
Organoid Technology and Its Role for Theratyping Applications in Cystic Fibrosis


1
Department of Medicine, Division of General Pathology, University of Verona, 37134 Verona, Italy
2
Cystic Fibrosis Centre, Azienda Ospedaliera Universitaria Integrata Verona, 37126 Verona, Italy
*
Authors to whom correspondence should be addressed.
Children 2023, 10(1), 4; https://doi.org/10.3390/children10010004
Submission received: 17 November 2022
/
Revised: 14 December 2022
/
Accepted: 19 December 2022
/
Published: 20 December 2022
(This article belongs to the Special Issue Cystic Fibrosis in Children)
Abstract
:Cystic fibrosis (CF) is a autosomal recessive, multisystemic disease caused by different mutations in the CFTR gene encoding CF transmembrane conductance regulator. Although symptom management is important to avoid complications, the approval of CFTR modulator drugs in the clinic has demonstrated significant improvements by targeting the primary molecular defect of CF and thereby preventing problems related to CFTR deficiency or dysfunction. CFTR modulator therapies have positively changed the patients’ quality of life, especially for those who start their use at the onset of the disease. Due to early diagnosis with the implementation of newborn screening programs and considerable progress in the treatment options, nowadays pediatric mortality was dramatically reduced. In any case, the main obstacle to treat CF is to predict the drug response of patients due to genetic complexity and heterogeneity. Advances in 3D culture systems have led to the extrapolation of disease modeling and individual drug response in vitro by producing mini organs called “organoids” easily obtained from nasal and rectal mucosa biopsies. In this review, we focus primarily on patient-derived intestinal organoids used as in vitro model for CF disease. Organoids combine high-validity of outcomes with a high throughput, thus enabling CF disease classification, drug development and treatment optimization in a personalized manner.
1. Introduction
Until a few years ago, treatments for Cystic Fibrosis (CF) were mainly based on relieving symptoms: physiotherapy to enhance airway clearance and combat lung infections and inflammation, nutritional status management and, in case of end-stage lung disease, lung transplantation. The fundamental current standards for CF therapy include pancreatic enzyme supplementation, fat-soluble vitamins and high-calorie ingestion to minimize pancreatic insufficiency and intestinal malabsorption, anti-inflammatory drugs, antibiotics and mucolytics [1,2,3]. Over the last decade, new therapeutic strategies have been proposed and a new class of drugs named CFTR modulators has been included in the therapeutic care of patients that are currently qualified for treatment. The development of new CF therapies has brought benefits in preventing disease complications, improving individual patient well-being and increasing survival rates. In fact, until the 1960s, CF was a fatal and incurable disease in infancy and today most people with CF are reaching adulthood. Pediatric mortality was dramatically reduced and the survival of CF patients has continuously improved with many individuals living up to 40–50 years in some countries today. Indeed, despite care being based on well-established guidelines, there are many health status disparities among CF patients according to healthcare systems, adherence to therapies, treatment type (route of administration, duration, number of daily medications, etc.), as well as patient socio-personal characteristics and genetic background [4]. Poor treatment adherence has been reported in CF and may lead to worse health outcomes and greater healthcare use. Because each patient is different in terms of lifestyle and social and economic aspects, individual motivational support and personalized educational/training courses can help the patient understand the importance of adhering to the therapeutic regimen to obtain the best clinical benefits. Developing a stronger relationship between patients, families/caregivers, and clinical CF researchers could be the first step to improving the therapeutic compliance of patients and relatives, especially critical in the case of children, and setting up a patient-oriented research infrastructure that promotes the translation of research into clinical impact.
The extensive knowledge obtained in this field has greatly modified the practices of care and outlook for CF pediatric patients. During the last 10 years, the use of newborn bloodspot screening (NBS) for the early diagnosis of children with CF has become widely adopted. Earlier diagnosis and CFTR-targeted therapies have led to efficient improvement of the quality of life. The increasing availability of CFTR-targeted drugs that may halt or severely reduce the disease progression and potentially interrupt the pathological sequences leading to CF organ complications provides the rationale for proposing early treatment (including during pregnancy) to reduce or prevent long-term consequences of the disease. As a result, a larger number of CF-affected children could start a treatment path even a few weeks after the birth, a well before showing symptoms or irreversible organ damage. Indeed, children who had access to earlier diagnosis, which means earlier access to medical management and intervention, show better health outcomes at an older age when compared to those children who had a later CF diagnosis [5,6]. The evidence supporting the clinical benefits of NBS programs have been extensively reviewed [7,8,9,10,11].
Furthermore, Szczesniak et al. [12] showed how lung function decline can be used as a parameter to identify individuals who have a higher potential to obtain an advantage whether from new therapies or those already available. For the possibility that the clinical manifestation of CF could be prevented, modulator therapy is increasingly used in younger children and even infants [13,14,15,16]. Over the last decade, significant efforts into high-throughput screening (HTS) of small molecule libraries have enabled the identification of CFTR modulators. CFTR modulator drugs have been described for the first time in 2003 [17]. Ivacaftor (Vertex Pharmaceuticals, MA, US) received a marketing authorization valid throughout the EU on 23 July 2012 (FDA approval on 31 January 2012) thus opening a new era in the treatment of this severe disease. The CFTR modulators currently available in clinic for CF are: ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, elexacaftor/tezacaftor/ivacaftor (Vertex Pharmaceuticals, MA, USA) and are currently revolutionizing the management of patients with CF, particularly those with at least one F508del variant (up to 85% of patients worldwide). These drugs primarily target CFTR variants that present a gating defect (class III variants) or a processing defect (Class II variants), but data in vitro and in vivo indicate how these drugs can be effective in other types of variants that affect CFTR function and/or processing. Table 1, Table 2, Table 3 and Table 4 summarize the current status of indications from the Food and Drugs Administration (FDA) approved CFTR modulators and the array of CFTR variants for which they are approved.
Since 2018, when the combinations Tezacaftor/Ivacaftor and Tezacaftor/Ivacaftor/Elexavaftor were marketed and introduced into clinical practice, the number of mutations that are responsive to modulators has steadily increased. Figure 1 shows how many mutations are targeted by a single CFTR modulator or by their combination. All variants initially approved for Ivacaftor treatment subsequently became eligible for treatment with Ivacaftor combined with one or two correctors. To date, approximately 50% of the mutations identified as responsive to modulators can be targeted by Ivacaftor or the combinations Tezacaftor/Ivacaftor and Tezacaftor/Ivacaftor/Elexacaftor. The number of responsive variants approved for Symkevi and Kaftrio are 2 and 32 respectively. On the basis of the clinical picture and the tolerability of the patient, it is the clinician’s responsibility to choose the therapy.
Alongside conventional therapies, CFTR modulators represent an important advance in the management of CF, as instead of treating the consequences of CFTR dysfunction, they target the underlying cause associated with CFTR mutations [18].
Nevertheless, the broad range of CFTR mutation classes with various intracellular consequences, epigenetics, modifying genes, individual responsiveness/tolerance to drugs, and still unknown reasons lead to a huge variability in the clinical phenotype of CF. Indeed, CF shows a huge variability between patients, for which several possible explanations can be proposed. Firstly, in the CFTR gene itself, over 2100 CFTR variants have been reported in the Cystic Fibrosis Mutation Database (accessed on 31 October 2022: https://cftr2.org/). Only ~20% of them are proven as CF disease-causing variants. For those mutations for which the functional defect has not yet been thoroughly studied prediction of the clinical manifestations is particularly challenging [19,20]. Secondly, other variables include genetic modifiers that regulate CFTR or other genes’ function (in epithelial or non-epithelial cells), and interactions with the environment [21,22]. As a consequence, phenotypic variations and variability in response to CFTR modulators can be observed even in patients with identical CF mutations. For this reason, each CF patient is unique in terms of response, compliance and tolerance to drugs and disease progression. Although it is clear that in vitro systems cannot recapitulate the complex interactions occurring between drugs and the whole organisms, nevertheless the availability of a patient’s avatar reproducing at least its genomic background at the cellular level represent a significant step forward in this direction. The available data suggest indeed that the capability of the channel to respond to agonist/s and the efficacy of modulators in correcting the molecular defect can be properly investigated using any of these models.
