr. Rev. 2019, 40, 1285–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.F.; Bashamboo, A.; Lucas-Herald, A.; McElreavey, K. Understanding the genetic aetiology in patients with XY DSD. Br. Med. Bull. 2013, 106, 67–89. [Google Scholar] [CrossRef] [PubMed]
- Bashamboo, A.; McElreavey, K. Human sex-determination and disorders of sex-development (DSD). Semin. Cell Dev. Biol. 2015, 45, 77–83. [Google Scholar] [CrossRef]
- Sinha, A.; Sharma, S.; Gulati, A.; Sharma, A.; Agarwala, S.; Hari, P.; Bagga, A. Frasier syndrome: Early gonadoblastoma and cyclosporine responsiveness. Pediatr. Nephrol. 2010, 25, 2171–2174. [Google Scholar] [CrossRef]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Peco-Antić, A.; Ozaltin, F.; Parezanović, V.; Milosevski-Lomić, G.; Zdravković, V. Proteinuria in Frasier syndrome. Srp. Arh. Za Celok. Lek. 2013, 141, 685–688. [Google Scholar] [CrossRef]
- Chiba, Y.; Inoue, C.N. Once-Daily Low-Dose Cyclosporine A Treatment with Angiotensin Blockade for Long-Term Remission of Nephropathy in Frasier Syndrome. Tohoku J. Exp. Med. 2019, 247, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Pleskacova, J.; Hersmus, R.; Oosterhuis, J.W.; Setyawati, B.A.; Faradz, S.M.; Cools, M.; Wolffenbuttel, K.P.; Lebl, J.; Drop, S.L.; Looijenga, L.H. Tumor risk in disorders of sex development. Sex. Dev. 2010, 4, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Dumić, M.; Jukić, S.; Batinica, S.; Ille, J.; Filipović-Grcić, B. Bilateral gonadoblastoma in a 9-month-old infant with 46,XY gonadal dysgenesis. J. Endocrinol. Investig. 1993, 16, 291–293. [Google Scholar] [CrossRef]
