

Molecular Genetic Analysis of Ukrainian Families with Congenital Cataracts
 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Study Design
2.2. Clinical Analysis
2.3. Whole-Exome Sequencing and Bioinformatics Analysis
2.4. Sanger Sequencing
3. Results
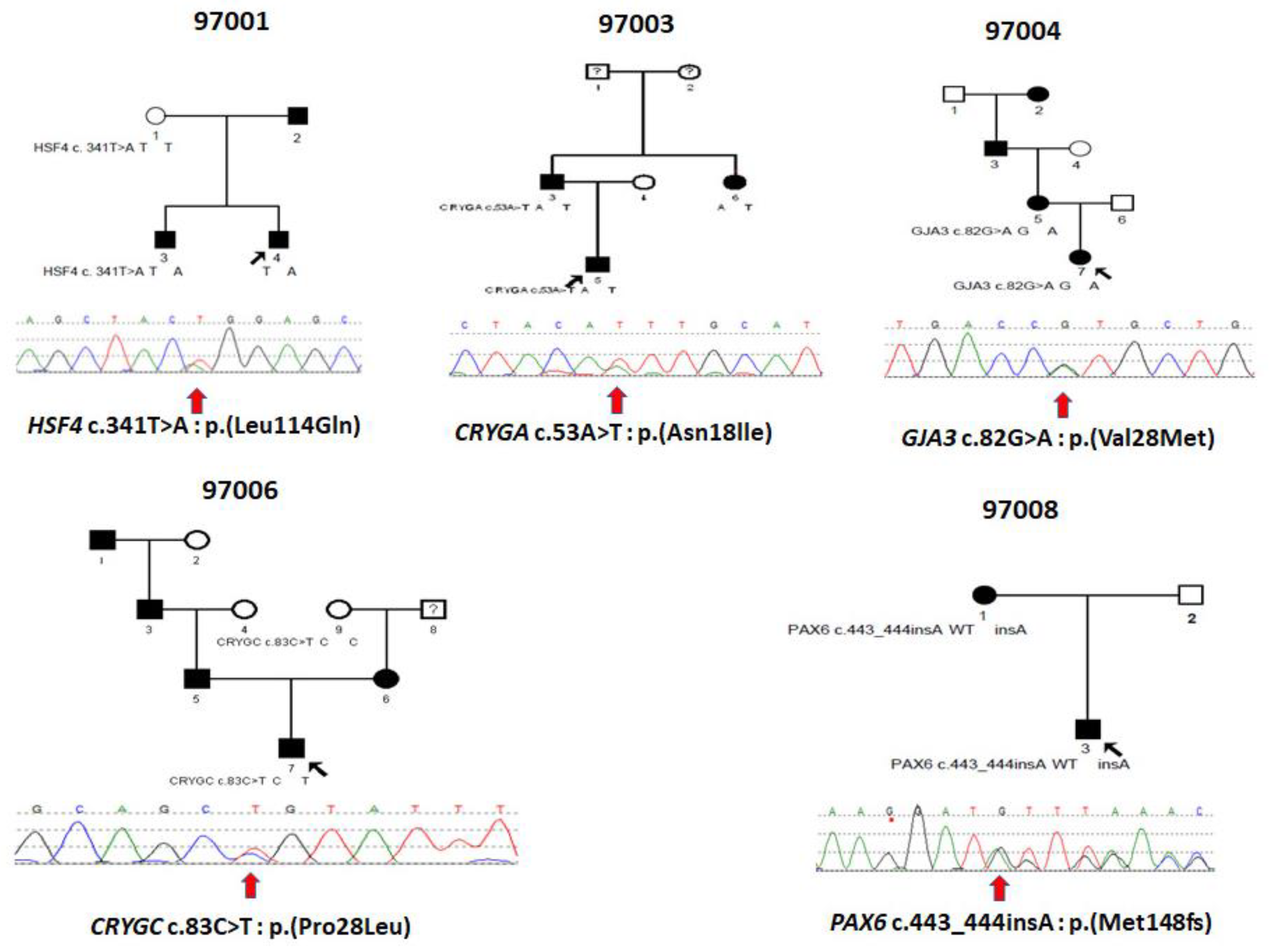
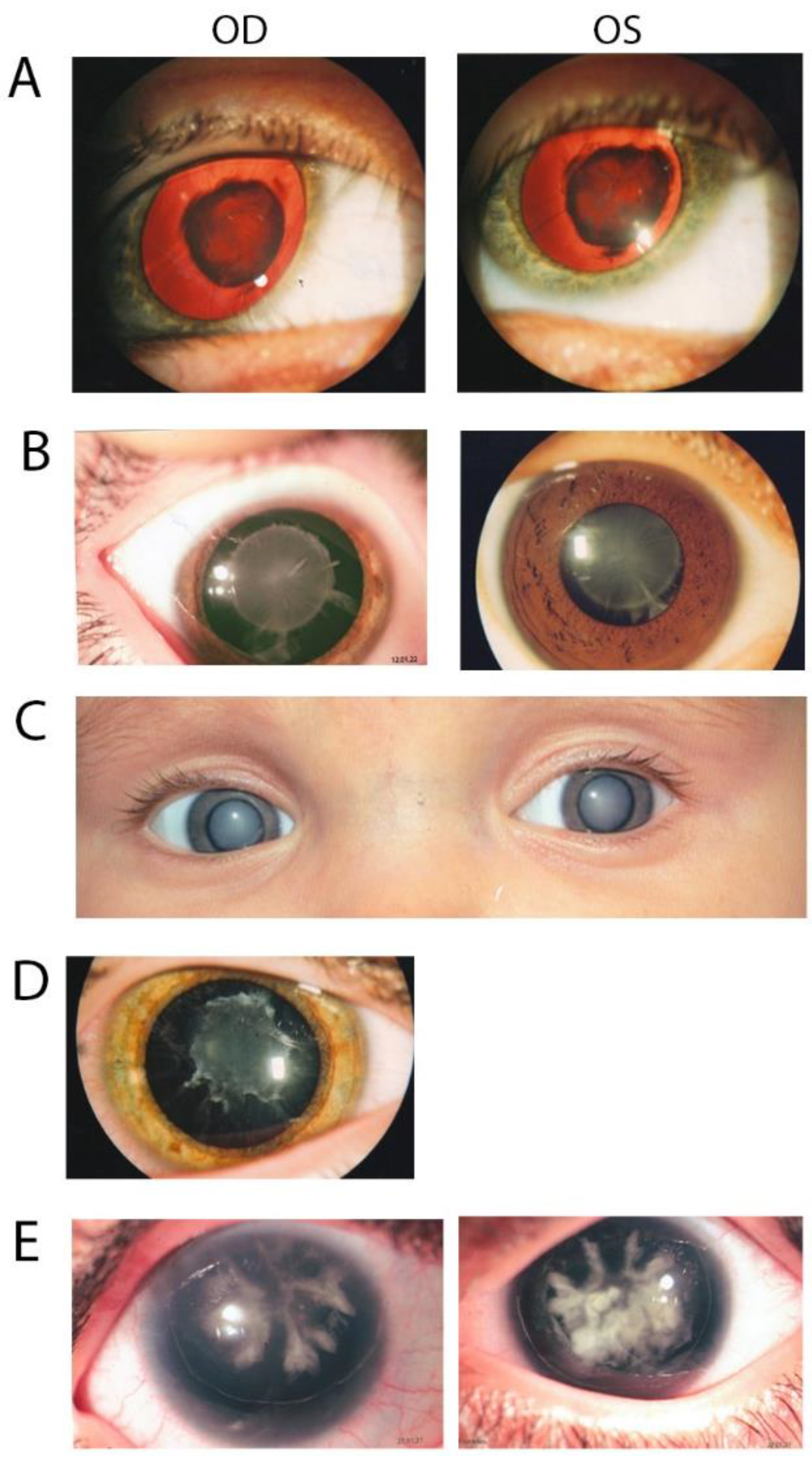
3.1. Family 97001
3.2. Family 97003
3.3. Family 97004
3.4. Family 97006
3.5. Family 97008
4. Discussion
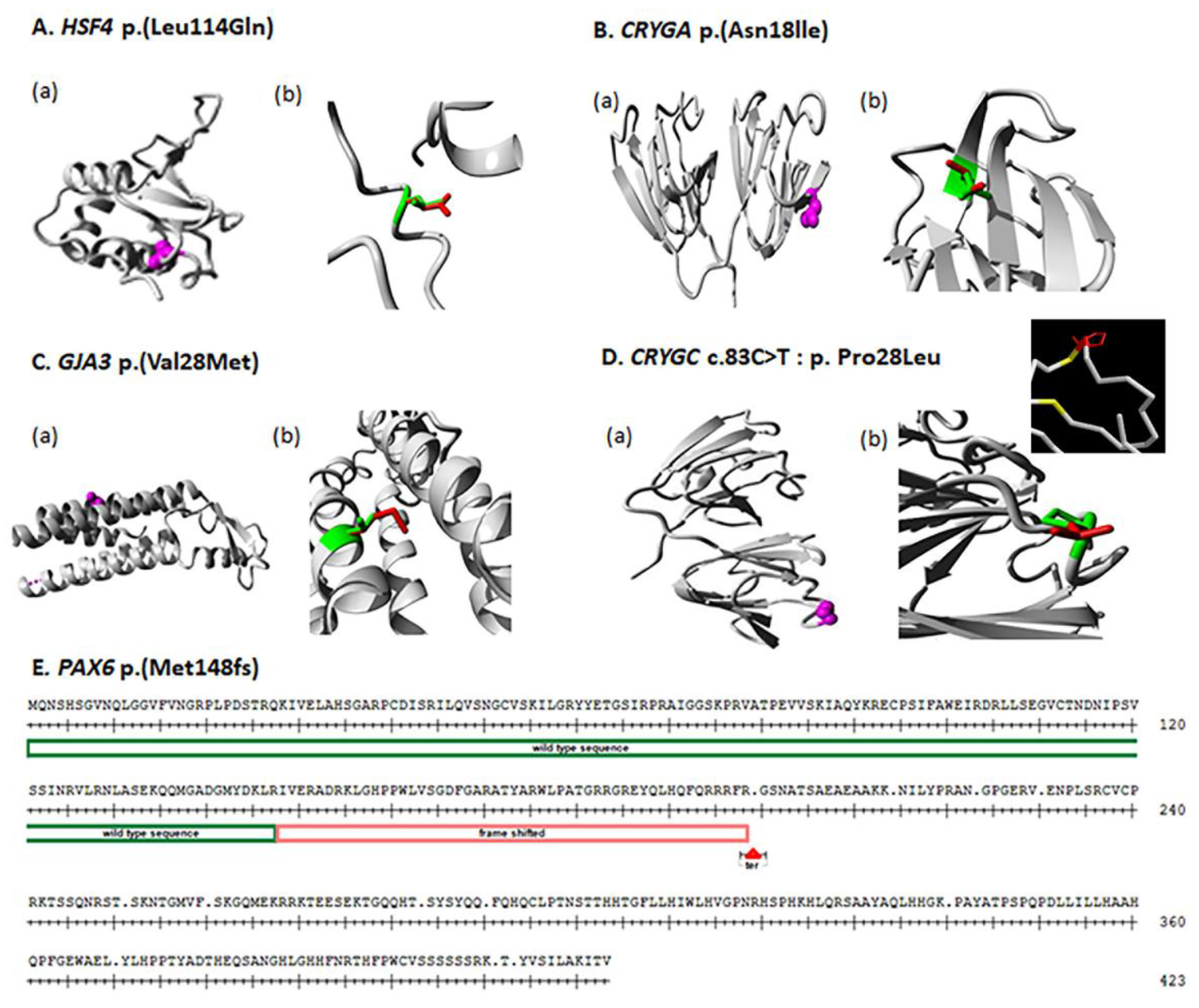
4.1. HSF4
4.2. CRYGA
4.3. GJA3
4.4. CRYGC
4.5. PAX6
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benedek, G.B. Theory of Transparency of the Eye. Appl. Opt. 1971, 10, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Delaye, M.; Tardieu, A. Short-range order of crystallin proteins accounts for eye lens transparency. Nature 1983, 302, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-C. Couching for Cataract in China. Surv. Ophthalmol. 2010, 55, 393–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leffler, C.T.; Klebanov, A.; Samara, W.A.; Grzybowski, A. The history of cataract surgery: From couching to phacoemulsification. Ann. Transl. Med. 2020, 8, 1551. [Google Scholar] [CrossRef] [PubMed]
- Hejtmancik, J.F.; Smaoui, N. Molecular Genetics of Cataract. In Developments in Ophthalmology; Wissinger, B., Kohl, S., Langenbeck, U., Eds.; S. Karger: Basel, Switzerland, 2003; pp. 67–82. [Google Scholar]
- Foster, P.J.; Wong, T.Y.; Machin, D.; Johnson, G.J.; Seah, S.K.L. Risk factors for nuclear, cortical and posterior subcapsular cataracts in the Chinese population of Singapore: The Tanjong Pagar Survey. Br. J. Ophthalmol. 2003, 87, 1112–1120. [Google Scholar] [CrossRef] [Green Version]