The main treatment barrier for CF is to predict the drug response of patients because of genetic complexity and heterogeneity. The most frequent mutations in large groups of patients have been well described with regards to clinical effects and CFTR-targeted treatment options associated with them but this is not the case for rare, orphan mutations identified in only a few patients worldwide, described in such a small patient population that classical clinical trial studies are not feasible [19]. Therefore, there is an urgent need to evaluate the individual drug response in patient-derived model systems that reflect pharmacological treatment efficacy in vivo. The ultimate goal is to avoid a try-and-error approach for expensive treatments with potential side effects.
2. Alternatives to Conventional Clinical Trials
Since developing a specific drug for each of the CF causing variants is infeasible, other than probably not even necessary, they have been classified according to their molecular mechanisms of the defects and their response to modulators [23,24]. Through a personalized medicine approach, it has been possible to prescribe already commercially available drugs to patients with less common CF mutations, considering that patients with mutations belonging to the same group can be treated by the same therapeutic scheme. Due to the theratyping process and considerable progress in the treatment options, a larger population of individuals with CF (Table 2, Table 3 and Table 4) may benefit from the drug and potentially be cured, although there is still a long way to go for ultra-rare and orphan mutations. Updated information can be retrieved at https://www.vertextreatmentshcp.com/eligibility-tool.
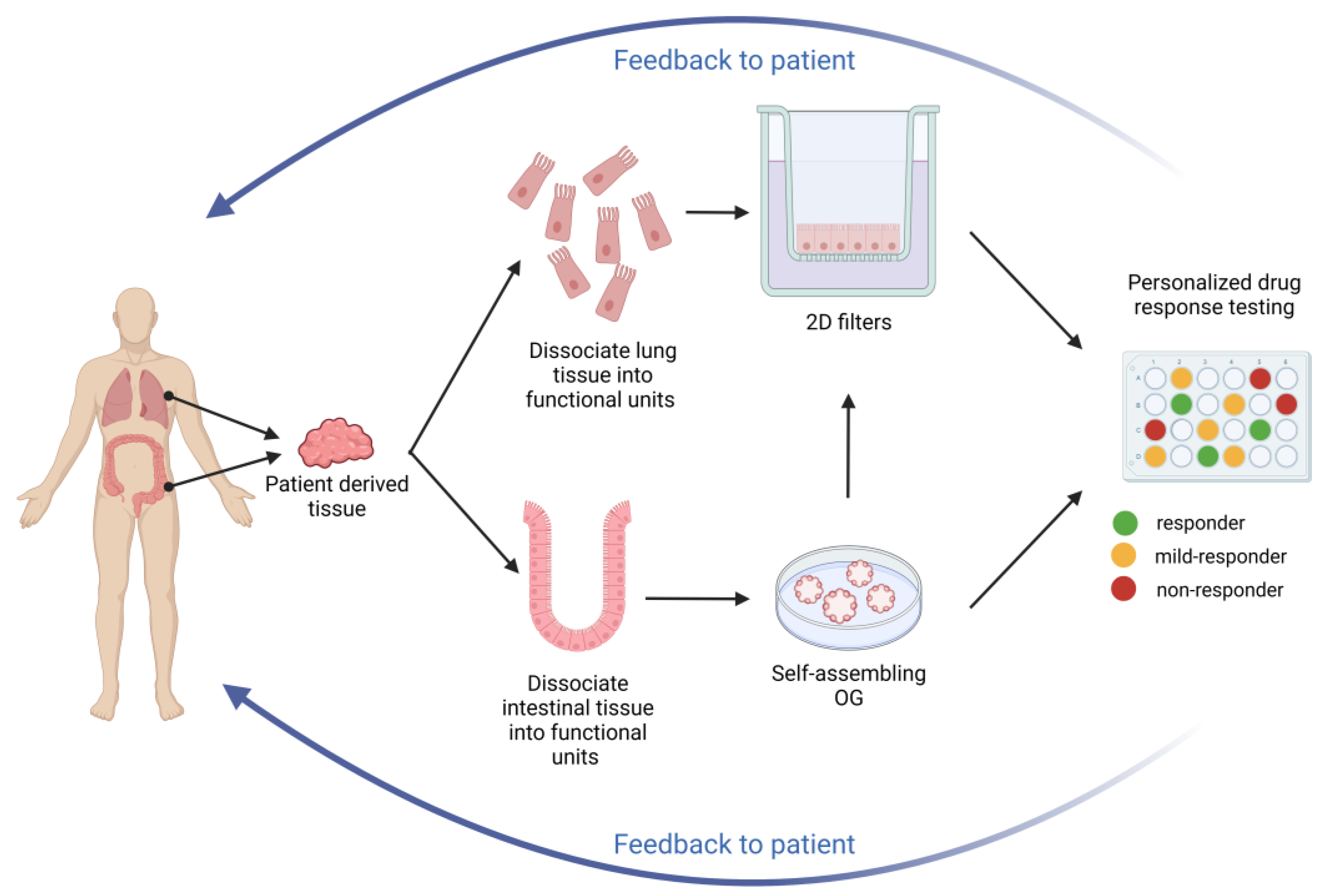
Considering the current high price of CFTR-targeting molecules and variability in the clinical phenotype of CF, even in patients with identical or similar genotypes, it would be ethically more correct to test the efficacy of drugs before administering them to the patient [25] because there are people who benefit from the drug, but there are also individuals who do not obtain an advantage and even those who have adverse effects. To identify effective treatments and avoid undesirable effects, an emerging alternative to genotype-based drugs in CF is personalized/precision medicine, i.e., to determine whether rare CFTR mutations respond to existing (or new) CFTR modulators by pre-evaluating them directly on the patient’s tissues ex vivo, which is now also termed theratyping. This approach proposes the advantage of directly selecting the best drug, or drug combination, for that individual and his/her combined mutations by pre-assessing the efficacy of CFTR-target drugs directly on specimens obtained from CF subject and as a consequence could diminish the approval of treatments that could be ineffective or harmful and create a high expectation to the patient and family members. Predictions based on patient-derived materials as a starting point are nonetheless a more achievable approach than the clinical trial of each CFTR modulator in an N-of-1 trial [26].
Children have the same right to evidence-based therapy as adults but data extrapolation from adults may be inappropriate or misleading [27], therefore it is important to elaborate well-accepted patient-oriented research tools to predict CF treatment response in the pediatric population as well.
3. Cell Models for Studying CF Disease Pathogenesis and Therapy
Experimental models based on the use of in vitro cell cultures have allowed for obtaining many and key information on the biological activity of the CFTR protein and its molecular defects, moreover they have permitted the screening of molecules with different pharmacological activities and to evaluate their pharmacological effects suggesting that cell response in vitro could be predict the clinical impact.
Immortalized epithelial cell lines such as A549, BEAS-2B, Calu-3, CFBE41o- and 16HBE14o-, are suitable models, easy to culture and expand [28,29,30]. However, these immortalized cell lines are derived from lung tumor cells or have been transformed and therefore lack original lung cell characteristics and have some disadvantages due to immortalization strategy that can induce genetic instability, karyotype anomalies and altered gene expression [31,32]. Of note is the fact that drug therapy recovery intervention of CFTR mutations is greatly affected by the cell background [33,34,35].
Fischer Rat Thyroid (FRT) cells that ectopically express CFTR cDNA are the pre-clinical, high-throughput model that has been mostly used to successfully develop CFTR modulators. The recent FDA approval for label extension of ivacaftor and ivacaftor/tezacaftor/elexacaftor to patients with different mutations was based primarily on laboratory evidence of efficacy in FRT cells [36,37]. However, this cellular model has intrinsic limitations: FRT cells were developed from Fischer rat thyroid gland, as such its protein folding machinery is not human and this condition might affect the response to treatment [34]. It is equally clear that the same model cannot be minimally predictive of variations in intronic sequences, and that the transfection under an exogenous promoter might alter a proper protein level and processing. As a consequence, the observed effects might not closely match the in vivo situation.
