
Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability

1
Division of Pediatric Neurology, Department of Pediatrics, McGill University, Montreal, QC H4A 3J1, Canada
2
National Neuroscience Institute, King Fahad Medical City, Riyadh 12231, Saudi Arabia
3
Division of Child Neurology, Department of Neurology and Neurosurgery, Federal University of São Paulo, São Paulo 04024-002, Brazil
4
Research Institute of the McGill University Health Center, Montreal, QC H4A 3J1, Canada
*
Author to whom correspondence should be addressed.
Children 2023, 10(3), 414; https://doi.org/10.3390/children10030414
Submission received: 29 December 2022
/
Revised: 11 February 2023
/
Accepted: 16 February 2023
/
Published: 21 February 2023
(This article belongs to the Special Issue Genetic Diagnosis in Children with Developmental Delay)
Abstract
:Global Developmental Delay (GDD) and Intellectual Disability (ID) are two of the most common presentations encountered by physicians taking care of children. GDD/ID is classified into non-syndromic GDD/ID, where GDD/ID is the sole evident clinical feature, or syndromic GDD/ID, where there are additional clinical features or co-morbidities present. Careful evaluation of children with GDD and ID, starting with detailed history followed by a thorough examination, remain the cornerstone for etiologic diagnosis. However, when initial history and examination fail to identify a probable underlying etiology, further genetic testing is warranted. In recent years, genetic testing has been shown to be the single most important diagnostic modality for clinicians evaluating children with non-syndromic GDD/ID. In this review, we discuss different genetic testing currently available, review common underlying copy-number variants and molecular pathways, explore the recent evidence and recommendations for genetic evaluation and discuss an approach to the diagnosis and management of children with non-syndromic GDD and ID.
1. Introduction
Global developmental delay (GDD) and intellectual disability (ID) are frequent referrals for clinical evaluation by pediatricians, neurologists, and geneticists. GDD is defined as a delay of two standard deviations below the mean in two or more developmental domains (e.g., motor [gross/fine], speech/language [expressive, receptive, mixed], cognition, personal–social, activities of daily living) [1] and is used in reference to children below the age of 5 years. ID refers to deficits in intellectual (such as in reasoning, verbal comprehension, abstract thought, comprehending instructions and rules, memory, problem-solving and learning from experience) or adaptive functioning evident in early childhood [1]. Neurodevelopmental disorders (NDD) are a group of brain-based conditions that first manifest in early childhood, that are characterized by evident impairments or limitations in personal, social, academic or occupational functioning; that encompass GDD and ID; and that include other entities such as autism, cerebral palsy, developmental language impairment, developmental coordination disorder, learning disability and attention deficit and hyperactivity disorder [1].
In the United States and Canada, 3–5% of children meet the criteria for GDD, and 2–3% eventually fulfill the criteria of ID when they are more than 7 years of age and when standardized intelligent quotient (IQ) testing can reliably diagnose ID [2]. The prevalence of ID is variable across different global populations, spectrum of severity and socioeconomic levels [3,4,5]. In a recent study, the global prevalence of ID was found to have decreased from 1.74% in 1990 to 1.39% in 2019 [5]. Given the overall increase in the global population, the total number of individuals with ID was larger overtime [5]. There is a higher proportion of individuals with ID in the regions of low–middle socio-demographic index compared with high socio-demographic index (five–six times higher) [5]. The higher prevalence of ID in developing countries and low socioeconomic areas was postulated to be due to higher exposure to risk factors and environmental influences [5].
Many children first diagnosed with GDD will later be diagnosed with ID, and conversely, many children with ID were first diagnosed with GDD. Approximately two-thirds of individuals have mild to moderate ID, and one-third have severe to profound ID. GDD/ID is often classified as either syndromic or non-syndromic (or isolated), whereby patients with syndromic GDD/ID present with additional clinical features (e.g., dysmorphic features, epilepsy, ataxia or focal neurological/motor deficit), malformations or co-morbidities (e.g., congenital malformations, growth abnormalities or involvement of other systems), including metabolic manifestations, while patients with non-syndromic GDD/ID have GDD/ID as the sole clinical feature [3,6]. The distinction between syndromic and non-syndromic GDD/ID can be relatively arbitrary, as additional manifestations may be subtle or overlooked, or may evolve and become more evident over time.
The etiology of GDD/ID is very heterogenous and encompasses both genetic and acquired causes. Hypoxia, infections and exposure to various environmental toxins (e.g., alcohol, illicit drugs and heavy metals) during pregnancy, in the perinatal or postnatal period underly between 11 and 55% of GDD/ID [7,8,9,10,11]. Genetic causes, identified in up to 47% [10,11], include chromosomal abnormalities/rearrangements (e.g., aneuploidies), small copy number variants (CNV) and monogenic causes with more than 1000 involved genes thus far identified [12,13,14]. All types of inheritance patterns (autosomal dominant, autosomal recessive and X-linked) are associated with GDD/ID. In a large cohort of individuals from an outbred population with unexplained NDDs, where GDD/ID constituted the most common phenotype, de novo pathogenic variants (i.e., absent in the parents) were found in 42–48% of affected individuals [15], and autosomal recessive causes were responsible for only 3.6% [15]. In contrast, autosomal recessive causes underlie larger proportions of GDD/ID in populations where consanguinity is common, estimated at 51 to 80% [16,17,18,19].
A specific and timely molecular diagnosis in GDD/ID is of great value to patients, families and treating physicians, as it allows a definite diagnosis for the patient, an end of the often-prolonged diagnostic odyssey, improved prognostication, as well as more accurate genetic counseling, estimation of recurrence risk and prenatal/preimplantation genetic diagnosis [20]. In addition, a specific diagnosis can result in improved care and management, with a modification of targeted surveillance efforts and an avoidance of unnecessary investigations. Furthermore, it may improve a patient’s and family’s access to community services and the evolving network of rare diseases support groups [21].
Guidelines for the genetic evaluation of individuals with GDD/ID are similar, though not homogenous. Most of the recommendations were developed prior to the wide-spread clinical availability of next-generation sequencing-based tests such as exome sequencing and comprehensive gene panels. Almost all professional organizations offering guidelines (American College of Medical Genetics, American Academy of Neurology, Child Neurology Society, American Academy of Pediatrics, American Academy of Child and Adolescent Psychiatry) have consistently recommended chromosomal microarray (CMA) and Fragile X testing in as a first-tier test for children with GDD/ID, especially when no specific syndrome or disorder can be readily delineated [22,23,24,25]. However, there are wide inconsistencies with recommendations related to biochemical screening for inherited metabolic disorders (IMDs) associated with GDD/ID [10,24,26,27].
In this review, we will discuss the available genetic tests, their yields as well as the common molecular pathways underlying GDD/ID disorders. We will also outline an approach to genetic testing and management in children with non-syndromic GDD/ID.
2. Genetic and Metabolic Investigations in Children with GDD/ID
2.1. Chromosomal Microarray
Chromosomal microarray (CMA) uses comparative genomic hybridization or single nucleotide polymorphism (SNP) testing to detect chromosomal copy number variants, i.e., gains and losses of chromosomal material. CMA can detect CNVs as small as 20–50 kb and has almost completely replaced the use of karyotyping. However, CMA cannot detect balanced chromosomal rearrangements and has a limited ability to detect low level mosaicism [28,29,30]. CNVs of uncertain established significance can be difficult to interpret. Furthermore, CMA may also detect incidental findings, unrelated to the primary indication. Finally, there are also individual susceptibility CNVs, with documented variable expressivity and/or incomplete penetrance, which are also frequently inherited [31,32,33]. The clinical utility of these susceptibility CNVs is not yet well-established [32].
The diagnostic yield of CMA in NDD overall, including GDD/ID, ranges between 10 and 20% [34,35,36,37]. The diagnostic yield of CMA is lower in individuals with mild vs. moderate to severe ID, with reported yields of 12–19% vs. 20–30% [38,39,40]. A few studies have explored CMA diagnostic yield specifically in non-syndromic GDD/ID, with a reported yield of 10.9% [41]. Note that karyotype analysis is needed to detect balanced translocations, with a diagnostic yield of 3% in cases of developmental disabilities of unknown cause [42]. All current society guidelines recommend CMA testing as a first line [22,23,24,25].
2.2. Exome Sequencing (ES) and Comprehensive GDD/ID Gene Panels
Exome sequencing is a massively parallel gene sequencing approach, also termed next generation sequencing (NGS), that allows examination of the DNA sequences of most of the protein-encoding exons (~1.5–2% of the genome) of an individual. ES has a few important limitations. Coverage of the exome is not complete and may vary between laboratories and technologies; therefore, not all exons are examined, potentially affecting testing yield. In addition, ES does not reliably detect mosaic variants, exon-level deletions, repetitive sequences, intronic or non-coding variants, mitochondrial DNA, epigenic variants or balanced rearrangements [43].
A recent metanalysis reviewed the diagnostic yield of ES in NDDs and found an overall diagnostic yield of 36% [28], which is well above that of CMA. The yield was 54% in syndromic NDDs and 31% in isolated NDDs [28]. While several studies examined the impact of various clinical features on the diagnostic yield of ES, none reported any statistically significant differences [44,45,46]. Higher yields were observed with abnormal head size (microcephaly and macrocephaly), developmental epileptic encephalopathy and a younger age at presentation [44,47,48,49,50]. In many studies, the yield was equivalent in syndromic and non-syndromic ID [44,45,46]. Periodic re-analysis of ES (every 1–3 years) can enhance diagnostic yield over time by 10–16% [51,52,53].
More recently, small exon-level insertion/deletion (Indel) calling is being incorporated into ES bioinformatic pipelines and has been shown to further increase diagnostic yield. In a recent study, exome-based single nucleotide variant (SNV) and Indel calling combined with exome-based CNV analysis in ES data from patients with NDD, revealed an overall diagnostic yield of 54.0% (35.1% from SNV/Indel and 18.9% from exome based CNV) [54]. A similar study explored diagnostic yield in unexplained DD/ID using exome-based exon-level Indel and CNV analysis, and reported an overall diagnostic yield of 58.8% (41.2% from SNV/Indel constitute and 17.6% CNV) [55].