- Haargaard, B.; Wohlfahrt, J.; Fledelius, H.C.; Rosenberg, T.; Melbye, M. Incidence and Cumulative Risk of Childhood Cataract in a Cohort of 2.6 Million Danish Children. Investig. Opthalmol. Vis. Sci. 2004, 45, 1316–1320. [Google Scholar] [CrossRef] [Green Version]
- Haargaard, B.; Wohlfahrt, J.; Rosenberg, T.; Fledelius, H.C.; Melbye, M. Risk Factors for Idiopathic Congenital/Infantile Cataract. Investig. Opthalmol. Vis. Sci. 2005, 46, 3067–3073. [Google Scholar] [CrossRef] [Green Version]
- Merin, S. Inherited Cataracts. In Inherited Eye Diseases; Merin, S., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1991; pp. 86–120. [Google Scholar]
- Chornobai, M.O. The level of pediatric cataract in regions according to data of Ministry of Health of Ukraine for 2007–2016 years. J. Ophthalmol. 2017, 6, 479. [Google Scholar] [CrossRef]
- Foster, A.; Gilbert, C.; Rahi, J. Epidemiology of cataract in childhood: A global perspective. J. Cataract. Refract. Surg. 1997, 23 (Suppl. 1), 601–604. [Google Scholar] [CrossRef]
- Shiels, A.; Bennett, T.M.; Hejtmancik, J.F. Cat-Map: Putting cataract on the map. Mol. Vis. 2010, 16, 2007–2015. [Google Scholar]
- Shoshany, N.; Hejtmancik, F.; Shiels, A.; Datiles, M.B. Congenital and Hereditary Cataracts: Epidemiology and Genetics. In Pediatric Cataract Surgery and IOL Implantation; Kraus, C.L., Ed.; Springer: New York, NY, USA, 2020; pp. 3–24. [Google Scholar] [CrossRef]
- Devi, R.R.; Reena, C.; Vijayalakshmi, P. Novel mutations in GJA3 associated with autosomal dominant congenital cataract in the Indian population. Mol. Vis. 2005, 11, 846–852. [Google Scholar] [PubMed]
- Zhai, Y.; Li, J.; Yu, W.; Zhu, S.; Yu, Y.; Wu, M.; Sun, G.; Gong, X.; Yao, K. Targeted Exome Sequencing of Congenital Cataracts Related Genes: Broadening the Mutation Spectrum and Genotype-Phenotype Correlations in 27 Chinese Han Families. Sci. Rep. 2017, 7, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, H.; Huang, C.; Zhang, J.; Liu, Z.; Zhang, Z.; Xu, H.; You, Y.; Hu, J.; Li, X.; Wang, W. A Novel HSF4 Gene Mutation Causes Autosomal-Dominant Cataracts in a Chinese Family. G3 Genes Genomes Genet. 