To evaluate individual CFTR modulators’ responses, several assays using CF patient-derived materials have been implemented and are widely used [38,39,40,41,42,43]. Ex-vivo individual-derived specimens, such as human bronchial epithelia (HBE), human nasal epithelial (HNE), intestinal organoids (OGs) and nasal as well as lung spheroids resemble parental organ epithelium morphology and functionality and reflect the complete genetic background of the subjects. These features permit us to come closer to evaluating the response in individual genetic backgrounds and are expected to better predict the clinical effectiveness of the given treatment.
Primary HBE cells are typically obtained by invasive procedures (bronchoscopy or lung transplantation) from lungs with advanced/end-stage disease that may or may not reflect cells’ behavior in early disease. They are usually available in a limited number of severely ill patients so cannot be used for large-scale or theratyping studies.
HNE cells seem to be a good surrogate for human bronchial epithelial cells. They are collected by minimal-invasive procedures such as nasal brushing or scraping of the lower turbinates. The current gold standard for modeling the primarily affected CF lung epithelium is air–liquid interface (ALI) culture of human nasal epithelial cells [39]. However, this nasal cell culture approach has some limitations: it requires a high number of cells, lengthy differentiation protocols, cells have limited ability to expand and HNE-derived cells are not necessarily representing the features of lower airways [44].
Nevertheless, in the last few years, several research groups have explored several approaches that allow for isolation, expansion, and differentiation of primary nasospheroids [40,45,46,47,48]. Recently a standardized protocol was proposed [49]. Nasal brush biopsies collection from infants through to adults is well known across most centers and can be performed with risk comparable to that of a nasopharyngeal swab for virus detection. In addition, the demonstration of the correlation between CFTR modulator responses in nasal and intestinal OGs provides early evidence that CFTR functional assay in nasal airway OGs can also be used to predict modulator efficacy in a genotype-dependent manner [48,50,51].
Another tissue representative of CF disease is the gastrointestinal tract which is affected in utero or early after birth by diseases such as meconium ileus and pancreatic insufficiency, the latter featuring typical pancreatic cysts after which the disease was named. Interestingly when nasal and intestinal mucosa were compared as in vivo biomarkers of CFTR function to distinguish people with CF (pwCF) and healthy controls, Intestinal Current Measurement (ICM) was found superior to Nasal Potential Difference (NPD) and ICM demonstrated substantially greater power than NPD to detect low levels of residual CFTR function [52,53] suggesting a potential superiority of intestinal over nasal mucosa for theratyping applications. In 2009, Sato et al. developed the basis for intestinal organoid technology [54] that recapitalized in-vivo tissue architecture forming three dimensional structures that can develop in a crypt-like epithelium [55]. In 2011, human intestinal OG cultures were described by the same group [56]. Organoid culture protocol requires a delicate balance of several growth factors such as Wnt, R-spondin and Noggin plus a specific basement membrane matrix. Intestinal OGs could be greatly expanded in vitro over long periods without losing their stemness and biobanked for future use without a need for genetic modifications or further patient inconvenience for repeated biopsies [56]. Human intestinal OG can be grown from intestinal crypt fragments isolated following a rectal biopsy procedure that causes only limited discomfort to patients being painless, usually well accepted by patients and feasible in people of all age groups (including newborns) without a need for anesthesia/sedation [57]. ICM in rectal biopsies have been included for decades in the diagnostic algorithm for CF and CFTR related disorders [58,59], in particular to aid establish or refute a diagnosis of CF in patients with equivocal sweat test or genetic testing results [60], and in many cases rectal samples can be used for generation of rectal OGs after ICM.
Intestinal OGs remain the most advanced three-dimensional in vitro model for CF to date. Other than being a primary target organ in CF it is worth noting that CFTR represents the dominant channel responsible for ion and fluid secretion in gastrointestinal cells, which make intestinal OGs valuable models to investigate CFTR function and modulation [61,62,63]. Moreover, while the airways are significantly affected, the intestine is not significantly affected by chronic inflammation and infection with CF pathogens. Furthermore, the lack of significant chronic organ damage and remodeling in the intestine is a factor that reduces the chance to have CFTR channel function affected independently of the presence of CFTR variants [64]. Finally, intestinal OGs develop fast from the biopsy which results in a shorter time for readout, likely derived from the exceptionally high cell turnover in the intestinal epithelium that renews itself within 3–5 days.
4. Disease Modeling of Intestinal OG in Infants and Children
In vitro human intestinal OGs provide a unique development model that can be applied to study intestinal prematurity diseases such as necrotizing enterocolitis, short bowel syndrome, Hirschsprung’s disease, infectious disease in the intestine and genetic diseases such as CF [65].
Intestinal OGs are robust tools for studying genetic diseases due to their genetic and phenotypic stability such as CF and cancer where genetics can influence disease severity, prognosis, and drug efficacy [66,67].
Certainly, research in the CF field provides an appropriate example where OG technology has generated a relevant impact. Here, patient-derived intestinal OGs have been used for disease modeling, drug screening, and personalized medicine and have provided advantages in the development of CFTR-modulating drugs. Intestinal OGs have demonstrated high validity and high-throughput potential to predict drug efficacy in individuals with CF [68,69].
The use of OG in high-throughput screening enables pre-clinical testing of many compounds on patient-specific tissues. In vitro tests based on patient-derived rectal OGs can facilitate rapid access to new treatment or can assist in the identification of variants that are currently not registered for treatment but can potentially be responsive to CFTR-targeting drugs. This general approach is underway within the HIT-CF program (e.g., Human Individualized Therapy: HIT-CF program in Europe, www.hitcf.org) using patient-derived rectal OG to assess cellular responses to various CFTR modulators. This European project aims to develop personalized treatments for PwCF allowing those with rare CFTR mutations access to treatment.
The potential of intestinal OGs as a pre-clinical model and for personalized medicine has been demonstrated in different studies [70,71,72]. The first study to directly compare OG response and clinical phenotype prospectively included 34 newborns with CF. Newborns were clustered into low responders or high responders. Low response in OG was related to increased pulmonary and pancreatic disease parameters at the age of 1 year. In CF children, intestinal OG response corresponds with clinical phenotypes at 1 year of age as well as in vivo sweat Cl- concentration (SCC) and ICM. Interestingly, in cases where SCC and ICM disagreed, FIS appeared to correctly align with the clinical indicators allowing the in vivo residual CFTR function to be accurately estimated for each patient [73].
Notably, Berkers et al. reported a strong correlation between in vivo and in vitro response of CFTR modulators indicating how OG can play a key role in personalized medicine approach [74]. The predictive value of intestinal OG response was demonstrated to significantly correlate with the most important therapeutic endpoints: ICM, reduction in SCC, and improvements in lung function measured as the volume of air that can forcibly be blown out in one second, after full inspiration and expressed as predicted percent FEV1 (ppFEV1) after administering the CFTR modulator therapies to patients. In addition, in vitro CFTR modulator responses in OG displayed excellent accuracy for stratifying drug responders from non-drug responders [68,74] suggesting the organoid model is suitable for guiding label extension or compassionate use for PwCF with rare genotypes [71,75].
Data from Pranke et al., also indicate that in vitro analysis of nasal epithelium may correlate with in vivo outcomes [76] and airway epithelium cultures obtained from nasal brushing of a CF child have been demonstrated to be a useful model for theratyping [77,78]. Although bronchoalveolar lavage has been proposed for this purpose, a simple nasal brushing is a more easily performed, well tolerated, and minimally invasive technique. It is therefore possible to study upper airway epithelial cells in very young CF infants to obtain information on the state of inflammation, infection, CFTR conductance and modulator drugs’ responsiveness [79]. Whether they represent a faithful model for lower airway epithelia, and whether this truly represents a requisite for theratyping applications require further studies [44].