- Matsuoka, D.; Noda, S.; Kamiya, M.; Hidaka, Y.; Shimojo, H.; Yamada, Y.; Miyamoto, T.; Nozu, K.; Iijima, K.; Tsukaguchi, H. Immune-complex glomerulonephritis with a membranoproliferative pattern in Frasier syndrome: A case report and review of the literature. BMC Nephrol. 2020, 21, 362. [Google Scholar] [CrossRef]
- Merhi, Z.; Pollack, S.E. Pituitary origin of persistently elevated human chorionic gonadotropin in a patient with gonadal failure. Fertil. Steril. 2013, 99, 293–296. [Google Scholar] [CrossRef]
- Kitsiou-Tzeli, S.; Deligiorgi, M.; Malaktari-Skarantavou, S.; Vlachopoulos, C.; Megremis, S.; Fylaktou, I.; Traeger-Synodinos, J.; Kanaka-Gantenbein, C.; Stefanadis, C.; Kanavakis, E. Sertoli cell tumor and gonadoblastoma in an untreated 29-year-old 46,XY phenotypic male with Frasier syndrome carrying a WT1 IVS9+4C>T mutation. Hormones (Athens) 2012, 11, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Mestrallet, G.; Bertholet-Thomas, A.; Ranchin, B.; Bouvier, R.; Frappaz, D.; Cochat, P. Recurrence of a dysgerminoma in Frasier syndrome. Pediatr Transplant 2011, 15, e53–e55. [Google Scholar] [CrossRef]
- Andrade, J.G.R.d.; Guaragna, M.S.; Soardi, F.C.; Guerra-Júnior, G.; Mello, M.P.d.; Maciel-Guerra, A.T. Clinical and genetic findings of five patients with WT1-related disorders. Arq. Bras. Endocrinol Metabol. 2008, 52, 1236–1243. [Google Scholar] [CrossRef] [Green Version]