2014, 4, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, R.L.; O’Sullivan, J.; Ashworth, J.; Bhaskar, S.; Williams, S.; Biswas, S.; Kehdi, E.; Ramsden, S.C.; Clayton-Smith, J.; Black, G.C.; et al. Personalized Diagnosis and Management of Congenital Cataract by Next-Generation Sequencing. Ophthalmology 2014, 121, 2124–2137.e2. [Google Scholar] [CrossRef]
- Li, D.; Wang, S.; Ye, H.; Tang, Y.; Qiu, X.; Fan, Q.; Rong, X.; Liu, X.; Chen, Y.; Yang, J.; et al. Distribution of gene mutations in sporadic congenital cataract in a Han Chinese population. Mol. Vis. 2016, 22, 589–598. [Google Scholar]
- Bu, L.; Jin, Y.; Shi, Y.; Chu, R.; Ban, A.; Eiberg, H.; Andres, L.; Jiang, H.; Zheng, G.; Qian, M.; et al. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat. Genet. 2002, 31, 276–278. [Google Scholar] [CrossRef]
- Shi, Y.; Shi, X.; Jin, Y.; Miao, A.; Bu, L.; He, J.; Jiang, H.; Lu, Y.; Kong, X.; Hu, L. Mutation screening of HSF4 in 150 age-related cataract patients. Mol. Vis. 2008, 14, 1850–1855. [Google Scholar]
- Cao, Z.; Zhu, Y.; Liu, L.; Wu, S.; Liu, B.; Zhuang, J.; Tong, Y.; Chen, X.; Xie, Y.; Nie, K.; et al. Novel mutations in HSF4 cause congenital cataracts in Chinese families. BMC Med. Genet. 2018, 19, 150. [Google Scholar] [CrossRef]
- Berry, V.; Pontikos, N.; Moore, A.; Ionides, A.C.W.; Plagnol, V.; Cheetham, M.E.; Michaelides, M. A novel missense mutation in HSF4 causes autosomal-dominant congenital lamellar cataract in a British family. Eye 2018, 32, 806–812. [Google Scholar] [CrossRef]