5. CFTR Bioassay in Intestinal Organoids
Predicting individual patient response necessary to optimize the application of a given treatment, based on the use of currently available and future CFTR modulators, require a robust and standardized pre-clinical test [80].
The majority of data describing the readout for quantifying the CFTR function and how it can be recovered by CFTR modulators in intestinal organoids derived from the Forskolin-Induced Swelling test (FIS). This test was described first by Dekkers and collaborators in 2013 [43]. Exposing intestinal OG to Forskolin causes the cells to rapidly increase their levels of cyclic adenosine monophosphate (cAMP), resulting in the opening of the CFTR channel. As chloride ions move through the channel and into the lumen, the OGs increase in size due to luminal water intake. The FIS phenotype is absent in human and mouse OG lacking functional CFTR gene products (e.g., two disease-causing mutations or CFTR knockout) and is inhibited by chemical CFTR inhibitors, supporting full CFTR-dependency of the FIS readout [43]. The FIS assay allows for considerable throughput since initially it had been set up in 96-well plates [43], but recently it was developed into a 384-well-based high-throughput screening format.
FIS quantitatively correlates with CFTR function and genotype: healthy control OGs appear already swollen and a further increase in swelling appear marginal due to the presence of liquid in the lumen that cannot accumulate over physiological limits. Absent FIS is present in patients with CFTR null alleles, is decreased in variants classified as mild, and is strongly reduced in OG derived from severe variants [81,82].
FIS of patient-derived OGs is very helpful to measure patient-specific CFTR activity, comparing CFTR function between individuals presenting with different and even identical CFTR mutations, and quantifying individual CFTR modulator response. Some studies found, however, that in healthy control OG FIS rate is negatively affected by fluid-filled lumens before forskolin treatment leading to an underestimation of CFTR function. The OG initial starting area must be similar to obtain accurate and reliable swelling results. Therefore, to compare healthy donor-derived OG and CF OGs, another test was introduced: the steady-state lumen area (SLA) assay in which lumen surface area is measured as a percentage of total OG size [68]. Whereas CF OGs have SLA between 0% and 10% of the total OG area, healthy control SLA is between 40% and 80%. Steady-state differences in luminal OG area exist between healthy control and CF OGs independent of forskolin. Healthy control rectal OGs have large fluid-filled lumens, suggesting the presence of functional CFTR and physiological cAMP-dependent signaling during standard culture conditions resulting in the luminal transport of salt and fluid. CF OHs have limited luminal volume or do not have lumens that are easily recognizable during visual inspection.
FIS and SLA are complementary assays. FIS of CF and healthy controls are not directly comparable therefore it is more suited to only compare CF conditions. SLA facilitates comparison between CF and healthy control OG but has a limited resolution to discriminate at lower CFTR function levels associated with severe CF disease.
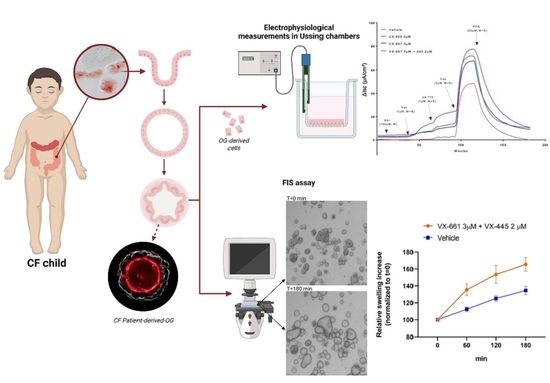
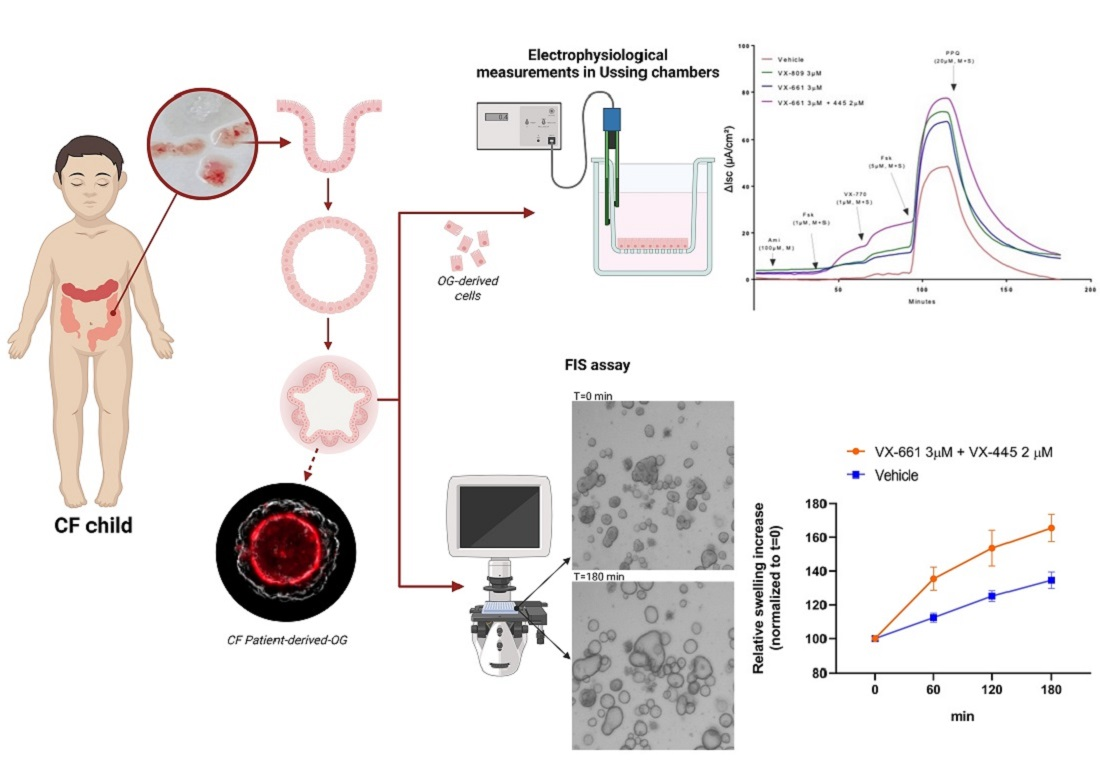
Despite these useful features, a limitation of FIS assay is that the selective delivery of compounds to the apical or basolateral compartment in OG growing as 3D structures is complicated. Apical stimulation can be performed in OG by microinjection, but this is especially challenging for CF OGs because of their limited luminal volume. More recent research has described protocols to generate two-dimensional monolayers grown on porous membrane filters from dissociated 3D OG [83]. A monolayer of intestinal OGs provides easy access to the apical side and allows for the assessment of CFTR rescue and function by traditional electrophysiological measurements in Ussing chambers [84,85]. Furthermore, Ussing chamber measurements in 2D monolayers allow us to separately measure CFTR-mediated Cl− and HCO3- transport [85]. This is of great importance as modulator drugs capable of restoring CFTR folding have demonstrated a different impact on CFTR-dependent Cl- transport compared to HCO3-, suggesting the occurrence of different behavior of rescued CFTR on different genotypes that might differentially affect chloride and bicarbonate transport [86,87]. This observation might have an impact on the predicted in vivo efficacy of a given modulator in the specific CFTR variant and cannot be detected by FIS assay which is unable to distinguish whether the swelling is associated with chloride only or other anions.
Epithelial monolayers of intestinal OG may be a valuable tool to evaluate ion transport of different channels/transporters in different culture conditions that can be precisely modified for the purpose other than assisting in diagnosis and precision medicine testing: Indeed, CFTR-dependent intestinal epithelial ion transport measured on rectal organoid-derived monolayers of subjects carrying distinct CF genotypes correlates well with donor-matched native ICM and FIS of 3D intestinal OG [88]. The high dynamic range of the response of monolayers derived from intestinal organoids demonstrated suitable to identify none/very low to high residual CFTR function and WT-CFTR currents might be used as a reference value for comparing the efficacy of modulator drugs on various CFTR variants [85].