Comprehensive NGS-based GDD/ID gene panels simultaneously sequence multiple genes (usually over 2000) associated with GDD/ID. A few studies have explored the diagnostic yield of comprehensive GDD/ID gene panels with reported figures of 11–39% [56,57,58,59]. Studies that compared “simulated” panels to ES demonstrated slightly lower diagnostic yields in panels vs. ES [45,60]. Multi-gene panels are usually performed on an exome back-bone, where variants are reported in only selected genes from the exome. Analysis of the remainder of the ES data is at times possible, depending on the providing clinical laboratory.
Trio testing refers to the testing of the proband and both biological parents to help identify and interpret suspected gene variants in the proband. Several studies have reported higher yield from trio testing than from proband-only testing [28,49,61,62,63,64]. Furthermore, additional advantages of trio-based testing include decreased resources for analysis, variant testing in parents and earlier time to definitive diagnosis.
Many of the professional practice guidelines do not currently formally recommend ES testing in GDD/ID, as many were published before ES and NGS testing was readily available in clinical practice. However, ES and comprehensive gene panels are routinely used as an integral part of the genetic evaluation of individuals with GDD/ID. The recent 2021 ACMG guidelines strongly recommend ES as first or second line testing in individuals with GDD/ID [21,28].
Therefore, ES, or comprehensive GDD/ID gene panels, should be considered in the specialty evaluation of children with GDD/ID, given their high yield (above all other testing). Early use of ES in the diagnostic journey, when possible, is recommended. Use will likely be dependent on locally available supports (including financial) and testing resources.
2.3. Genome Sequencing
Genome sequencing (GS) is an NGS approach that determines the sequence of most of the DNA of the entire genome of an individual. The main advantage of GS over ES is the ability to query intronic regions, and its superior ability compared with ES to detect structural rearrangements including small and large CNVs [43,65]. GS is not yet clinically available in most centers, and its use is still mainly restricted to research paradigms. One study demonstrated that the addition of GS to the investigation of patients with GDD/ID following unrevealing initial testing (either CMA, ES or both) had a diagnostic yield of 21% [65]. The yield was 64% if only a CMA had been previously performed and 14% if ES was performed [65].
GS is not yet recommended by any professional organization given its limited availability. However, it is likely that GS will supplant ES and CMV in the foreseeable future as decreasing testing costs and increased accessibility to clinicians emerge.
2.4. Fragile X Syndrome Testing
Fragile X syndrome (FXS) is an X-linked triplet repeat expansion disorder caused by the unstable expansion of CGG repeats in the 5′untranslated region of the FMR1 gene. FXS is one of the most common monogenic causes of GDD/ID with a prevalence of 1.4:10,000 males and 0.9:10,000 females. Though it is characterized by distinctive clinical features, including distinctive facial dysmorphisms (long face, large ears, prominent jaw) and macro-orchidism, these may be subtle or only apparent with entry into puberty. Fragile X testing in a cohort of 2486 individuals with NDD demonstrated a yield of 1.2% [66]. Furthermore, 96% of the FXS-positive cases had clinical features of FXS or a positive family history [66]. The yield has been shown to be significantly higher when testing is restricted to males with NDD with characteristic physical/behavioral features or family history, at 9.5–17% [67,68,69].
FXS testing is widely available. It is important to note that FXS cannot be diagnosed by CMA, ES or gene panels. Given the fact that it is an X-linked condition, prompt diagnosis is critical as it will have important impact on recurrence risk and genetic counseling.
Most professional organizations presently recommend testing for FXS as first line in all individuals with GDD/ID.
2.5. Metabolic/Biochemical Screening for Inherited Metabolic Diseases
Inherited metabolic disorders (IMDs) are genetic disorders that result in metabolic defects due to a deficiency of enzymes, membrane transporters or other functional proteins. Consideration of IMDs when evaluating individuals with GDD/ID is important, especially when early detection can lead to specific treatments that improve clinical outcomes. Over 100 IMDs associated with GDD/ID have potential treatment [26,27]. Furthermore, since these conditions are largely autosomal recessive, diagnosis has as impact on genetic counseling, as risk of recurrence in subsequent pregnancies is often elevated in a Mendelian fashion.
Biochemical screening should be performed in any individual displaying red flags suggestive of an IMD. These include, but are not limited to, developmental plateau or regression in the context of an abnormal exam, altered level of consciousness, observed movement disorders (such as chorea, dystonia, ataxia, and myoclonus), hepatomegaly, splenomegaly, drug-resistant seizures, coarse facial features and multisystemic involvement crossing embryonic origin of affected tissues. Individuals with these features should also be urgently referred to biochemical genetic specialists. Screening metabolic testing may include serum ammonia, lactate, copper, ceruloplasmin, homocysteine, plasma amino acids, urine organic acids, purines, pyrimidines, creatine metabolites, oligosaccharides, and glycosaminoglycan.
There is no current consensus regarding whether biochemical screening for treatable IMDs should be performed in children with non-syndromic GDD/ID in the absence of red flags. Both the Canadian Pediatric Society and the American Academy of Pediatrics recommend the systematic biochemical screening in all children with GDD/ID [10,24,26], while others recommended screening only if clinical features were suggestive of IMDs [23,70,71].
The diagnostic yield of metabolic screening in non-syndromic GDD/ID is extremely low, estimated at 0.25%−0.42% in a non-consanguineous population [71,72]. A review of the implementation of Treatable Intellectual Disability Endeavor (TIDE) screen protocol for cases of GDD/ID without clinical features suggestive of IMDs showed no significant increase in the diagnosis of IMDs, despite a four-fold increase in testing [71].
Given the low yield, routine screening for IMDs is not presently recommended without the presence of clinical features suggestive of IMDs, though should be considered if newborn screening was not previously performed.
3. Common CNVs in Non-Syndromic GDD/ID
Several microdeletions and duplications have been associated with a wide range of phenotypic features in patients with both syndromic and non-syndromic GDD/ID. A list of common CNVs associated with non-syndromic GDD/ID is provided in Table 1.
In addition, several of the microdeletion syndromes that are associated with syndromic GDD/ID, such as Prader–Willi and Angelman syndromes (deletion of 15q11-q13), Williams–Beuren (deletion of 7q11.23) and Smith–Magenis syndrome (deletion of 17p12) [73], may present milder phenotypes with unrecognizable syndromic features. For example, in a recent study that identified pathogenic CNVs in 33 patients with non-syndromic GDD/ID, one individual was found with 15q11.2 deletion but absent characteristic features of Angelman/Prader–Willi syndromes. The same study also showed other frequent aneusomies in chromosomes 15, 16 and X [41].

{kind=link}
Table 1.
Common microdeletions and microduplications associated with non-syndromic GDD/ID.
| Chromosome Region | Deletion or Duplication | Main Clinical Features | Candidate Genes | References |
|---|---|---|---|---|
| 15q11.2 | Deletion/Duplication | ID, schizophrenia, epilepsy | TUBGCP5, CYFIP1, NIPA2, NIPA1 | [74,75,76,77,78,79,80] |
| 15q13.3 | Deletion | ID, epilepsy, schizophrenia, ASD | CHRNA7 | [74,81,82,83,84,85,86] |
| 16p11.2 (distal) | Deletion | ID, ASD, obesity, schizophrenia | SH2B1 | [87,88] |
| 16p11.2 (proximal) | Deletion/Duplication | ID, language delay, ASD, obesity | MVP, CDIPT1, SEZ6L2, ASPHD1, KCTD13 | [89,90,91,92,93,94,95,96] |
| 16p12 | Deletion | Intellectual disability | UQCRC2, EEF2K, POLR3E, CDR2 | [41,97] |
| Xq28 | Duplication | Males: hypotonia, severe GDD and ID, progressive spasticity, seizures, ASD Females: milder phenotype | RAB39B, CLIC2, IRAK1, MECP2, GDI1 | [41,98,99,100,101] |
4. Common Pathways Underlying NDDs
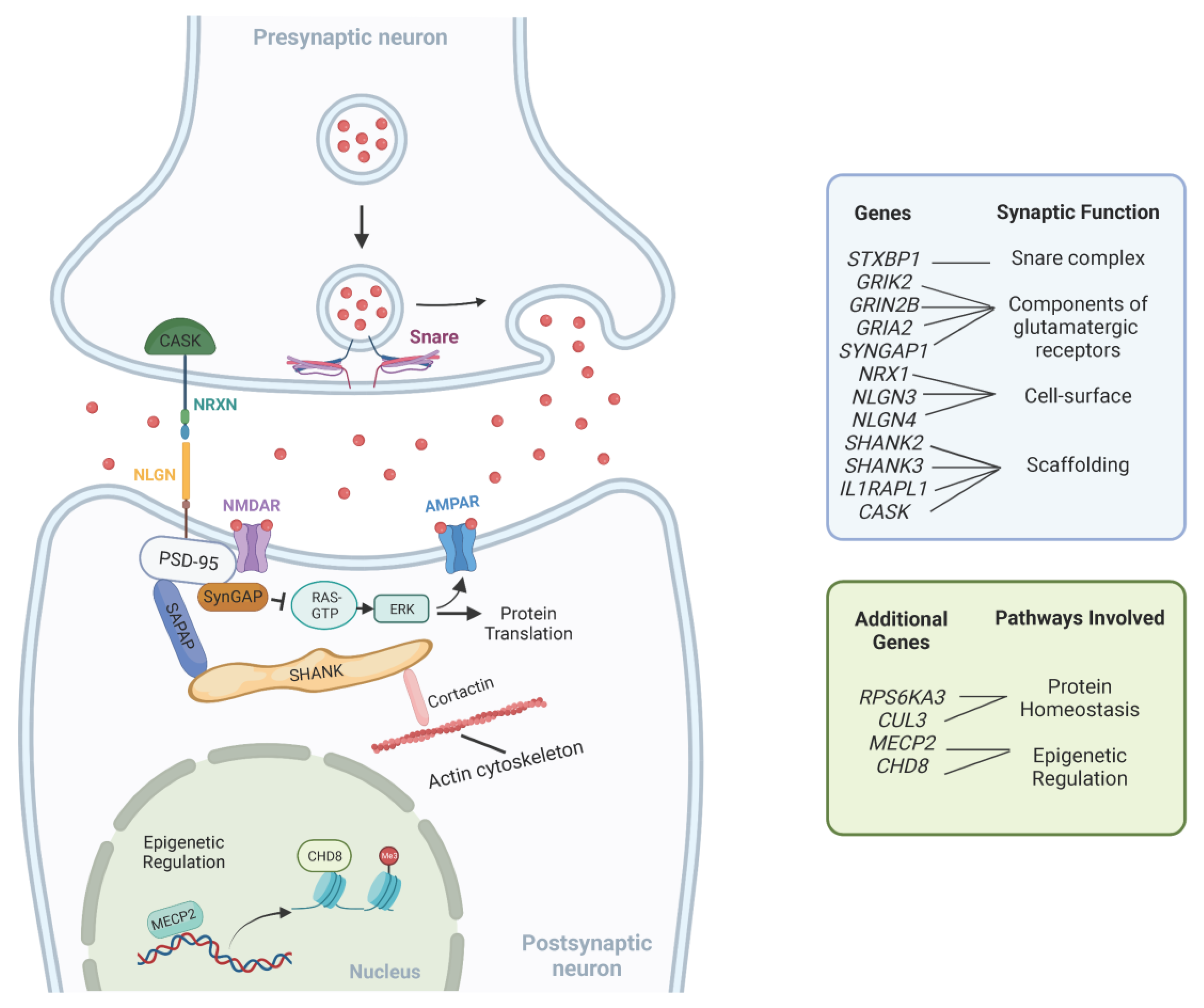
With the current expanding number of genes associated with NDDs, one interesting approach toward a better understanding of their complex genetic heterogeneity is to understand the common pathways in which GDD/ID genes are acting. Since patients with non-syndromic GDD/ID present intellectual impairment as their main clinical feature, it is logical that causative genes function in processes related to learning and memory. Key cellular mechanisms involved in GDD/ID pathogenesis include those related to synaptic structure and signaling, protein homeostasis and epigenetic regulation of transcription [102]. Furthermore, altered neural circuits in multiple brain areas, including the cerebral cortex, basal ganglia and thalamus, have been described in ID/GDD [103]. A summary of these pathways is provided in Table 2. Figure 1 illustrates the interplay between these pathways.