- Aucella, F.; Bisceglia, L.; De Bonis, P.; Gigante, M.; Caridi, G.; Barbano, G.; Mattioli, G.; Perfumo, F.; Gesualdo, L.; Ghiggeri, G.M. WT1 mutations in nephrotic syndrome revisited. High prevalence in young girls, associations and renal phenotypes. Pediatr. Nephrol. 2006, 21, 1393–1398. [Google Scholar] [CrossRef]
- Chan, W.K.Y.; To, K.F.; But, W.M.; Lee, K.W. Frasier syndrome: A rare cause of delayed puberty. Hong Kong Med. J. 2006, 12, 225–227. [Google Scholar]
- Ito, S.-i.; Hataya, H.; Ikeda, M.; Takata, A.; Kikuchi, H.; Hata, J.-i.; Morikawa, Y.; Kawamura, S.; Honda, M. Alport syndrome-like basement membrane changes in Frasier syndrome: An electron microscopy study. Am. J. Kidney Dis. 2003, 41, 1110–1115. [Google Scholar] [CrossRef]
- Melo, K.F.S.; Martin, R.M.; Costa, E.M.F.; Carvalho, F.M.; Jorge, A.A.; Arnhold, I.J.P.; Mendonca, B.B. An unusual phenotype of Frasier syndrome due to IVS9 +4C>T mutation in the WT1 gene: Predominantly male ambiguous genitalia and absence of gonadal dysgenesis. J. Clin. Endocrinol. Metab. 2002, 87, 2500–2505. [Google Scholar] [CrossRef]
- Bönte, A.; Schröder, W.; Denamur, E.; Querfeld, U. Absent pubertal development in a child with chronic renal failure: The case of Frasier syndrome. Nephrol. Dial. Transplant. 