Another CFTR bioassay with intestinal OG that can be used as a complementary diagnostic test is rectal organoid morphology analysis (ROMA). ROMA is based on the measurement of two morphological parameters of the organoids: (1) the intensity ratio (IR) that evaluates the presence or absence of a central lumen, and (2) the circularity index (CI) that measures the roundness of the organoids [89]. As outcomes, the test allows for discrimination of CF patients from healthy subjects and it evaluates if there is a response of OG to modulator treatments. The functional recovery of the CFTR protein is reflected in a more open central lumen and a more circular shape, measured by IR and CI respectively. Current strategies for theratyping are summarized in Figure 2.
6. Limitations of Organoid-Based Assays
The most feasible personalized biomarker platforms for testing CFTR function and CFTR modulator responsiveness in the foreseeable future will likely be intestinal and nasal cells and derived OG as they represent key targets and easily accessible sites for cell procurement, with the former currently representing a more robust and reproducible platform based on available data in the literature [90]. In addition to the general limitations of any single tissue-based organoid assay (e.g., lacking host environment-related signal factors) already discussed in previous reviews [91,92], additional specific limitations are currently associated with technical issues such as the need to develop common Standard Operating Procedures (SOPs) among laboratories, the need to train highly skilled personnel and the costs associated to tissue procurement, processing and analysis other than the need to introduce common platforms permitting reference samples and data exchange among laboratories. Rather important is the development of quality control, standardization, and compliance with technical standards possibly via certification. The recognition of the use of primary cells as suitable biomarkers for theratyping applications might eventually be required by certifying agencies, such as the European Medicines Agency (EMA) and the FDA, to enter the approval procedure of future medications or to include new CFTR variants in the approved list. All of these issues are summarized in Table 5.
7. Conclusions
The common goal is to give new hope to all PwCF by changing the general perception of CF as a lethal disease. Prospects aim to identify new CFTR modulators and to extend eligibility for drugs now adopted for clinical use to patients with CFTR variants without approved treatment so that all CF patients can eventually be treated with a matched therapy. All these new perspectives will hopefully bring existing and novel CFTR modulators faster to patients with rare mutations and in the long run also bring the most efficacious drug to individual patients who carry more common mutations. The possibility to develop and biobank patient’s “avatars”, such as OGs, that can be developed at birth, recovered, and analyzed once new drugs might become available (a highly likely possibility in the foreseeable future) without the need for resampling ensures previously unthinkable opportunities to match the treatment with the individual molecular asset and develop a much needed theratyping approach, especially for those patients whose variants are so rare to require an N-of-1 approach.
It remains to establish the relationship between the results obtained from patient-derived materials and the long-term results, but as with any new approach, these data will only be collected as they become available.
Author Contributions
Conceptualization, J.C., C.S. and P.M.; resources, C.S., P.M.; writing—original draft preparation, J.C.; writing—review and editing, C.S., P.M.; visualization, J.C.; supervision, C.S., and P.M.; All authors have read and agreed to the published version of the manuscript.
Funding
Research activity was supported by the Italian Cystic Fibrosis Research Foundation (FFC), grant numbers FFC #13/2018 (Delegazione FFC di Tradate Gallarate) to C.S., and #9/2020 (Delegazione FFC di Pesaro, Rivarolo Canavese e Gruppo sostegno FFC di Fidenza) to P.M.; Lega Italiana Fibrosi Cistica onlus (C.S. and P.M.); and American CFF, grant number Assael 08A0 (P.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All datasets utilized are indicated throughout the text.
Conflicts of Interest
The authors declare no conflict of interest. P.M. acted as paid expert testimony for Vertex Pharmaceuticals, MA, USA.
References
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS Best Practice Guidelines: The 2018 Revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanazio, R.A.; Filho, L.V.R.F.S.; Vergara, A.A.; Ribeiro, A.F.; Riedi, C.A.; Procianoy, E.; Adde, F.V.; Reis, F.J.C.; Ribeiro, J.D.; Torres, L.A.; et al. Brazilian Guidelines for the Diagnosis and Treatment of Cystic Fibrosis. J. Bras. Pneumol. 2017, 43, 219–245. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E. Managing Cystic Fibrosis: Strategies That Increase Life Expectancy and Improve Quality of Life. Am. J. Respir. Crit. Care Med. 2011, 183, 1463–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, S.; Mainz, J.G.; Gala, S.; Tabori, H.; Grossoehme, D. Adherence to Therapies in Cystic Fibrosis: A Targeted Literature Review. Expert Rev. Respir. Med. 2017, 11, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.W.; White, T.B. Newborn Screening for Cystic Fibrosis: An Opportunity to Improve Care and Outcomes. J. Pediatr. 2005, 147, S2–S5. [Google Scholar] [CrossRef] [PubMed]
- Coffey, M.J.; Whitaker, V.; Gentin, N.; Junek, R.; Shalhoub, C.; Nightingale, S.; Hilton, J.; Wiley, V.; Wilcken, B.; Gaskin, K.J.; et al. Differences in Outcomes between Early and Late Diagnosis of Cystic Fibrosis in the Newborn Screening Era. J. Pediatr. 2017, 181, 137–145. [Google Scholar] [CrossRef]
- Wagener, J.S.; Zemanick, E.T.; Sontag, M.K. Newborn Screening for Cystic Fibrosis. Curr. Opin. Pediatr. 2012, 24, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, M. Newborn Screening for Cystic Fibrosis: The Motion against—Voices in the Wilderness. Paediatr. Respir. Rev. 2008, 9, 295–300. [Google Scholar] [CrossRef]
- McKay, K.; Wilcken, B. Newborn Screening for Cystic Fibrosis Offers an Advantage over Symptomatic Diagnosis for the Long Term Benefit of Patients: The Motion For. Paediatr. Respir. Rev. 2008, 9, 290–294. [Google Scholar] [CrossRef]
- Southern, K.W.; Mérelle, M.M.E.; Dankert-Roelse, J.E.; Nagelkerke, A. Newborn Screening for Cystic Fibrosis. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Grosse, S.D.; Rosenfeld, M.; Devine, O.J.; Lai, H.J.; Farrell, P.M. Potential Impact of Newborn Screening for Cystic Fibrosis on Child Survival: A Systematic Review and Analysis. J. Pediatr. 2006, 149, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Szczesniak, R.D.; Li, D.; Su, W.; Brokamp, C.; Pestian, J.; Seid, M.; Clancy, J.P. Phenotypes of Rapid Cystic Fibrosis Lung Disease Progression during Adolescence and Young Adulthood. Am. J. Respir. Crit. Care Med. 2017, 196, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Chilvers, M.; Higgins, M.; Tian, S.; et al. An Open-Label Extension Study of Ivacaftor in Children with CF and a CFTR Gating Mutation Initiating Treatment at Age 2–5 Years (KLIMB). J. Cyst. Fibros. 2019, 18, 838–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, M.; Wainwright, C.E.; Higgins, M.; Wang, L.T.; McKee, C.; Campbell, D.; Tian, S.; Schneider, J.; Cunningham, S.; Davies, J.C.; et al. Ivacaftor Treatment of Cystic Fibrosis in Children Aged 12 to <24 Months and with a CFTR Gating Mutation (ARRIVAL): A Phase 3 Single-Arm Study. Lancet Respir. Med. 2018, 6, 545–553. [Google Scholar] [CrossRef]