4.1. Synaptic Signaling Dysregulation
Many genes associated with GDD/ID encode molecules with important roles in the assembly, structure, and function of the neuronal synapse.
Synaptic cell-surface proteins, such as the neurexins (NRXN) and neuroligins (NLGN), have important roles in the assembly of functional synapses, and post-synaptic scaffolding proteins, such as members of the SHANK family, have an important role in organizing the architecture and molecular composition of synapses [102]. Their encoding genes, NRX1, NLGN3, NLGN4, SHANK2 and SHANK3, respectively, have been previously associated with a wide range of NDDs, including GDD, ID and autism spectrum disorder (ASD) [104,105,106].
Impaired glutamate excitatory signaling has also been associated with NDDs. The two main ionotropic glutamatergic channels found in the post-synaptic density are α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl D-Aspartate (NMDA) receptors. Mutations in genes encoding components of glutamatergic receptors such as GRIK2, GRIN2B and GRIA2 have been associated with non-syndromic ID and other neurodevelopmental abnormalities, including ASD [107,108,109].
The brain-specific synaptic Ras/Rap GTPase-activating protein, encoded by SYNGAP1, is another critical component of the postsynaptic density [102]. It suppresses the signaling of pathways linked to the NMDA receptor, such as the Ras/ERK pathway [110,111,112]. De novo heterozygous loss-of-function mutations in SYNGAP1 are associated with non-syndromic ID, ASD and epilepsy [113].
The membrane-associated guanylate kinase (MAGUK) proteins are another group of multidomain scaffolding proteins in the postsynaptic density. Impaired synapse formation due to incorrect localization of the MAGUK family protein PSD-95 (DLG4) was reported in association with mutations in the interleukin 1 receptor accessory protein-like 1 (IL1RAPL1) [3]. Pathogenic variants in this gene were also previously associated with non-syndromic ID and ASD [114]. Mutations in another MAGUK family protein gene, CASK, which encodes a calcium/calmodulin-dependent serine protein kinase, were also previously reported in patients with non-syndromic ID [3,115].
NDDs have also been associated with disorders in neurotransmitter release at the pre-synaptic membrane. The SNARE complex is responsible for mediating the fusion of the vesicle with the pre-synaptic membrane and pathogenic variants in STXBP1, one of its subunits, are classically associated with severe infantile epileptic encephalopathy [110,116]. However, variants in this gene have also been reported in individuals with isolated ID [117].
4.2. Protein Homeostasis
Protein homeostasis is a key regulatory process of synaptic plasticity and assembly in cortical and basal ganglia circuits [103].
The phosphatidylinositol 3-kinase (PI3K)-mTOR pathway is major signaling cascade implicated with the emerge of well-known syndromic ID and ASD-related syndromes, such as tuberous sclerosis, Cowden and Fragile X [110].
The ERK/MAPK is another important signaling cascade required for synaptic plasticity [3]. Besides the previously mentioned SynGAP protein that negatively regulates the pathway, mutations in the RPS6KA3 gene, a member of the ribosomal S6 kinase family that functions as a downstream effector of the ERK signaling pathway, are also related to non-syndromic ID [118].
Other findings highlight that deficient protein synthesis is a common mechanism in non-syndromic NDDs. De novo loss-of-function mutations in CUL3, which encodes a component of the Cullin-RING ligase (CRL) complex that controls proteasomal degradation of specific target proteins, have been associated with non-syndromic ASD [119,120]. In mouse models, protein translation deficiencies have also been linked to autism-associated variants in the synaptic adhesion molecule neuroligin-3 (NLGN3). The loss of NLGN3 was accompanied by a disruption of homeostasis in the ventral tegmental area [121].
4.3. Epigenetic Regulation
Increasing evidence supports the dysregulation of transcriptional and epigenetic control of gene expression as mechanisms underlying both syndromic and non-syndromic NDDs [103]. DNA methylation and histone post-translational modification (e.g., acetylation, methylation and phosphorylation) are the main molecular changes implicated in the epigenetic regulation of transcription [110].
MECP2 encodes methyl CpG-binding protein 2 and is believed to act as a transcriptional modulator by binding methylated CpG DNA [110]. Although traditionally associated with Rett syndrome, mutations in MECP2 are also described in sporadic cases of autism [122,123]. Mutations in chromatin regulators, such as the chromodomain helicase DNA-binding protein 8 (CHD8) gene, have also been associated with ID and ASD [124].
4.4. Thalamic and Peripheral Circuits
Altered tactile sensitivity is a common feature in individuals with GDD/ID and autistic features. Thalamic circuits are responsible for processing sensory signaling from the periphery to the cerebral cortex. Following that idea, autistic symptoms found in individuals with GDD/ID are now being related to thalamic dysfunction [103].
Loss of function mutations in the patched domain containing protein 1 (PTCHD1) were previously associated with an X-linked neurodevelopmental disorder with a strong propensity to autistic behaviors [125], suggesting that this protein has a significant role in cognitive and attentional control. Interestingly, enriched expression of the PTCHD1 in the thalamic reticular nucleus of mice was associated with inhibitory inputs to the thalamus [103]. In addition, another study showed that mice with ASD-linked genes (including previously mentioned MECP2 and SHANK3) deleted selectively from mechanosensory neurons presented altered tactile sensitivity and discrimination [126]. SHANK3 and MECP2 variants have also been shown to be responsible for alterations in basal ganglia circuits in animal models [127,128,129,130].
5. Diagnostic Approach to the Evaluation of Children with Non-Syndromic GDD/ID
An overview of the evaluation of individuals with non-syndromic GDD/ID is presented in Table 3.
Careful history taking and thorough clinical examination is the first step in the evaluation of children with GDD/ID, with particular attention to the identification of an acquired etiology and recognition of clinical features that could be suggestive of a syndromic cause. A detailed three-generation family history, prenatal and birth history as well as neurodevelopmental histories should be obtained. If a clear acquired cause is identified, further genetic testing is not warranted. If a known syndrome is recognized based on the clinical features (e.g., Rett syndrome or Zellweger syndrome), then targeted genetic or metabolic testing should be ordered first. It is important to highlight that individuals with an underlying genetic disorder are at a higher risk of also having an additional acquired neurologic insult; for example, children with neuromuscular disorders and hypotonia are at a higher risk of neonatal asphyxia. Therefore, clinicians should keep in mind that both acquired and genetic etiologies may co-occur in some patients.
Formal visual and hearing assessment should be performed in all children with GDD/ID due to the high frequency of primary sensory impairments and their potential for amelioration and remediation [10,131,132,133].
Brain imaging should be performed in the presence of abnormal head circumference, rapid change in head circumference over time, focal neurological findings or focal epilepsy [134]. Brain imaging is more likely to detect abnormalities when MRI is performed in individuals with GDD/ID with additional clinical or neurological signs [135,136]. In one study, abnormal imaging findings were observed in 41% of individuals when selective imaging was performed compared with a yield of 14% with non-selective screening [137]. MRI brain is generally preferred when compared with CT brain given its higher sensitivity [23,24,132]. Most professional organizations recommend brain imaging especially when other neurological findings are present [24]. The addition of MR Spectroscopy can be considered to aid in the diagnosis of suspected mitochondrial disorders or cerebral creatine deficiency syndrome.
Screening for IMDs is not presently recommended for non-syndromic GDD/ID in the absence of red flags. Metabolic screening should be considered in children who have not undergone newborn metabolic screening, as performed in many countries.