2000, 15, 1688–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denamur, E.; Bocquet, N.; Baudouin, V.; Da Silva, F.; Veitia, R.; Peuchmaur, M.; Elion, J.; Gubler, M.C.; Fellous, M.; Niaudet, P.; et al. WT1 splice-site mutations are rarely associated with primary steroid-resistant focal and segmental glomerulosclerosis. Kidney Int. 2000, 57, 1868–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuhara, K.; Tajima, S.; Nakae, J.; Sasaki, S.; Tochimaru, H.; Abe, S.; Fujieda, K. A Japanese case with Frasier syndrome caused by the splice junction mutation of WT1 gene. Endocr. J. 1999, 46, 639–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demmer, L.; Primack, W.; Loik, V.; Brown, R.; Therville, N.; McElreavey, K. Frasier syndrome: A cause of focal segmental glomerulosclerosis in a 46,XX female. J. Am. Soc. Nephrol. 1999, 10, 2215–2218. [Google Scholar] [CrossRef]
- Barbosa, A.S.; Hadjiathanasiou, C.G.; Theodoridis, C.; Papathanasiou, A.; Tar, A.; Merksz, M.; Györvári, B.; Sultan, C.; Dumas, R.; Jaubert, F.; et al. The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms’ tumor. Hum. Mutat. 1999, 13, 146–153. [Google Scholar] [CrossRef]
- Kikuchi, H.; Takata, A.; Akasaka, Y.; Fukuzawa, R.; Yoneyama, H.; Kurosawa, Y.; Honda, M.; Kamiyama, Y.; Hata, J. Do intronic mutations affecting splicing of WT1 exon 9 cause Frasier syndrome? J. Med. Genet. 1998, 35, 45–48. [Google Scholar] [CrossRef]
- Jeanpierre, C.; Denamur, E.; Henry, I.; Cabanis, M.O.; Luce, S.; Cécille, A.; Elion, J.; Peuchmaur, M.; Loirat, C.; Niaudet, P.; et al. Identification of constitutional WT1 mutations, in patients with isolated diffuse mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a computerized mutation database. Am. J. Hum. Genet. 1998, 62, 824–833. [Google Scholar] [CrossRef] [Green Version]