- Milla, C.E.; Ratjen, F.; Marigowda, G.; Liu, F.; Waltz, D.; Rosenfeld, M. Lumacaftor/Ivacaftor in Patients Aged 6–11 Years with Cystic Fibrosis and Homozygous for F508del-CFTR. Am. J. Respir. Crit. Care Med. 2017, 195, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Springsteel, M.F.; Galietta, L.J.V.; Ma, T.; By, K.; Berger, G.O.; Yang, H.; Dicus, C.W.; Choung, W.; Quan, C.; Shelat, A.A.; et al. Benzoflavone Activators of the Cystic Fibrosis Transmembrane Conductance Regulator: Towards a Pharmacophore Model for the Nucleotide-Binding Domain. Bioorg. Med. Chem. 2003, 11, 4113–4120. [Google Scholar] [CrossRef]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the Disease Liability of Variants in the Cystic Fibrosis Transmembrane Conductance Regulator Gene. Nat. Genet. 2013, 45, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Sosnay, P.R.; Salinas, D.B.; White, T.B.; Ren, C.L.; Farrell, P.M.; Raraigh, K.S.; Girodon, E.; Castellani, C. Applying Cystic Fibrosis Transmembrane Conductance Regulator Genetics and CFTR2 Data to Facilitate Diagnoses. J. Pediatr. 2017, 181, S27–S32. [Google Scholar] [CrossRef]
- Kraiczy, J.; Nayak, K.M.; Howell, K.J.; Ross, A.; Forbester, J.; Salvestrini, C.; Mustata, R.; Perkins, S.; Andersson-Rolf, A.; Leenen, E.; et al. DNA Methylation Defines Regional Identity of Human Intestinal Epithelial Organoids and Undergoes Dynamic Changes during Development. Gut 2019, 68, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Kerschner, J.L.; Gosalia, N.; Neems, D.; Gorsic, L.K.; Safi, A.; Crawford, G.E.; Kosak, S.T.; Leir, S.-H.; Harris, A. Differential Contribution of Cis -Regulatory Elements to Higher Order Chromatin Structure and Expression of the CFTR Locus. Nucleic Acids Res. 2016, 44, 3082–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielenski, J.; Tsui, L.-C. Cystic Fibrosis: Genotypic And Phenotypic Variations. Annu. Rev. Genet. 1995, 29, 777–807. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.J.; Smith, A.E. Molecular Mechanisms of CFTR Chloride Channel Dysfunction in Cystic Fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Marson, F.A.L. Personalized or Precision Medicine? The Example of Cystic Fibrosis. Front. Pharmacol. 2017, 8, 390. [Google Scholar] [CrossRef]
- Schork, N.J. Personalized Medicine: Time for One-Person Trials. Nature 2015, 520, 609–611. [Google Scholar] [CrossRef] [Green Version]
- Dobra, R.; Bentley, S.; Edmondson, C.; Ovens, M.; Saunders, C.; Short, C.; Wilson, G.; Davies, J.C.; Bush, A. Going the Extra Mile: Why Clinical Research in Cystic Fibrosis Must Include Children. Children 2022, 9, 1080. [Google Scholar] [CrossRef]
- Gruenert, D.C.; Willems, M.; Cassiman, J.J.; Frizzell, R.A. Established Cell Lines Used in Cystic Fibrosis Research. J. Cyst. Fibros. 2004, 3, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Bednarski, C.; Tomczak, K.; vom Hövel, B.; Weber, W.-M.; Cathomen, T. Targeted Integration of a Super-Exon into the CFTR Locus Leads to Functional Correction of a Cystic Fibrosis Cell Line Model. PLoS ONE 2016, 11, e0161072. [Google Scholar] [CrossRef] [Green Version]
- Bellec, J.; Bacchetta, M.; Losa, D.; Anegon, I.; Chanson, M.; Nguyen, T. CFTR Inactivation by Lentiviral Vector-Mediated RNA Interference and CRISPR-Cas9 Genome Editing in Human Airway Epithelial Cells. Curr. Gene Ther. 2015, 15, 447–459. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Collnot, E.-M.; Baldes, C.; Becker, U.; Laue, M.; Kim, K.-J.; Lehr, C.-M. Towards an in Vitro Model of Cystic Fibrosis Small Airway Epithelium: Characterisation of the Human Bronchial Epithelial Cell Line CFBE41o-. Cell Tissue Res. 2006, 323, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.S.; Randell, S.H.; Stewart, S.A.; Elenbaas, B.; Hartwell, K.A.; Brooks, M.W.; Fleming, M.D.; Olsen, J.C.; Miller, S.W.; Weinberg, R.A.; et al. Immortalization and Transformation of Primary Human Airway Epithelial Cells by Gene Transfer. Oncogene 2002, 21, 4577–4586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M.; Bacalhau, M.; Ramalho, S.S.; Silva, I.A.L.; Ferreira, F.C.; Carlile, G.W.; Thomas, D.Y.; Farinha, C.M.; Hanrahan, J.W.; Amaral, M.D. Rescue of Mutant CFTR Trafficking Defect by the Investigational Compound MCG1516A. Cells 2022, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Galietta, L.J.V. Influence of Cell Background on Pharmacological Rescue of Mutant CFTR. Am. J. Physiol.-Cell Physiol. 2010, 298, C866–C874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostedgaard, L.S.; Rogers, C.S.; Dong, Q.; Randak, C.O.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Welsh, M.J. Processing and Function of CFTR-ΔF508 Are Species-Dependent. Proc. Natl. Acad. Sci. USA 2007, 104, 15370–15375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durmowicz, A.G.; Lim, R.; Rogers, H.; Rosebraugh, C.J.; Chowdhury, B.A. The U.S. Food and Drug Administration’s Experience with Ivacaftor in Cystic Fibrosis. Establishing Efficacy Using In Vitro Data in Lieu of a Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Vertex Pharmaceuticals Inc. Highlights of Prescribing Information: Trikafta® (Elexacaftor/Tezacaftor/Ivacaftor). 2021. Available online: https://pi.vrtx.com/files/uspi_elexacaftor_tezacaftor_ivacaftor.pdf (accessed on 31 October 2022).
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR Function in Nasal Epithelial Cells from Cystic Fibrosis Patients Predicts Improvement of Respiratory Function by CFTR Modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef] [Green Version]
- Gianotti, A.; Delpiano, L.; Caci, E. In Vitro Methods for the Development and Analysis of Human Primary Airway Epithelia. Front. Pharmacol. 2018, 9, 1176. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term Expanding Human Airway Organoids for Disease Modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Vonk, A.M.; van Mourik, P.; Ramalho, A.S.; Silva, I.A.L.; Statia, M.; Kruisselbrink, E.; Suen, S.W.F.; Dekkers, J.F.; Vleggaar, F.P.; Houwen, R.H.J.; et al. Protocol for Application, Standardization and Validation of the Forskolin-Induced Swelling Assay in Cystic Fibrosis Human Colon Organoids. STAR Protoc. 2020, 1, 100019. [Google Scholar] [CrossRef]
- de Courcey, F.; Zholos, A.V.; Atherton-Watson, H.; Williams, M.T.S.; Canning, P.; Danahay, H.L.; Elborn, J.S.; Ennis, M. Development of Primary Human Nasal Epithelial Cell Cultures for the Study of Cystic Fibrosis Pathophysiology. Am. J. Physiol.-Cell Physiol. 2012, 303, C1173–C1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A Functional CFTR Assay Using Primary Cystic Fibrosis Intestinal Organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Scudieri, P.; Musante, I.; Venturini, A.; Guidone, D.; Genovese, M.; Cresta, F.; Caci, E.; Palleschi, A.; Poeta, M.; Santamaria, F.; et al. Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 2020, 9, 2090. [Google Scholar] [CrossRef] [PubMed]