Genetic testing should be performed in all children with GDD/ID in whom an initial evaluation did not uncover an etiology. All individuals should undergo CMA testing and ES. When ES is not available, a comprehensive GDD/ID gene panel is a good substitute. Trio testing is preferred given easier analysis and no need to obtain additional samples for variant segregation. FXS testing also should be performed in all patients. CMA and FXS testing are generally recommended as first-tier as they are easily accessible, less costly and can be ordered by primary providers. ES, given its higher cost and more limited accessibility to non-specialists and non-geneticists, is usually ordered as a second-tier test upon specialty evaluation.
6. Overview of Management Principles for Children with GDD/ID
The comprehensive care of individuals with GDD/ID is based on a family-centered, multidisciplinary approach. Multiple medical expertises and different subspecialists, including pediatricians, pediatric neurologists, geneticists, psychiatrists, family practitioners, physiatrists and orthopedic surgeons, amongst others, are often necessary to provide ideal care [138]. Furthermore, the involvement of occupational therapists, physiotherapists, psychologists, special educators, nurses and social workers is fundamental, and enables the minimization of impairments and possible complications, and the maximization of the activity, participation, health and well-being of patients [139]. The challenges and needs of the patients evolve over their lifespan [140].
Medical issues that are relatively frequently encountered in children with GDD/ID include epilepsy, behavioral challenges, sleep disturbances, movement disorders and orthopedic deformities [140].
General principles for antiepileptic drug treatment include selection of the drug based on seizure type, avoidance of polypharmacy and minimal use of sedating or cognitively depressing antiepileptic drugs, as well as those that can exacerbate pre-existing behavioral challenges (i.e., Levetiracetam) [110].
Behavioral challenges possibly represent the most difficult issue for caregivers. Psychologic/psychiatric intervention is frequently the first line of approach recommended, but pharmacologic treatment can be necessary for handling disorders of attention and impulsivity (with stimulant or non-stimulant alternatives), agitation, opposition, disinhibition and aggression towards both others and self, in which cases atypical antipsychotics have shown a good response [110].
Sleep disturbances are highly prevalent in children with neurodevelopmental disabilities, and significantly impact the children and the family’s quality of life. Proper sleep hygiene is a first step, followed by the use of melatonin, and, if needed, other medications such as gabapentin, clonidine and trazodone [141].
Movement disorders may include prominent spasticity with possible progression to orthopedic deformities. Physiotherapy is often a good preventive measure, but in those cases with refractory pain or functional limitations intramuscular botulin toxin injections and oral antispasmodic agents could be helpful.
7. Conclusions
While detailed history and clinical examination remains a cornerstone in the evaluation of children with GDD/ID, genetic testing is essential and key in individuals in whom the etiology is not readily apparent. In the era of genomic first approach, the underlying genetic causes of non-syndromic GDD/ID can be explored with a wide variety of diagnostic assessments and result in high etiologic yields, allowing a specific genetic diagnosis. As technologies progress and evidence emerges, approaches regarding genetic testing will evolve over time. Reaching a specific genetic diagnosis provides many advantages to patients and families, including an end of the diagnostic odyssey, precise genetic counseling and optimized longitudinal management and targeted surveillance. Though targeted treatments and gene therapies are limited in GDD/ID, advances in this area are intensively being investigated and their application in the clinical setting is anxiously awaited.
Author Contributions
Writing—original draft preparation, R.A.; writing—review and editing, R.A, M.M., M.I.S. and M.S.; supervision, M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This review received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
M Srour holds a salary award from the Fonds de Recherche de Santé Quebec.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Diagnostic and Statistical Manual of Mental Disorders DSM-5. Available online: https://doi.org/10.1176/appi.books.9780890425596 (accessed on 29 November 2022).
- Michelson, D.J.; Clark, R.D. Optimizing Genetic Diagnosis of Neurodevelopmental Disorders in the Clinical Setting. Clin. Lab. Med. 2020, 40, 231–256. [Google Scholar] [CrossRef]
- Kaufman, L.; Ayub, M.; Vincent, J.B. The Genetic Basis of Non-Syndromic Intellectual Disability: A Review. J. Neurodev. Disord. 2010, 2, 182–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiurazzi, P.; Pirozzi, F. Advances in Understanding—Genetic Basis of Intellectual Disability. F1000Research 2016, 5, 599. [Google Scholar] [CrossRef]
- Nair, R.; Chen, M.; Dutt, A.S.; Hagopian, L.; Singh, A.; Du, M. Significant Regional Inequalities in the Prevalence of Intellectual Disability and Trends from 1990 to 2019: A Systematic Analysis of GBD 2019. Epidemiol. Psychiatr. Sci. 2022, 31, e91. [Google Scholar] [CrossRef] [PubMed]
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld, M.A.W.; Kleefstra, T.; Kramer, J.M.; et al. Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. Am. J. Hum. Genet. 2016, 98, 149–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichenberg, A.; Cederlöf, M.; McMillan, A.; Trzaskowski, M.; Kapra, O.; Fruchter, E.; Ginat, K.; Davidson, M.; Weiser, M.; Larsson, H.; et al. Discontinuity in the Genetic and Environmental Causes of the Intellectual Disability Spectrum. Proc. Natl. Acad. Sci. USA 2016, 113, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Modabbernia, A.; Mollon, J.; Boffetta, P.; Reichenberg, A. Impaired Gas Exchange at Birth and Risk of Intellectual Disability and Autism: A Meta-Analysis. J. Autism. Dev. Disord. 2016, 46, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- McDermott, S.; Bao, W.; Tong, X.; Cai, B.; Lawson, A.; Aelion, C.M. Are Different Soil Metals near the Homes of Pregnant Women Associated with Mild and Severe Intellectual Disability in Children? Dev. Med. Child Neurol. 2014, 56, 888–897. [Google Scholar] [CrossRef] [Green Version]
- Bélanger, S.A.; Caron, J. Evaluation of the Child with Global Developmental Delay and Intellectual Disability. Paediatr. Child Health 2018, 23, 403–419. [Google Scholar] [CrossRef]
- Jimenez-Gomez, A.; Standridge, S.M. A Refined Approach to Evaluating Global Developmental Delay for the International Medical Community. Pediatr. Neurol. 2014, 51, 198–206. [Google Scholar] [CrossRef]
- Aspromonte, M.C.; Bellini, M.; Gasparini, A.; Carraro, M.; Bettella, E.; Polli, R.; Cesca, F.; Bigoni, S.; Boni, S.; Carlet, O.; et al. Characterization of Intellectual Disability and Autism Comorbidity through Gene Panel Sequencing. Hum. Mutat. 2019, 40, 1346–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.Y.; Jang, J.H.; Park, J.; Lee, I.G. Targeted Next-Generation Sequencing of Korean Patients With Developmental Delay and/or Intellectual Disability. Front. Pediatr. 2018, 6, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, C.; Race, V.; Keldermans, L.; Bauters, M.; Van Esch, H. Challenges in Molecular Diagnosis of X-Linked Intellectual Disability. Br. Med. Bull. 2020, 133, 36–48. [Google Scholar] [CrossRef]
- Deciphering Developmental Disorders Study. Prevalence and Architecture of de Novo Mutations in Developmental Disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Monies, D.; Abouelhoda, M.; Assoum, M.; Moghrabi, N.; Rafiullah, R.; Almontashiri, N.; Alowain, M.; Alzaidan, H.; Alsayed, M.; Subhani, S.; et al. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 2019, 104, 1182–1201. [Google Scholar] [CrossRef] [Green Version]
- Harripaul, R.; Vasli, N.; Mikhailov, A.; Rafiq, M.A.; Mittal, K.; Windpassinger, C.; Sheikh, T.I.; Noor, A.; Mahmood, H.; Downey, S.; et al. Mapping Autosomal Recessive Intellectual Disability: Combined Microarray and Exome Sequencing Identifies 26 Novel Candidate Genes in 192 Consanguineous Families. Mol. Psychiatry 2018, 23, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep Sequencing Reveals 50 Novel Genes for Recessive Cognitive Disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef]
- Anazi, S.; Maddirevula, S.; Faqeih, E.; Alsedairy, H.; Alzahrani, F.; Shamseldin, H.E.; Patel, N.; Hashem, M.; Ibrahim, N.; Abdulwahab, F.; et al. Clinical Genomics Expands the Morbid Genome of Intellectual Disability and Offers a High Diagnostic Yield. Mol. Psychiatry 2017, 22, 615–624. [Google Scholar] [CrossRef]