- Ke, T.; Wang, Q.K.; Ji, B.; Wang, X.; Liu, P.; Zhang, X.; Tang, Z.; Ren, X.; Liu, M. Novel HSF4 Mutation Causes Congenital Total White Cataract in a Chinese Family. Am. J. Ophthalmol. 2006, 142, 298–303.e2. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Q.; Zhou, L.-X.; Tang, Z.-H. A novel HSF4 mutation in a Chinese family with autosomal dominant congenital cataract. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Mikkelsen, A.; Nu¨rnberg, P.; Nu¨rnberg, G.; Anjum, I.; Eiberg, H.; Rosenberg, T. Comprehensive Mutational Screening in a Cohort of Danish Families with Hereditary Congenital Cataract. Investig. Opthalmol. Vis. Sci. 2009, 50, 3291–3303. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, J.; Cao, Y.; You, Y.; Zhao, X. Novel mutations identified in Chinese families with autosomal dominant congenital cataracts by targeted next-generation sequencing. BMC Med. Genet. 2019, 20, 196. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Malka, S.; Harding, P.; Palma, J.; Dunbar, H.; Moosajee, M. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 578–589. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Q.; Cabrera, P.E.; Zhong, Z.; Sun, W.; Jiao, X.; Chen, Y.; Govindarajan, G.; Naeem, M.A.; Khan, S.N.; et al. Molecular Genetic Analysis of Pakistani Families With Autosomal Recessive Congenital Cataracts by Homozygosity Screening. Investig. Opthalmol. Vis. Sci. 2017, 58, 2207–2217. [Google Scholar] [CrossRef]
- Behnam, M.; Imagawa, E.; Chaleshtori, A.R.S.; Ronasian, F.; Salehi, M.; Miyake, N.; Matsumoto, N. A novel homozygous mutation in HSF4 causing autosomal recessive congenital cataract. J. Hum. Genet. 2016, 61, 177–179. [Google Scholar] [CrossRef]
- Forshew, T.; Johnson, C.A.; Khaliq, S.; Pasha, S.; Willis, C.; Abbasi, R.; Tee, L.; Smith, U.; Trembath, R.; Mehdi, S.Q.; et al. Locus heterogeneity in autosomal recessive congenital cataracts: Linkage to 9q and germline HSF4 mutations. Hum. Genet. 2005, 117, 452–459. [Google Scholar] [CrossRef]