- Sette, G.; Lo Cicero, S.; Blaconà, G.; Pierandrei, S.; Bruno, S.M.; Salvati, V.; Castelli, G.; Falchi, M.; Fabrizzi, B.; Cimino, G.; et al. Theratyping Cystic Fibrosis in Vitro in ALI Culture and Organoid Models Generated from Patient-Derived Nasal Epithelial Conditionally Reprogrammed Stem Cells. Eur. Respir. J. 2021, 58, 2100908. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Chung, M.-I.; Fioret, B.; Gao, X.; Katsura, H.; Hogan, B.L.M. Lung Organoids: Current Uses and Future Promise. Development 2017, 144, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Anderson, J.D.; Deng, L.; Mackay, S.; Bailey, J.; Kersh, L.; Rowe, S.M.; Guimbellot, J.S. Human Nasal Epithelial Organoids for Therapeutic Development in Cystic Fibrosis. Genes 2020, 11, 603. [Google Scholar] [CrossRef]
- Amatngalim, G.D.; Rodenburg, L.W.; Aalbers, B.L.; Raeven, H.H.; Aarts, E.M.; Sarhane, D.; Spelier, S.; Lefferts, J.W.; Silva, I.A.; Nijenhuis, W.; et al. Measuring Cystic Fibrosis Drug Responses in Organoids Derived from 2D Differentiated Nasal Epithelia. Life Sci. Alliance 2022, 5, e202101320. [Google Scholar] [CrossRef]
- Golec, A.; Pranke, I.; Scudieri, P.; Hayes, K.; Dreano, E.; Dunlevy, F.; Hatton, A.; Downey, D.G.; Galietta, L.; Sermet, I. Isolation, Cultivation, and Application of Primary Respiratory Epithelial Cells Obtained by Nasal Brushing, Polyp Samples, or Lung Explants. STAR Protoc. 2022, 3, 101419. [Google Scholar] [CrossRef]
- Wong, S.L.; Awatade, N.T.; Astore, M.A.; Allan, K.M.; Carnell, M.J.; Slapetova, I.; Chen, P.; Setiadi, J.; Pandzic, E.; Fawcett, L.K.; et al. Molecular Dynamics and Theratyping in Airway and Gut Organoids Reveal R352Q-CFTR Conductance Defect. Am. J. Respir Cell Mol. Biol. 2022, 67, 99–111. [Google Scholar] [CrossRef]
- Silva, I.A.L.; Railean, V.; Duarte, A.; Amaral, M.D. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421. [Google Scholar] [CrossRef]
- Clancy, J.P.; Szczesniak, R.D.; Ashlock, M.A.; Ernst, S.E.; Fan, L.; Hornick, D.B.; Karp, P.H.; Khan, U.; Lymp, J.; Ostmann, A.J.; et al. Multicenter Intestinal Current Measurements in Rectal Biopsies from CF and Non-CF Subjects to Monitor CFTR Function. PLoS ONE 2013, 8, e73905. [Google Scholar] [CrossRef] [PubMed]
- Bagheri-Hanson, A.; Nedwed, S.; Rueckes-Nilges, C.; Naehrlich, L. Intestinal Current Measurement versus Nasal Potential Difference Measurements for Diagnosis of Cystic Fibrosis: A Case–Control Study. BMC Pulm. Med. 2014, 14, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures in Vitro without a Mesenchymal Niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Clevers, H. Growing Self-Organizing Mini-Guts from a Single Intestinal Stem Cell: Mechanism and Applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; van Es, J.H.; van den Brink, S.; van Houdt, W.J.; Pronk, A.; van Gorp, J.; Siersema, P.D.; et al. Long-Term Expansion of Epithelial Organoids From Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Servidoni, M.F.; Sousa, M.; Vinagre, A.M.; Cardoso, S.R.; Ribeiro, M.A.; Meirelles, L.R.; de Carvalho, R.B.; Kunzelmann, K.; Ribeiro, A.F.; Ribeiro, J.D.; et al. Rectal Forceps Biopsy Procedure in Cystic Fibrosis: Technical Aspects and Patients Perspective for Clinical Trials Feasibility. BMC Gastroenterol. 2013, 13, 91. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, K. Cystic Fibrosis: Terminology and Diagnostic Algorithms. Thorax 2006, 61, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Sermet-Gaudelus, I.; Girodon, E.; Vermeulen, F.; Solomon, G.M.; Melotti, P.; Graeber, S.Y.; Bronsveld, I.; Rowe, S.M.; Wilschanski, M.; Tümmler, B.; et al. ECFS Standards of Care on CFTR-Related Disorders: Diagnostic Criteria of CFTR Dysfunction. J. Cyst. Fibros. 2022, 21, 922–936. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15. [Google Scholar] [CrossRef] [Green Version]
- Kunzelmann, K.; Mall, M. Electrolyte Transport in the Mammalian Colon: Mechanisms and Implications for Disease. Physiol. Rev. 2002, 82, 245–289. [Google Scholar] [CrossRef]
- Mall, M.; Wissner, A.; Seydewitz, H.H.; Kuehr, J.; Brandis, M.; Greger, R.; Kunzelmann, K. Defective Cholinergic Cl—Secretion and Detection of K + Secretion in Rectal Biopsies from Cystic Fibrosis Patients. Am. J. Physiol.-Gastrointest. Liver Physiol. 2000, 278, G617–G624. [Google Scholar] [CrossRef] [PubMed]
- Veeze, H.J.; Sinaasappel, M.; Bijman, J.; Bouquet, J.; De Jonge, H.R. Ion Transport Abnormalities in Rectal Suction Biopsies from Children with Cystic Fibrosis. Gastroenterology 1991, 101, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Vitzthum, C.; Mall, M.A. Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis. J. Pers. Med. 2021, 11, 384. [Google Scholar] [CrossRef] [PubMed]
- Chusilp, S.; Li, B.; Lee, D.; Lee, C.; Vejchapipat, P.; Pierro, A. Intestinal Organoids in Infants and Children. Pediatr. Surg. Int. 2020, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [Green Version]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human Primary Liver Cancer–Derived Organoid Cultures for Disease Modeling and Drug Screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; de Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; van de Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing Responses to CFTR-Modulating Drugs Using Rectal Organoids Derived from Subjects with Cystic Fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- Beekman, J.M. Individualized Medicine Using Intestinal Responses to CFTR Potentiators and Correctors: Individualized Medicine Using Intestinal Responses. Pediatr. Pulmonol. 2016, 51, S23–S34. [Google Scholar] [CrossRef]
- de Poel, E.; Lefferts, J.W.; Beekman, J.M. Intestinal Organoids for Cystic Fibrosis Research. J. Cyst. Fibros. 2020, 19, S60–S64. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, A.S.; Fürstová, E.; Vonk, A.M.; Ferrante, M.; Verfaillie, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekman, J.M.; Sarouk, I.; et al. Correction of CFTR Function in Intestinal Organoids to Guide Treatment of Cystic Fibrosis. Eur. Respir. J. 2021, 57, 1902426. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Gogorza Gondra, R.A.; Kruisselbrink, E.; Vonk, A.M.; Janssens, H.M.; de Winter-de Groot, K.M.; van der Ent, C.K.; Beekman, J.M. Optimal Correction of Distinct CFTR Folding Mutants in Rectal Cystic Fibrosis Organoids. Eur. Respir. J. 2016, 48, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Winter-de Groot, K.M.; Janssens, H.M.; van Uum, R.T.; Dekkers, J.F.; Berkers, G.; Vonk, A.; Kruisselbrink, E.; Oppelaar, H.; Vries, R.; Clevers, H.; et al. Stratifying Infants with Cystic Fibrosis for Disease Severity Using Intestinal Organoid Swelling as a Biomarker of CFTR Function. Eur. Respir. J. 2018, 52, 1702529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aalbers, B.L.; Brunsveld, J.E.; van der Ent, C.K.; van den Eijnden, J.C.; Beekman, J.M.; Heijerman, H.G.M. Forskolin Induced Swelling (FIS) Assay in Intestinal Organoids to Guide Eligibility for Compassionate Use Treatment in a CF Patient with a Rare Genotype. J. Cyst. Fibros. 2022, 21, 254–257. [Google Scholar] [CrossRef]
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might Brushed Nasal Cells Be a Surrogate for CFTR Modulator Clinical Response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; Wachter, E.; et al. Two CFTR Mutations within Codon 970 Differently Impact on the Chloride Channel Functionality. Hum. Mutat. 2019, 40, 742–748. [Google Scholar] [CrossRef]