- Tan, T.Y.; Dillon, O.J.; Stark, Z.; Schofield, D.; Alam, K.; Shrestha, R.; Chong, B.; Phelan, D.; Brett, G.R.; Creed, E.; et al. Diagnostic Impact and Cost-Effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. JAMA Pediatr. 2017, 171, 855–862. [Google Scholar] [CrossRef]
- Manickam, K.; McClain, M.R.; Demmer, L.A.; Biswas, S.; Kearney, H.M.; Malinowski, J.; Massingham, L.J.; Miller, D.; Yu, T.W.; Hisama, F.M.; et al. Exome and Genome Sequencing for Pediatric Patients with Congenital Anomalies or Intellectual Disability: An Evidence-Based Clinical Guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.; Hudgins, L. Professional Practice and Guidelines Committee Array-Based Technology and Recommendations for Utilization in Medical Genetics Practice for Detection of Chromosomal Abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelson, D.J.; Shevell, M.I.; Sherr, E.H.; Moeschler, J.B.; Gropman, A.L.; Ashwal, S. Evidence Report: Genetic and Metabolic Testing on Children with Global Developmental Delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2011, 77, 1629–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeschler, J.B.; Shevell, M. Committee on Genetics Comprehensive Evaluation of the Child with Intellectual Disability or Global Developmental Delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhle, R.A.; Reed, H.E.; Vo, L.C.; Mehta, S.; McGuire, K.; Veenstra-VanderWeele, J.; Pedapati, E. Clinical Diagnostic Genetic Testing for Individuals With Developmental Disorders. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 910–913. [Google Scholar] [CrossRef]
- van Karnebeek, C.D.M.; Stockler, S. Treatable Inborn Errors of Metabolism Causing Intellectual Disability: A Systematic Literature Review. Mol. Genet. Metab. 2012, 105, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Hoytema van Konijnenburg, E.M.M.; Wortmann, S.B.; Koelewijn, M.J.; Tseng, L.A.; Houben, R.; Stöckler-Ipsiroglu, S.; Ferreira, C.R.; van Karnebeek, C.D.M. Treatable Inherited Metabolic Disorders Causing Intellectual Disability: 2021 Review and Digital App. Orphanet J. Rare Dis. 2021, 16, 170. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-Analysis and Multidisciplinary Consensus Statement: Exome Sequencing Is a First-Tier Clinical Diagnostic Test for Individuals with Neurodevelopmental Disorders. Genet. Med. 2019, 21, 2413–2421. [Google Scholar] [CrossRef] [Green Version]
- Savatt, J.M.; Myers, S.M. Genetic Testing in Neurodevelopmental Disorders. Front. Pediatr. 2021, 9, 526779. [Google Scholar] [CrossRef]
- Waggoner, D.; Wain, K.E.; Dubuc, A.M.; Conlin, L.; Hickey, S.E.; Lamb, A.N.; Martin, C.L.; Morton, C.C.; Rasmussen, K.; Schuette, J.L.; et al. Yield of Additional Genetic Testing after Chromosomal Microarray for Diagnosis of Neurodevelopmental Disability and Congenital Anomalies: A Clinical Practice Resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 1105–1113. [Google Scholar] [CrossRef]
- Gillentine, M.A.; Lupo, P.J.; Stankiewicz, P.; Schaaf, C.P. An Estimation of the Prevalence of Genomic Disorders Using Chromosomal Microarray Data. J. Hum. Genet. 2018, 63, 795–801. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Coe, B.P.; Eichler, E.E.; Cuckle, H.; Shaffer, L.G. Estimates of Penetrance for Recurrent Pathogenic Copy-Number Variations. Genet. Med. 2013, 15, 478–481. [Google Scholar] [CrossRef]
- Smajlagić, D.; Lavrichenko, K.; Berland, S.; Helgeland, Ø.; Knudsen, G.P.; Vaudel, M.; Haavik, J.; Knappskog, P.M.; Njølstad, P.R.; Houge, G.; et al. Population Prevalence and Inheritance Pattern of Recurrent CNVs Associated with Neurodevelopmental Disorders in 12,252 Newborns and Their Parents. Eur. J. Hum. Genet. 2021, 29, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.; Kim, Y.; Han, E.; Park, J.; Chae, H.; Kwon, A.; Choi, H.; Kim, J.; Son, J.O.; Lee, S.J.; et al. Chromosomal Microarray Analysis as a First-Tier Clinical Diagnostic Test in Patients With Developmental Delay/Intellectual Disability, Autism Spectrum Disorders, and Multiple Congenital Anomalies: A Prospective Multicenter Study in Korea. Ann. Lab. Med. 2019, 39, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.S.W.; Chan, K.Y.K.; Leung, K.K.P.; Au, P.K.C.; Tam, W.-K.; Li, S.K.M.; Luk, H.-M.; Kan, A.S.Y.; Chung, B.H.Y.; Lo, I.F.M.; et al. Experience of Chromosomal Microarray Applied in Prenatal and Postnatal Settings in Hong Kong. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.; Doccini, V.; Bernardini, L.; Novelli, A.; Loddo, S.; Capalbo, A.; Filippi, T.; Carey, J.C. Confirmation of Chromosomal Microarray as a First-Tier Clinical Diagnostic Test for Individuals with Developmental Delay, Intellectual Disability, Autism Spectrum Disorders and Dysmorphic Features. Eur. J. Paediatr. Neurol. 2013, 17, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Bartnik, M.; Wiśniowiecka-Kowalnik, B.; Nowakowska, B.; Smyk, M.; Kędzior, M.; Sobecka, K.; Kutkowska-Kaźmierczak, A.; Klapecki, J.; Szczałuba, K.; Castañeda, J.; et al. The Usefulness of Array Comparative Genomic Hybridization in Clinical Diagnostics of Intellectual Disability in Children. Dev. Period Med. 2014, 18, 307–317. [Google Scholar]
- Fan, Y.; Wu, Y.; Wang, L.; Wang, Y.; Gong, Z.; Qiu, W.; Wang, J.; Zhang, H.; Ji, X.; Ye, J.; et al. Chromosomal Microarray Analysis in Developmental Delay and Intellectual Disability with Comorbid Conditions. BMC Med. Genom. 2018, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- D’Arrigo, S.; Gavazzi, F.; Alfei, E.; Zuffardi, O.; Montomoli, C.; Corso, B.; Buzzi, E.; Sciacca, F.L.; Bulgheroni, S.; Riva, D.; et al. The Diagnostic Yield of Array Comparative Genomic Hybridization Is High Regardless of Severity of Intellectual Disability/Developmental Delay in Children. J. Child Neurol. 2016, 31, 691–699. [Google Scholar] [CrossRef]
- Oğuz, S.; Arslan, U.E.; Kiper, P.Ö.Ş.; Alikaşifoğlu, M.; Boduroğlu, K.; Utine, G.E. Diagnostic Yield of Microarrays in Individuals with Non-Syndromic Developmental Delay and Intellectual Disability. J. Intellect. Disabil. Res. 2021, 65, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D.; Quality Standards Subcommittee of the American Academy of Neurology; et al. Practice Parameter: Evaluation of the Child with Global Developmental Delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Corominas, J.; Smeekens, S.P.; Nelen, M.R.; Yntema, H.G.; Kamsteeg, E.-J.; Pfundt, R.; Gilissen, C. Clinical Exome Sequencing-Mistakes and Caveats. Hum. Mutat. 2022, 43, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, D.; Heeley, J.; Vineyard, M.; Manwaring, L.; Toler, T.L.; Fassi, E.; Fiala, E.; Brown, S.; Goss, C.W.; Willing, M.; et al. The Exome Clinic and the Role of Medical Genetics Expertise in the Interpretation of Exome Sequencing Results. Genet. Med. 2017, 19, 1040–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, O.J.; Lunke, S.; Stark, Z.; Yeung, A.; Thorne, N.; Melbourne Genomics Health Alliance; Gaff, C.; White, S.M.; Tan, T.Y. Exome Sequencing Has Higher Diagnostic Yield Compared to Simulated Disease-Specific Panels in Children with Suspected Monogenic Disorders. Eur. J. Hum. Genet. 2018, 26, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Gieldon, L.; Mackenroth, L.; Kahlert, A.-K.; Lemke, J.R.; Porrmann, J.; Schallner, J.; von der Hagen, M.; Markus, S.; Weidensee, S.; Novotna, B.; et al. Diagnostic Value of Partial Exome Sequencing in Developmental Disorders. PLoS ONE 2018, 13, e0201041. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.; Carlson, M. Whole Exome Sequencing in Pediatric Neurology Patients: Clinical Implications and Estimated Cost Analysis. J. Child Neurol. 2016, 31, 887–894. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular Findings among Patients Referred for Clinical Whole-Exome Sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [Green Version]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical Application of Whole-Exome Sequencing across Clinical Indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Palmer, E.E.; Schofield, D.; Shrestha, R.; Kandula, T.; Macintosh, R.; Lawson, J.A.; Andrews, I.; Sampaio, H.; Johnson, A.M.; Farrar, M.A.; et al. Integrating Exome Sequencing into a Diagnostic Pathway for Epileptic Encephalopathy: Evidence of Clinical Utility and Cost Effectiveness. Mol. Genet. Genomic. Med. 2018, 6, 186–199. [Google Scholar] [CrossRef]
- Wenger, A.M.; Guturu, H.; Bernstein, J.A.; Bejerano, G. Systematic Reanalysis of Clinical Exome Data Yields Additional Diagnoses: Implications for Providers. Genet. Med. 2017, 19, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Al-Nabhani, M.; Al-Rashdi, S.; Al-Murshedi, F.; Al-Kindi, A.; Al-Thihli, K.; Al-Saegh, A.; Al-Futaisi, A.; Al-Mamari, W.; Zadjali, F.; Al-Maawali, A. Reanalysis of Exome Sequencing Data of Intellectual Disability Samples: Yields and Benefits. Clin. Genet. 2018, 94, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Basel-Salmon, L.; Orenstein, N.; Markus-Bustani, K.; Ruhrman-Shahar, N.; Kilim, Y.; Magal, N.; Hubshman, M.W.; Bazak, L. Improved Diagnostics by Exome Sequencing Following Raw Data Reevaluation by Clinical Geneticists Involved in the Medical Care of the Individuals Tested. Genet. Med. 2019, 21, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Zhang, Z.; Shi, P.; Martin, D.M.; Kong, X. Incorporation of Exome-Based CNV Analysis Makes Trio-WES a More Powerful Tool for Clinical Diagnosis in Neurodevelopmental Disorders: A Retrospective Study. Hum. Mutat. 2021, 42, 990–1004. [Google Scholar] [CrossRef]