- Sajjad, N.; Goebel, I.; Kakar, N.; Cheema, A.M.; Kubisch, C.; Ahmad, J. A novel HSF4gene mutation (p.R405X) causing autosomal recessive congenital cataracts in a large consanguineous family from Pakistan. BMC Med. Genet. 2008, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Da Wang, J.; Jia, H.Y.; Zhang, J.S.; Li, Y.; Xiong, Y.; Li, J.; Li, X.X.; Huang, Y.; Zhu, G.Y.; et al. Mutation profiles of congenital cataract genes in 21 northern Chinese families. Mol. Vis. 2018, 24, 471–477. [Google Scholar]
- Fernández-Alcalde, C.; Nieves-Moreno, M.; Noval, S.; Peralta, J.; Montaño, V.; del Pozo, Á.; Santos-Simarro, F.; Vallespín, E. Molecular and Genetic Mechanism of Non-Syndromic Congenital Cataracts. Mutation Screening in Spanish Families. Genes 2021, 12, 580. [Google Scholar] [CrossRef]
- Xu, W.; Xu, J.; Shi, C.; Wu, J.; Wang, H.; Wu, W.; Chen, X.; Hu, L. A novel cataract-causing mutation Ile82Met of gammaA crystallin trends to aggregate with unfolding intermediate. Int. J. Biol. Macromol. 2022, 211, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Astiazarán, M.C.; García-Montaño, L.A.; Sánchez-Moreno, F.; Matiz-Moreno, H.; Zenteno, J.C. Next generation sequencing-based molecular diagnosis in familial congenital cataract expands the mutational spectrum in known congenital cataract genes. Am. J. Med. Genet. A 2018, 176, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, Y.V.; Hejtmancik, J.F.; Wingfield, P.T. Energetics of Domain-Domain Interactions and Entropy Driven Association of beta-Crystallins. Biochemistry 2004, 43, 415–424. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, G.B.; Gagne, M.; Rappe, A.K. pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burley, S.K.; Petsko, G.A. Aromatic-Aromatic Interaction: A Mechanism of Protein Structure Stabilization. Science 1985, 229, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Shiels, A.; Hejtmancik, J.F. Mutations and mechanisms in congenital and age-related cataracts. Exp. Eye Res. 2017, 156, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francois, J. Genetics of cataract. Ophthalmologica 1982, 184, 61–71. [Google Scholar] [CrossRef]
- Haargaard, B.; Wohlfahrt, J.; Fledelius, H.C.; Rosenberg, T.; Melbye, M. A nationwide Danish study of 1027 cases of congenital/infantile cataracts: Etiological and clinical classifications. Ophthalmology 2004, 111, 2292–2298. [Google Scholar] [CrossRef]
- Wirth, M.G.; Russell-Eggitt, I.M.; Craig, J.E.; Elder, J.E.; Mackey, D.A. Aetiology of congenital and paediatric cataract in an Australian population. Br. J. Ophthalmol. 2002, 86, 782–786. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age at Diagnosed | Clinical Diagnosis | Other Eye Pathologies | Visual Acuity (OD/OS) | Surgery |
|---|---|---|---|---|---|---|
| 97001-3 | M | 6 y.o. | Congenital nuclear cataract | amblyopia, concomitant strabismus | 0.7/0.5 | Yes |
| 97001-4 | M | 3 y.o. | Congenital hereditary nuclear cataract | axial myopia, amblyopia, concomitant strabismus, lower eyelid tum(Entropon) | 0.7/0.5 | Yes |
| 97003-3 | M | 5 y.o. | Congenital cataract, nuclear pulverulent with sutural component and cortical riders | OD:High-grade myopia, complex myopic astigmatism; OS:Myopia is mild, myopic astigmatism | 0.02/0.4 | Yes |
| 97003-5 | M | 3 y.o. | Congenital hereditary cataract | 0.4/0.015 | Yes | |
| 97003-6 | F | 6 y.o. | Congenital cataract | OS:Medium-grade hypermetropia, Hypermetropic astgmatism | 1.0/0.3 | Yes |
| 97004-5 | F | 7 y.o. | Congenital cataract | 0.85/0.85 | Yes | |
| 97004-7 | F | 2 month | Congenital cataract, total | 0.1/light perception | Yes | |