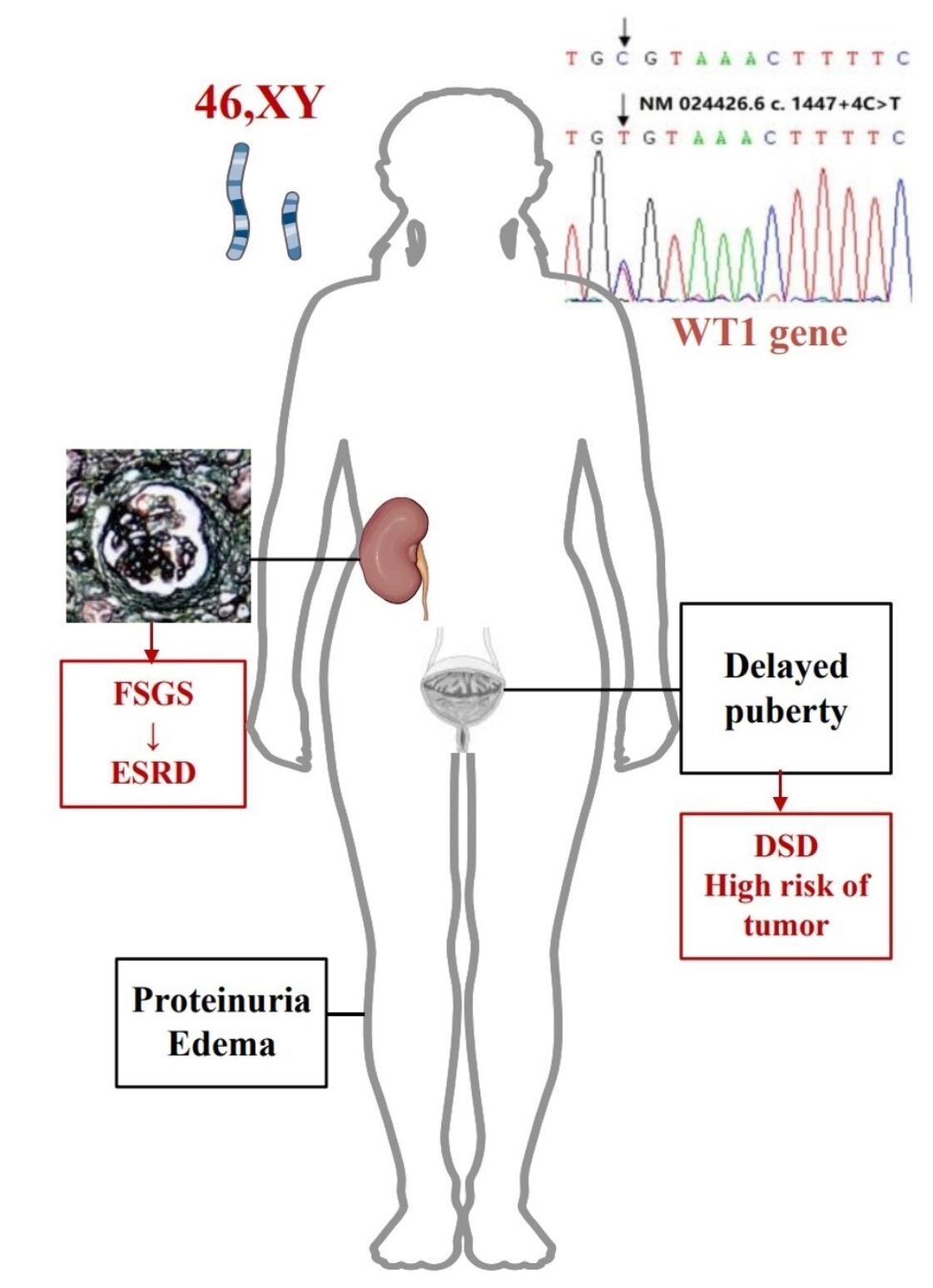{kind=link}
{kind=link}
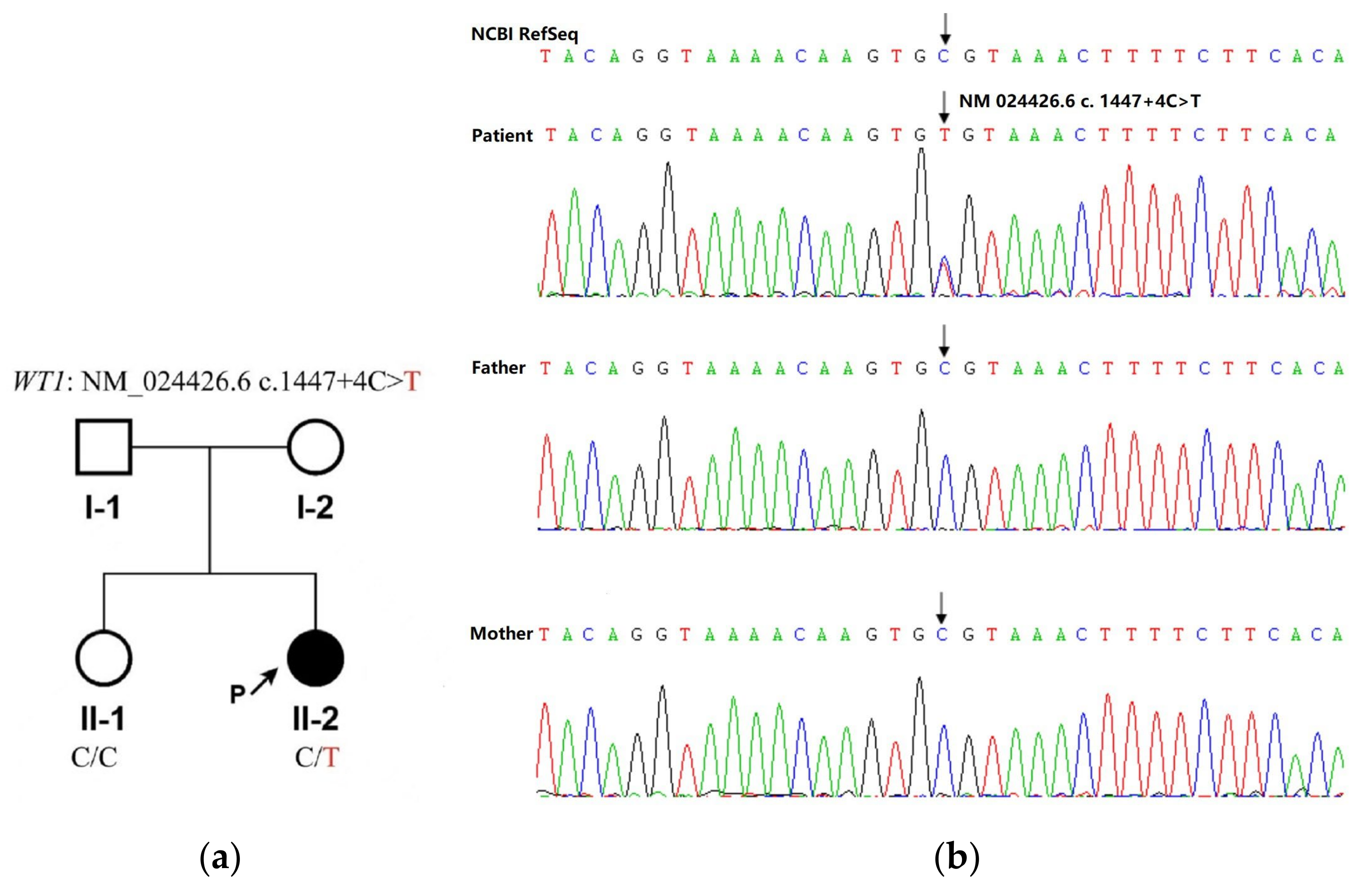{kind=link}
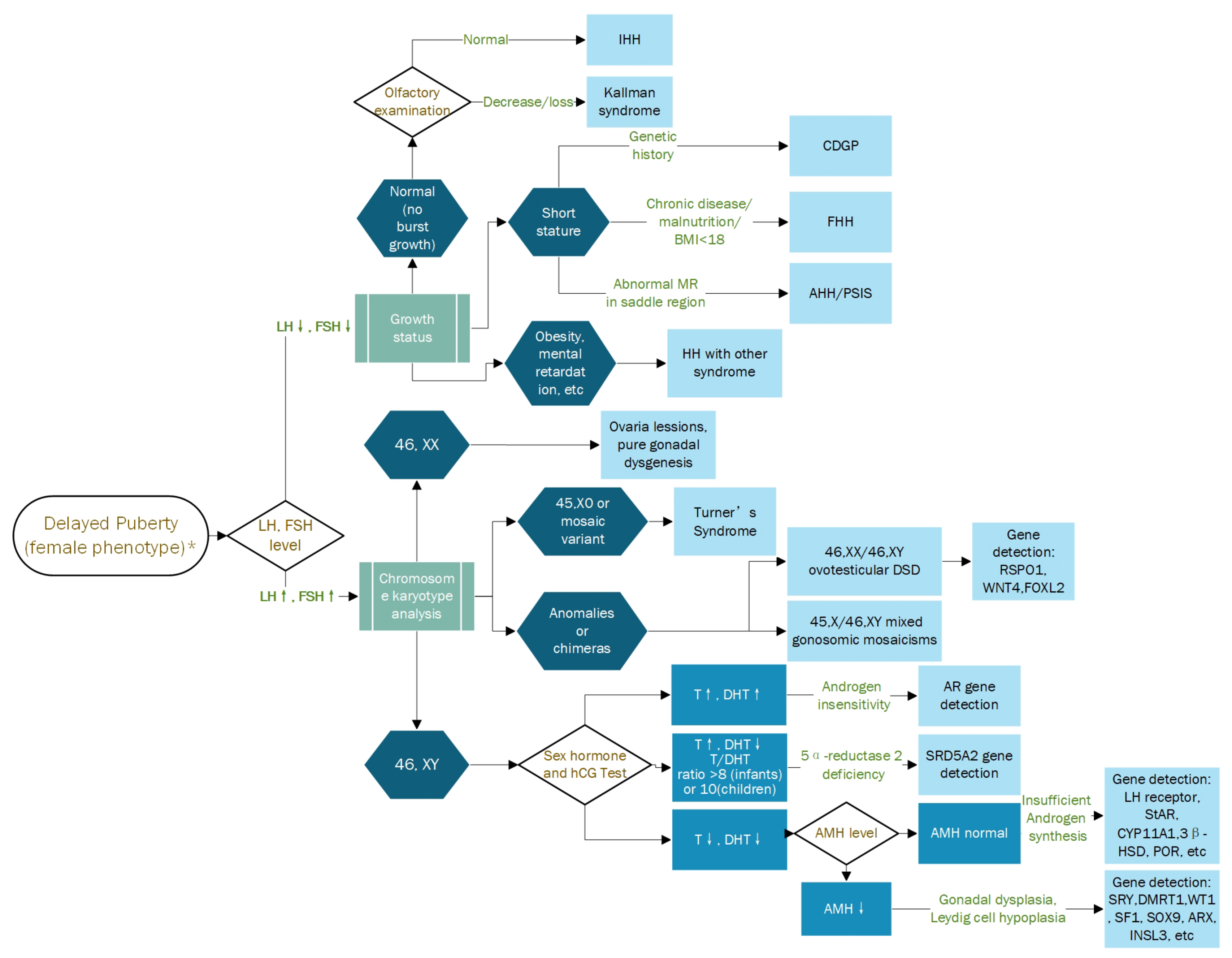{kind=link}
| Sex Hormones | Results 1 | Results 2 | Normal Range |
|---|---|---|---|
| PRL (ng/mL) | / | 39.60 | 6.0–29.9 |
| DHEAS (umol/L) | / | 1.310 | 1.77–9.99 |
| LH (IU/L) | 76.19 | 171.0 | 7.7–58.8 |
| FSH (IU/L) | 153.9 | >200.0 | 25.8–134.8 |
| E2 (pg/mL) | 5.3 | <5.0 | <138 |
| P (ng/mL) | 0.16 | 0.23 | <0.126 |
| T (ng/mL) | 0.06 | 0.025 | 0.03–0.27 |
| DHT (ng/mL) | / | 0.350 | / |
| AMH (ng/mL) | / | 1.05 | 2–6.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, Q.; Xie, X.; Geng, J.; Yang, X.; Li, W.; Zhang, Y. Frasier Syndrome: A 15-Year-Old Phenotypically Female Adolescent Presenting with Delayed Puberty and Nephropathy. Children 2023, 10, 577. https://doi.org/10.3390/children10030577
Shao Q, Xie X, Geng J, Yang X, Li W, Zhang Y. Frasier Syndrome: A 15-Year-Old Phenotypically Female Adolescent Presenting with Delayed Puberty and Nephropathy. Children. 2023; 10(3):577. https://doi.org/10.3390/children10030577
Chicago/Turabian StyleShao, Qing, Xinglei Xie, Jia Geng, Xiaoling Yang, Wei Li, and Yuwei Zhang. 2023. "Frasier Syndrome: A 15-Year-Old Phenotypically Female Adolescent Presenting with Delayed Puberty and Nephropathy" Children 10, no. 3: 577. https://doi.org/10.3390/children10030577