- Xiang, J.; Ding, Y.; Yang, F.; Gao, A.; Zhang, W.; Tang, H.; Mao, J.; He, Q.; Zhang, Q.; Wang, T. Genetic Analysis of Children With Unexplained Developmental Delay and/or Intellectual Disability by Whole-Exome Sequencing. Front. Genet 2021, 12, 738561. [Google Scholar] [CrossRef] [PubMed]
- Martínez, F.; Caro-Llopis, A.; Roselló, M.; Oltra, S.; Mayo, S.; Monfort, S.; Orellana, C. High Diagnostic Yield of Syndromic Intellectual Disability by Targeted Next-Generation Sequencing. J. Med. Genet. 2017, 54, 87–92. [Google Scholar] [CrossRef]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Tejada, M.-I.; Gecz, J.; Shaw, M.; Corbett, M.; Haan, E.; Thompson, E.; Friend, K.; et al. Targeted Next-Generation Sequencing Analysis of 1000 Individuals with Intellectual Disability. Hum. Mutat. 2015, 36, 1197–1204. [Google Scholar] [CrossRef] [Green Version]
- Pekeles, H.; Accogli, A.; Boudrahem-Addour, N.; Russell, L.; Parente, F.; Srour, M. Diagnostic Yield of Intellectual Disability Gene Panels. Pediatr. Neurol. 2019, 92, 32–36. [Google Scholar] [CrossRef]
- Chérot, E.; Keren, B.; Dubourg, C.; Carré, W.; Fradin, M.; Lavillaureix, A.; Afenjar, A.; Burglen, L.; Whalen, S.; Charles, P.; et al. Using Medical Exome Sequencing to Identify the Causes of Neurodevelopmental Disorders: Experience of 2 Clinical Units and 216 Patients. Clin. Genet. 2018, 93, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Snoeijen-Schouwenaars, F.M.; van Ool, J.S.; Verhoeven, J.S.; van Mierlo, P.; Braakman, H.M.H.; Smeets, E.E.; Nicolai, J.; Schoots, J.; Teunissen, M.W.A.; Rouhl, R.P.W.; et al. Diagnostic Exome Sequencing in 100 Consecutive Patients with Both Epilepsy and Intellectual Disability. Epilepsia 2019, 60, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef]
- Bowling, K.M.; Thompson, M.L.; Amaral, M.D.; Finnila, C.R.; Hiatt, S.M.; Engel, K.L.; Cochran, J.N.; Brothers, K.B.; East, K.M.; Gray, D.E.; et al. Genomic Diagnosis for Children with Intellectual Disability and/or Developmental Delay. Genome. Med. 2017, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Ewans, L.J.; Schofield, D.; Shrestha, R.; Zhu, Y.; Gayevskiy, V.; Ying, K.; Walsh, C.; Lee, E.; Kirk, E.P.; Colley, A.; et al. Whole-Exome Sequencing Reanalysis at 12 Months Boosts Diagnosis and Is Cost-Effective When Applied Early in Mendelian Disorders. Genet. Med. 2018, 20, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Ontario Health (Quality). Genome-Wide Sequencing for Unexplained Developmental Disabilities or Multiple Congenital Anomalies: A Health Technology Assessment. Ont. Health Technol. Assess. Ser. 2020, 20, 1–178. [Google Scholar]
- Sun, Y.; Peng, J.; Liang, D.; Ye, X.; Xu, N.; Chen, L.; Yan, D.; Zhang, H.; Xiao, B.; Qiu, W.; et al. Genome Sequencing Demonstrates High Diagnostic Yield in Children with Undiagnosed Global Developmental Delay/Intellectual Disability: A Prospective Study. Hum. Mutat. 2022, 43, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Borch, L.A.; Parboosingh, J.; Thomas, M.A.; Veale, P. Re-Evaluating the First-Tier Status of Fragile X Testing in Neurodevelopmental Disorders. Genet. Med. 2020, 22, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Christofolini, D.M.; Abbud, E.M.; Lipay, M.V.N.; Costa, S.S.; Vianna-Morgante, A.M.; Bellucco, F.T.S.; Nogueira, S.I.; Kulikowski, L.D.; Brunoni, D.; Juliano, Y.; et al. Evaluation of Clinical Checklists for Fragile X Syndrome Screening in Brazilian Intellectually Disabled Males: Proposal for a New Screening Tool. J. Intellect. Disabil. 2009, 13, 239–248. [Google Scholar] [CrossRef]
- Giangreco, C.A.; Steele, M.W.; Aston, C.E.; Cummins, J.H.; Wenger, S.L. A Simplified Six-Item Checklist for Screening for Fragile X Syndrome in the Pediatric Population. J. Pediatr. 1996, 129, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Lubala, T.K.; Lumaka, A.; Kanteng, G.; Mutesa, L.; Mukuku, O.; Wembonyama, S.; Hagerman, R.; Luboya, O.N.; Lukusa Tshilobo, P. Fragile X Checklists: A Meta-analysis and Development of a Simplified Universal Clinical Checklist. Mol. Genet. Genomic. Med. 2018, 6, 526–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, G.B.; Mendelsohn, N.J. Professional Practice and Guidelines Committee Clinical Genetics Evaluation in Identifying the Etiology of Autism Spectrum Disorders: 2013 Guideline Revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Vallance, H.; Sinclair, G.; Rakic, B.; Stockler-Ipsiroglu, S. Diagnostic Yield from Routine Metabolic Screening Tests in Evaluation of Global Developmental Delay and Intellectual Disability. Paediatr. Child Health 2021, 26, 344–348. [Google Scholar] [CrossRef]
- Sempere, A.; Arias, A.; Farré, G.; García-Villoria, J.; Rodríguez-Pombo, P.; Desviat, L.R.; Merinero, B.; García-Cazorla, A.; Vilaseca, M.A.; Ribes, A.; et al. Study of Inborn Errors of Metabolism in Urine from Patients with Unexplained Mental Retardation. J. Inherit. Metab. Dis. 2010, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Rosenfeld, J.A.; Shur, N.; Slavotinek, A.M.; Cox, V.A.; Hennekam, R.C.; Firth, H.V.; Willatt, L.; Wheeler, P.; Morrow, E.M.; et al. Further Clinical and Molecular Delineation of the 15q24 Microdeletion Syndrome. J. Med. Genet. 2012, 49, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietiläinen, O.P.H.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large Recurrent Microdeletions Associated with Schizophrenia. Nature 2008, 455, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Kovel, C.G.F.; Trucks, H.; Helbig, I.; Mefford, H.C.; Baker, C.; Leu, C.; Kluck, C.; Muhle, H.; von Spiczak, S.; Ostertag, P.; et al. Recurrent Microdeletions at 15q11.2 and 16p13.11 Predispose to Idiopathic Generalized Epilepsies. Brain 2010, 133, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mefford, H.C.; Cooper, G.M.; Zerr, T.; Smith, J.D.; Baker, C.; Shafer, N.; Thorland, E.C.; Skinner, C.; Schwartz, C.E.; Nickerson, D.A.; et al. A Method for Rapid, Targeted CNV Genotyping Identifies Rare Variants Associated with Neurocognitive Disease. Genome. Res. 2009, 19, 1579–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/Microduplication of Proximal 15q11.2 between BP1 and BP2: A Susceptibility Region for Neurological Dysfunction Including Developmental and Language Delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.; Sikkema-Raddatz, B.; Ruijvenkamp, C.A.L.; Dijkhuizen, T.; Bijlsma, E.K.; Gijsbers, A.C.J.; Hilhorst-Hofstee, Y.; Hordijk, R.; Verbruggen, K.T.; Kerstjens-Frederikse, W.S.M.; et al. Nine Patients with a Microdeletion 15q11.2 between Breakpoints 1 and 2 of the Prader-Willi Critical Region, Possibly Associated with Behavioural Disturbances. Eur. J. Med. Genet. 2009, 52, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.K.; Nygren, A.O.H.; El Shakankiry, H.M.; Schouten, J.P.; Al Khayat, A.I.; Ridha, A.; Al Ali, M.T. Detection of a Novel Familial Deletion of Four Genes between BP1 and BP2 of the Prader-Willi/Angelman Syndrome Critical Region by Oligo-Array CGH in a Child with Neurological Disorder and Speech Impairment. Cytogenet. Genome. Res. 2007, 116, 135–140. [Google Scholar] [CrossRef] [PubMed]
- von der Lippe, C.; Rustad, C.; Heimdal, K.; Rødningen, O.K. 15q11.2 Microdeletion—Seven New Patients with Delayed Development and/or Behavioural Problems. Eur. J. Med. Genet. 2011, 54, 357–360. [Google Scholar] [CrossRef]
- Helbig, I.; Mefford, H.C.; Sharp, A.J.; Guipponi, M.; Fichera, M.; Franke, A.; Muhle, H.; de Kovel, C.; Baker, C.; von Spiczak, S.; et al. 15q13.3 Microdeletions Increase Risk of Idiopathic Generalized Epilepsy. Nat. Genet. 2009, 41, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Mefford, H.C.; Li, K.; Baker, C.; Skinner, C.; Stevenson, R.E.; Schroer, R.J.; Novara, F.; De Gregori, M.; Ciccone, R.; et al. A Recurrent 15q13.3 Microdeletion Syndrome Associated with Mental Retardation and Seizures. Nat. Genet. 2008, 40, 322–328. [Google Scholar] [CrossRef] [PubMed]