| 97006-7 | M | 1 month | Congenital hereditary nuclear cataract | Microphthalmos, concomitant strabismus, nystagmus | 0.2/0.4 | Yes |
| 97008-1 | F | At the birth | Congenital cataract | congenital aniridia corneal degeneration;secondary glaucoma, partial optic atrophy; nystagmuscongenital aniridia | Can’t establish/Hm + 10.0 D | Yes |
| 97008-3 | M | At the birth | Congenital cataract, corraliform | congenital aniridia, secondary glaucoma, nystagmus | 0.12/0.1 | No |
| Fam | Gene | Nuc Change | AA Change | PP2 | Pro | MT | LRT | Sift | Type | ACMG | Note |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 97001 | HSF4 | c.341T>A | p.(Leu114Gln) | D | Del | DC | D | D | Het | PM2 | New |
| 97003 | CRYGA | c.53A>T | p.(Asn18lle) | B | Del | N | D | D | Het | PM2 | New |
| 97004 | GJA3 | c.82G>A | p.(Val28Met) | D | Del | DC | D | D | Het | PS1 | Ref [14] |
| 97006 | CRYGC | c.83C>T | p.(Pro28Leu) | B | Del | N | N | T | Het | PM2 | New |
| 97008 | PAX6 | c.443_444insA | p.(Met148fs) | N/A | N/A | N/A | N/A | N/A | Het | PVS1 | New |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, X.; Viswanathan, M.; Bobrova, N.F.; Romanova, T.V.; Hejtmancik, J.F. Molecular Genetic Analysis of Ukrainian Families with Congenital Cataracts. Children 2023, 10, 51. https://doi.org/10.3390/children10010051
Jiao X, Viswanathan M, Bobrova NF, Romanova TV, Hejtmancik JF. Molecular Genetic Analysis of Ukrainian Families with Congenital Cataracts. Children. 2023; 10(1):51. https://doi.org/10.3390/children10010051
Chicago/Turabian StyleJiao, Xiaodong, Mariia Viswanathan, Nadiia Fedorivna Bobrova, Tatiana Viktorivna Romanova, and J. Fielding Hejtmancik. 2023. "Molecular Genetic Analysis of Ukrainian Families with Congenital Cataracts" Children 10, no. 1: 51. https://doi.org/10.3390/children10010051