- van Bon, B.W.M.; Mefford, H.C.; Menten, B.; Koolen, D.A.; Sharp, A.J.; Nillesen, W.M.; Innis, J.W.; de Ravel, T.J.L.; Mercer, C.L.; Fichera, M.; et al. Further Delineation of the 15q13 Microdeletion and Duplication Syndromes: A Clinical Spectrum Varying from Non-Pathogenic to a Severe Outcome. J. Med. Genet. 2009, 46, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shachar, S.; Lanpher, B.; German, J.R.; Qasaymeh, M.; Potocki, L.; Nagamani, S.C.S.; Franco, L.M.; Malphrus, A.; Bottenfield, G.W.; Spence, J.E.; et al. Microdeletion 15q13.3: A Locus with Incomplete Penetrance for Autism, Mental Retardation, and Psychiatric Disorders. J. Med. Genet. 2009, 46, 382–388. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Wing, K.; Sadighi Akha, E.; Knight, S.J.L.; Bölte, S.; Schmötzer, G.; Duketis, E.; Poustka, F.; Klauck, S.M.; Poustka, A.; et al. A 15q13.3 Microdeletion Segregating with Autism. Eur. J. Hum. Genet. 2009, 17, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Shen, Y.; Weiss, L.A.; Korn, J.; Anselm, I.; Bridgemohan, C.; Cox, G.F.; Dickinson, H.; Gentile, J.; Harris, D.J.; et al. Microdeletion/Duplication at 15q13.2q13.3 among Individuals with Features of Autism and Other Neuropsychiatric Disorders. J. Med. Genet. 2009, 46, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann-Gagescu, R.; Mefford, H.C.; Cowan, C.; Glew, G.M.; Hing, A.V.; Wallace, S.; Bader, P.I.; Hamati, A.; Reitnauer, P.J.; Smith, R.; et al. Recurrent 200-Kb Deletions of 16p11.2 That Include the SH2B1 Gene Are Associated with Developmental Delay and Obesity. Genet. Med. 2010, 12, 641–647. [Google Scholar] [CrossRef] [Green Version]
- Bochukova, E.G.; Huang, N.; Keogh, J.; Henning, E.; Purmann, C.; Blaszczyk, K.; Saeed, S.; Hamilton-Shield, J.; Clayton-Smith, J.; O’Rahilly, S.; et al. Large, Rare Chromosomal Deletions Associated with Severe Early-Onset Obesity. Nature 2010, 463, 666–670. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.R.; Green, T.; et al. Association between Microdeletion and Microduplication at 16p11.2 and Autism. N. Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, A.; Novelli, A.; Bernardini, L.; Igliozzi, R.; Parrini, B. Further Characterization of the New Microdeletion Syndrome of 16p11.2-P12.2. Am. J. Med. Genet. A 2009, 149A, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, E.K.; Gijsbers, A.C.J.; Schuurs-Hoeijmakers, J.H.M.; van Haeringen, A.; Fransen van de Putte, D.E.; Anderlid, B.-M.; Lundin, J.; Lapunzina, P.; Pérez Jurado, L.A.; Delle Chiaie, B.; et al. Extending the Phenotype of Recurrent Rearrangements of 16p11.2: Deletions in Mentally Retarded Patients without Autism and in Normal Individuals. Eur. J. Med. Genet. 2009, 52, 77–87. [Google Scholar] [CrossRef]
- Hempel, M.; Rivera Brugués, N.; Wagenstaller, J.; Lederer, G.; Weitensteiner, A.; Seidel, H.; Meitinger, T.; Strom, T.M. Microdeletion Syndrome 16p11.2-P12.2: Clinical and Molecular Characterization. Am. J. Med. Genet. A 2009, 149A, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Shinawi, M.; Liu, P.; Kang, S.-H.L.; Shen, J.; Belmont, J.W.; Scott, D.A.; Probst, F.J.; Craigen, W.J.; Graham, B.H.; Pursley, A.; et al. Recurrent Reciprocal 16p11.2 Rearrangements Associated with Global Developmental Delay, Behavioural Problems, Dysmorphism, Epilepsy, and Abnormal Head Size. J. Med. Genet. 2010, 47, 332–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemont, S.; Reymond, A.; Zufferey, F.; Harewood, L.; Walters, R.G.; Kutalik, Z.; Martinet, D.; Shen, Y.; Valsesia, A.; Beckmann, N.D.; et al. Mirror Extreme BMI Phenotypes Associated with Gene Dosage at the Chromosome 16p11.2 Locus. Nature 2011, 478, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, R.G.; Jacquemont, S.; Valsesia, A.; de Smith, A.J.; Martinet, D.; Andersson, J.; Falchi, M.; Chen, F.; Andrieux, J.; Lobbens, S.; et al. A New Highly Penetrant Form of Obesity Due to Deletions on Chromosome 16p11.2. Nature 2010, 463, 671–675. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, S.E.; Makarov, V.; Kirov, G.; Addington, A.M.; McClellan, J.; Yoon, S.; Perkins, D.O.; Dickel, D.E.; Kusenda, M.; Krastoshevsky, O.; et al. Microduplications of 16p11.2 Are Associated with Schizophrenia. Nat. Genet. 2009, 41, 1223–1227. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A Recurrent 16p12.1 Microdeletion Supports a Two-Hit Model for Severe Developmental Delay. Nat. Genet. 2010, 42, 203–209. [Google Scholar] [CrossRef]
- Andersen, E.F.; Baldwin, E.E.; Ellingwood, S.; Smith, R.; Lamb, A.N. Xq28 Duplication Overlapping the Int22h-1/Int22h-2 Region and Including RAB39B and CLIC2 in a Family with Intellectual and Developmental Disability. Am. J. Med. Genet. A 2014, 164A, 1795–1801. [Google Scholar] [CrossRef]
- Piton, A.; Redin, C.; Mandel, J.-L. XLID-Causing Mutations and Associated Genes Challenged in Light of Data from Large-Scale Human Exome Sequencing. Am. J. Hum. Genet. 2013, 93, 368–383. [Google Scholar] [CrossRef] [Green Version]
- Van Esch, H. MECP2 Duplication Syndrome. Mol. Syndromol. 2012, 2, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 Duplication Syndrome. Am. J. Med. Genet. A 2010, 152A, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef]
- Exposito-Alonso, D.; Rico, B. Mechanisms Underlying Circuit Dysfunction in Neurodevelopmental Disorders. Annu. Rev. Genet. 2022, 56, 391–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, Y.; Zhang, F.; Chiang, C.; Pillalamarri, V.; Blumenthal, I.; Talkowski, M.; Wu, B.-L.; Gusella, J.F. Molecular Analysis of a Deletion Hotspot in the NRXN1 Region Reveals the Involvement of Short Inverted Repeats in Deletion CNVs. Am. J. Hum. Genet. 2013, 92, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-Linked Genes Encoding Neuroligins NLGN3 and NLGN4 Are Associated with Autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, P.; Feng, G. SHANK Proteins: Roles at the Synapse and in Autism Spectrum Disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Motazacker, M.M.; Rost, B.R.; Hucho, T.; Garshasbi, M.; Kahrizi, K.; Ullmann, R.; Abedini, S.S.; Nieh, S.E.; Amini, S.H.; Goswami, C.; et al. A Defect in the Ionotropic Glutamate Receptor 6 Gene (GRIK2) Is Associated with Autosomal Recessive Mental Retardation. Am. J. Hum. Genet. 2007, 81, 792–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA Receptor GluA2 Subunit Defects Are a Cause of Neurodevelopmental Disorders. Nat. Commun. 2019, 10, 3094. [Google Scholar] [CrossRef] [Green Version]
- Srour, M.; AlHakeem, A.; Shevell, M. Chapter 19—Global Developmental Delay and Intellectual Disability. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 269–281. ISBN 978-0-12-813955-4. [Google Scholar]
- Komiyama, N.H.; Watabe, A.M.; Carlisle, H.J.; Porter, K.; Charlesworth, P.; Monti, J.; Strathdee, D.J.C.; O’Carroll, C.M.; Martin, S.J.; Morris, R.G.M.; et al. SynGAP Regulates ERK/MAPK Signaling, Synaptic Plasticity, and Learning in the Complex with Postsynaptic Density 95 and NMDA Receptor. J. Neurosci. 2002, 22, 9721–9732. [Google Scholar] [CrossRef]
- Tomoda, T.; Kim, J.H.; Zhan, C.; Hatten, M.E. Role of Unc51.1 and Its Binding Partners in CNS Axon Outgrowth. Genes. Dev. 2004, 18, 541–558. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, F.F.; Daoud, H.; Piton, A.; Gauthier, J.; Dobrzeniecka, S.; Krebs, M.-O.; Joober, R.; Lacaille, J.-C.; Nadeau, A.; Milunsky, J.M.; et al. De Novo SYNGAP1 Mutations in Nonsyndromic Intellectual Disability and Autism. Biol. Psychiatry 2011, 69, 898–901. [Google Scholar] [CrossRef]
- Yoshida, T.; Yasumura, M.; Uemura, T.; Lee, S.-J.; Ra, M.; Taguchi, R.; Iwakura, Y.; Mishina, M. IL-1 Receptor Accessory Protein-like 1 Associated with Mental Retardation and Autism Mediates Synapse Formation by Trans-Synaptic Interaction with Protein Tyrosine Phosphatase δ. J. Neurosci. 2011, 31, 13485–13499. [Google Scholar] [CrossRef] [Green Version]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A Systematic, Large-Scale Resequencing Screen of X-Chromosome Coding Exons in Mental Retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef]
- Verhage, M.; Sørensen, J.B. SNAREopathies: Diversity in Mechanisms and Symptoms. Neuron 2020, 107, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Gauthier, J.; Dobrzeniecka, S.; Lortie, A.; Mottron, L.; Vanasse, M.; D’Anjou, G.; Lacaille, J.C.; Rouleau, G.A.; Michaud, J.L. Intellectual Disability without Epilepsy Associated with STXBP1 Disruption. Eur. J. Hum. Genet. 2011, 19, 607–609. [Google Scholar] [CrossRef]
- Merienne, K.; Jacquot, S.; Pannetier, S.; Zeniou, M.; Bankier, A.; Gecz, J.; Mandel, J.L.; Mulley, J.; Sassone-Corsi, P.; Hanauer, A. A Missense Mutation in RPS6KA3 (RSK2) Responsible for Non-Specific Mental Retardation. Nat. Genet. 1999, 22, 13–14. [Google Scholar] [CrossRef]
- Nakashima, M.; Kato, M.; Matsukura, M.; Kira, R.; Ngu, L.-H.; Lichtenbelt, K.D.; van Gassen, K.L.I.; Mitsuhashi, S.; Saitsu, H.; Matsumoto, N. De Novo Variants in CUL3 Are Associated with Global Developmental Delays with or without Infantile Spasms. J. Hum. Genet. 2020, 65, 727–734. [Google Scholar] [CrossRef] [PubMed]
- da Silva Montenegro, E.M.; Costa, C.S.; Campos, G.; Scliar, M.; de Almeida, T.F.; Zachi, E.C.; Silva, I.M.W.; Chan, A.J.S.; Zarrei, M.; Lourenço, N.C.V.; et al. Meta-Analyses Support Previous and Novel Autism Candidate Genes: Outcomes of an Unexplored Brazilian Cohort. Autism. Res. 2020, 13, 199–206. [Google Scholar] [CrossRef]
- Hörnberg, H.; Pérez-Garci, E.; Schreiner, D.; Hatstatt-Burklé, L.; Magara, F.; Baudouin, S.; Matter, A.; Nacro, K.; Pecho-Vrieseling, E.; Scheiffele, P. Rescue of Oxytocin Response and Social Behaviour in a Mouse Model of Autism. Nature 2020, 584, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.W.; Yeung, W.L.; Ko, C.H.; Poon, P.M.; Tong, S.F.; Chan, K.Y.; Lo, I.F.; Chan, L.Y.; Hui, J.; Wong, V.; et al. Spectrum of Mutations in the MECP2 Gene in Patients with Infantile Autism and Rett Syndrome. J. Med. Genet. 2000, 37, E41. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.W.; Chahrour, M.H.; Coulter, M.E.; Jiralerspong, S.; Okamura-Ikeda, K.; Ataman, B.; Schmitz-Abe, K.; Harmin, D.A.; Adli, M.; Malik, A.N.; et al. Using Whole-Exome Sequencing to Identify Inherited Causes of Autism. Neuron 2013, 77, 259–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; Wamsley, B.; Klei, L.; Wang, L.; Hao, S.P.; et al. Rare Coding Variation Provides Insight into the Genetic Architecture and Phenotypic Context of Autism. Nat. Genet. 2022, 54, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Noor, A.; Degagne, B.; Baker, K.; Bok, L.A.; Brady, A.F.; Chitayat, D.; Chung, B.H.; Cytrynbaum, C.; Dyment, D.; et al. Phenotypic Spectrum Associated with PTCHD1 Deletions and Truncating Mutations Includes Intellectual Disability and Autism Spectrum Disorder. Clin. Genet. 2015, 88, 224–233. [Google Scholar] [CrossRef]
- Orefice, L.L.; Zimmerman, A.L.; Chirila, A.M.; Sleboda, S.J.; Head, J.P.; Ginty, D.D. Peripheral Mechanosensory Neuron Dysfunction Underlies Tactile and Behavioral Deficits in Mouse Models of ASDs. Cell 2016, 166, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, R.T.; Chantranupong, L.; Hakim, R.; Levasseur, J.; Wang, W.; Merchant, T.; Gorman, K.; Budnik, B.; Sabatini, B.L. Abnormal Striatal Development Underlies the Early Onset of Behavioral Deficits in Shank3B−/− Mice. Cell Rep. 2019, 29, 2016–2027.e4. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, R.T.; Wang, W.; Croney, D.M.; Kozorovitskiy, Y.; Sabatini, B.L. Early Hyperactivity and Precocious Maturation of Corticostriatal Circuits in Shank3B−/− Mice. Nat. Neurosci. 2016, 19, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Bariselli, S.; Tzanoulinou, S.; Glangetas, C.; Prévost-Solié, C.; Pucci, L.; Viguié, J.; Bezzi, P.; O’Connor, E.C.; Georges, F.; Lüscher, C.; et al. SHANK3 Controls Maturation of Social Reward Circuits in the VTA. Nat. Neurosci. 2016, 19, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Kao, F.-C.; Su, S.-H.; Carlson, G.C.; Liao, W. MeCP2-Mediated Alterations of Striatal Features Accompany Psychomotor Deficits in a Mouse Model of Rett Syndrome. Brain Struct. Funct. 2015, 220, 419–434. [Google Scholar] [CrossRef]
- van Karnebeek, C.D.; Stockler-Ipsiroglu, S. Early Identification of Treatable Inborn Errors of Metabolism in Children with Intellectual Disability: The Treatable Intellectual Disability Endeavor Protocol in British Columbia. Paediatr. Child Health 2014, 19, 469–471. [Google Scholar] [CrossRef] [Green Version]
- Silove, N.; Collins, F.; Ellaway, C. Update on the Investigation of Children with Delayed Development. J. Paediatr. Child Health 2013, 49, 519–525. [Google Scholar] [CrossRef]
- Tirosh, E.; Jaffe, M. Global Developmental Delay and Mental Retardation—A Pediatric Perspective. Dev. Disabil. Res. Rev. 2011, 17, 85–92. [Google Scholar] [CrossRef]
- Mithyantha, R.; Kneen, R.; McCann, E.; Gladstone, M. Current Evidence-Based Recommendations on Investigating Children with Global Developmental Delay. Arch. Dis. Child 2017, 102, 1071–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbruggen, K.T.; Meiners, L.C.; Sijens, P.E.; Lunsing, R.J.; van Spronsen, F.J.; Brouwer, O.F. Magnetic Resonance Imaging and Proton Magnetic Resonance Spectroscopy of the Brain in the Diagnostic Evaluation of Developmental Delay. Eur. J. Paediatr. Neurol. 2009, 13, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Batty, R.; Warren, D.; Hart, A.; Sharrard, M.; Mordekar, S.R.; Raghavan, A.; Connolly, D.J.A. The Use of MR Imaging and Spectroscopy of the Brain in Children Investigated for Developmental Delay: What Is the Most Appropriate Imaging Strategy? Eur. Radiol. 2011, 21, 1820–1830. [Google Scholar] [CrossRef] [PubMed]
- Shevell, M.I.; Majnemer, A.; Rosenbaum, P.; Abrahamowicz, M. Etiologic Yield of Subspecialists’ Evaluation of Young Children with Global Developmental Delay. J. Pediatr. 2000, 136, 593–598. [Google Scholar] [CrossRef]
- Shevell, M. The Role of the Pediatric Neurologist in the Care of Children with Neurodevelopmental Disabilities. Pediatr. Neurol. 2018, 88, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kostanjsek, N. Use of The International Classification of Functioning, Disability and Health (ICF) as a Conceptual Framework and Common Language for Disability Statistics and Health Information Systems. BMC Public Health 2011, 11 (Suppl. 4), S3. [Google Scholar] [CrossRef] [Green Version]
- Fehlings, D.L.; Penner, M.; Donner, E.J.; Shevell, M.I. Management of Common Comorbidities Associated with Neurodevelopmental Disorders. In Swaiman’s Pediatric Neurology, 6th ed.; Swaiman, K.F., Ashwal, S., Ferriero, D.M., Schor, N.F., Finkel, R.S., Gropman, A.L., Pearl, P.L., Shevell, M.I., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 472–477. ISBN 978-0-323-37101-8. [Google Scholar]
- Bruni, O.; Angriman, M.; Melegari, M.G.; Ferri, R. Pharmacotherapeutic Management of Sleep Disorders in Children with Neurodevelopmental Disorders. Expert Opin. Pharmacother. 2019, 20, 2257–2271. [Google Scholar] [CrossRef]
Figure 1.
Representation of common pathways underlying GDD/ID. This figure is not exhaustive and only shows a few examples of molecules involved in synaptic function, epigenetic regulation and protein homeostasis. Adapted from “Tripartite Glutamatergic Synapse”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates. Accessed on 10 February 2023.
Figure 1.
Representation of common pathways underlying GDD/ID. This figure is not exhaustive and only shows a few examples of molecules involved in synaptic function, epigenetic regulation and protein homeostasis. Adapted from “Tripartite Glutamatergic Synapse”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates. Accessed on 10 February 2023.

Table 2.
Summary of common pathways and examples of genes associated with non-syndromic GDD/ID and their respective functions.
Table 2.
Summary of common pathways and examples of genes associated with non-syndromic GDD/ID and their respective functions.
| Common Pathways | Genes | Function |
|---|---|---|
| Synaptic Signaling | NRX1 | Cell-surface receptors that bind neuroligins; required for efficient neurotransmission. |
| NLGN3 NLGLN4 | Mediate cell-to-cell interactions between neurons; linked to glutamatergic postsynaptic proteins. | |
| SHANK2 SHANK3 | Scaffolding and cell adhesion proteins; required for synaptic plasticity. | |
| GRI2K GRIN2B GRIA2 | Subunits of synaptic glutamate receptors; required for neurotransmission. | |
| SYNGAP1 | Part of the NMDA receptor complex; involved in negative regulation of ERK/MAPK pathway. | |
| IL1RAPL1 | Part of the interleukin 1 receptor; required for neuronal calcium-regulated vesicle release and dendrite differentiation. | |
| CASK | Part of the MAGUK family; scaffolding proteins. | |
| STXBP1 | Synaptic vesicle docking and fusion; required for efficient neurotransmission. | |
| Protein Homeostasis | RPS6KA3 | Part of the RSK (ribosomal S6 kinase) family of growth-factor-regulated serine/threonine kinases; involved in ERK/MAPK pathway. |
| CUL3 | Part of the ubiquitin-proteasome system; required for proteasomal degradation of unwanted proteins. | |
| Epigenetic Regulation | MECP2 | Chromatin-associated protein involved in methyl binding to control transcription; required for maturation of neurons. |
| CHD8 | ATP-dependent chromatin-remodeling factor that regulates transcription. |
Table 3.
Approach to evaluation of children with non-syndromic GDD/ID.
| Evaluation | Recommendation |
|---|---|
| 1. Detailed history including developmental and family history with thorough clinical examination |
|
| 2. First tier genetic testing |
|
| 3. Second tier genetic testing |
|
| 4. Further investigations |
|
ES: exome sequencing, FXS: Fragile X syndrome, GDD: global developmental delay, GS: genome sequencing, IMD: inherited metabolic disorders, TIDE: Treatable Intellectual Disability Endeavor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
AlMutiri, R.; Malta, M.; Shevell, M.I.; Srour, M. Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability. Children 2023, 10, 414. https://doi.org/10.3390/children10030414
AMA Style
AlMutiri R, Malta M, Shevell MI, Srour M. Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability. Children. 2023; 10(3):414. https://doi.org/10.3390/children10030414
Chicago/Turabian StyleAlMutiri, Rowim, Maisa Malta, Michael I. Shevell, and Myriam Srour. 2023. "Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability" Children 10, no. 3: 414. https://doi.org/10.3390/children10030414
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.