- Terlizzi, V.; Amato, F.; Castellani, C.; Ferrari, B.; Galietta, L.J.V.; Castaldo, G.; Taccetti, G. Ex Vivo Model Predicted in Vivo Efficacy of CFTR Modulator Therapy in a Child with Rare Genotype. Mol. Genet. Genom. Med. 2021, 9, e1656. [Google Scholar] [CrossRef]
- Mosler, K.; Coraux, C.; Fragaki, K.; Zahm, J.-M.; Bajolet, O.; Bessaci-Kabouya, K.; Puchelle, E.; Abély, M.; Mauran, P. Feasibility of Nasal Epithelial Brushing for the Study of Airway Epithelial Functions in CF Infants. J. Cyst. Fibros. 2008, 7, 44–53. [Google Scholar] [CrossRef]
- Lyman, G.H.; Moses, H.L. Biomarker Tests for Molecularly Targeted Therapies—The Key to Unlocking Precision Medicine. N. Engl. J. Med. 2016, 375, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR Protein Processing Defect in Vitro by the Investigational Drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF Airway Epithelial Cell Function in Vitro by a CFTR Potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, C.; VanDussen, K.L.; Miyoshi, H.; Stappenbeck, T.S. Development of a Primary Mouse Intestinal Epithelial Cell Monolayer Culture System to Evaluate Factors That Modulate IgA Transcytosis. Mucosal Immunol. 2014, 7, 818–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozuka, K.; He, Y.; Koo-McCoy, S.; Kumaraswamy, P.; Nie, B.; Shaw, K.; Chan, P.; Leadbetter, M.; He, L.; Lewis, J.G.; et al. Development and Characterization of a Human and Mouse Intestinal Epithelial Cell Monolayer Platform. Stem Cell Rep. 2017, 9, 1976–1990. [Google Scholar] [CrossRef] [Green Version]
- Ciciriello, F.; Bijvelds, M.J.C.; Alghisi, F.; Meijsen, K.F.; Cristiani, L.; Sorio, C.; Melotti, P.; Fiocchi, A.G.; Lucidi, V.; De Jonge, H.R. Theratyping of the Rare CFTR Variants E193K and R334W in Rectal Organoid-Derived Epithelial Monolayers. J. Pers. Med. 2022, 12, 632. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, L.; Baroni, D.; Moran, O. Lumacaftor-Rescued F508del-CFTR Has a Modified Bicarbonate Permeability. J. Cyst. Fibros. 2019, 18, 602–605. [Google Scholar] [CrossRef]
- Fiore, M.; Picco, C.; Moran, O. Correctors Modify the Bicarbonate Permeability of F508del-CFTR. Sci. Rep. 2020, 10, 8440. [Google Scholar] [CrossRef] [PubMed]
- Zomer-van Ommen, D.D.; de Poel, E.; Kruisselbrink, E.; Oppelaar, H.; Vonk, A.M.; Janssens, H.M.; van der Ent, C.K.; Hagemeijer, M.C.; Beekman, J.M. Comparison of Ex Vivo and in Vitro Intestinal Cystic Fibrosis Models to Measure CFTR-Dependent Ion Channel Activity. J. Cyst. Fibros. 2018, 17, 316–324. [Google Scholar] [CrossRef]
- Cuyx, S.; Ramalho, A.S.; Corthout, N.; Fieuws, S.; Fürstová, E.; Arnauts, K.; Ferrante, M.; Verfaillie, C.; Munck, S.; Boon, M.; et al. Rectal Organoid Morphology Analysis (ROMA) as a Promising Diagnostic Tool in Cystic Fibrosis. Thorax 2021, 76, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; de Boeck, K.; Amaral, M.; Davies, J.C.; de Boeck, K.; Drevinek, P.; Elborn, S.; Kerem, E.; Lee, T. Theranostics by Testing CFTR Modulators in Patient-Derived Materials: The Current Status and a Proposal for Subjects with Rare CFTR Mutations. J. Cyst. Fibros. 2019, 18, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.G.; Mallon, B.S.; Park, K.; Robey, P.G.; McKay, R.D.G.; Gottesman, M.M.; Zheng, W. Pluripotent Stem Cell Platforms for Drug Discovery. Trends Mol. Med. 2018, 24, 805–820. [Google Scholar] [CrossRef]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in Vitro Model of Human Development and Disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef]
Figure 1.
Venn Diagram showing the number and relative percentage of mutations identified so far as responsive to Ivacaftor (Iva) or to the combinations of the correctors Tezacaftor (Teza) and Elexacaftor (Elexa) with the potentiator (Iva).
Figure 1.
Venn Diagram showing the number and relative percentage of mutations identified so far as responsive to Ivacaftor (Iva) or to the combinations of the correctors Tezacaftor (Teza) and Elexacaftor (Elexa) with the potentiator (Iva).

Figure 2.
Theratyping strategies. Nasal and intestinal biopsies can be collected and utilized to derive long-term cultures that can be utilized for functional assays measuring CFTR function, as described in the text. The results can inform the clinician on the response of the specific CFTR variants and combinations expressed by the proband thus providing directions on the choice of the most appropriate treatment.
Figure 2.
Theratyping strategies. Nasal and intestinal biopsies can be collected and utilized to derive long-term cultures that can be utilized for functional assays measuring CFTR function, as described in the text. The results can inform the clinician on the response of the specific CFTR variants and combinations expressed by the proband thus providing directions on the choice of the most appropriate treatment.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of licensed CFTR modulator approved by FDA (https://www.vertexgps.com, accessed on 31 October 2022); worldwide data are available from https://news.vrtx.com, accessed on 31 October 2022.
Table 1.
Summary of licensed CFTR modulator approved by FDA (https://www.vertexgps.com, accessed on 31 October 2022); worldwide data are available from https://news.vrtx.com, accessed on 31 October 2022.
| Modulator | License Age and Characteristics | Mutations |
|---|---|---|
| Ivacaftor | Class III gating mutations (Table 2) | |
| Lumacaftor/Ivacaftor | Homozygous F508del | |
| Tezacaftor/Ivacaftor | Homozygous F508del or at least one copy of responsive mutations (Table 3) | |
| Ivacaftor/Tezacaftor/Elexacaftor | At least one F508del mutation or at least one copy of responsive mutations (Table 4) |
Table 2.
List of CFTR mutations eligible for the treatment with Ivacaftor and approved by the FDA. Available from https://www.vertexgps.com, accessed on 14 November 2022.
Table 2.
List of CFTR mutations eligible for the treatment with Ivacaftor and approved by the FDA. Available from https://www.vertexgps.com, accessed on 14 November 2022.
| 711 + 3A→G | D1152H | G194R | I807M | Q237H | R553Q | S1159F |
| 2789 + 5 G→A | D1270N | G314E | I1027T | Q359R | R668C | S1159P |
| 3272–26A→G | E56K | G551D | I1139V | Q1291R | R792G | S1251N |
| 3849 + 10kbC→T | E193K | G551S | K1060T | R74W | R933G | S1255P |
| A120T | E822K | G576A | L206W | R75Q | R1070Q | T338I |
| A234D | E831X | G970D | L320V | R117C | R1070W | T1053I |
| A349V | F311del | G1069R | L967S | R117G | R1162L | V232D |
| A455E | F311L | G1244E | L997F | R117H | R1283M | V562I |
| A1067T | F508C | G1249R | L1480P | R117L | S549N | V754M |
| D110E | F508C/S1251N | G1349D | M152V | R117P | S549R | V1293G |
| D110H | F1052V | H939R | M952I | R170H | S589N | W1282R |
| D192G | F1074L | H1375P | M952T | R347H | S737F | Y1014C |
| D579G | G178E | I148T | P67L | R347L | S945L | Y1032C |
| D924N | G178R | I175V | Q237E | R352Q | S977F |
Table 3.
List of CFTR mutations eligible for the treatment with Tezacaftor/Ivacaftor and approved by the FDA. Patients should carry F508del mutation in both alleles or at least one copy of the mutations listed here. Available from https://www.vertexgps.com, accessed on 14 November 2022.
Table 3.
List of CFTR mutations eligible for the treatment with Tezacaftor/Ivacaftor and approved by the FDA. Patients should carry F508del mutation in both alleles or at least one copy of the mutations listed here. Available from https://www.vertexgps.com, accessed on 14 November 2022.
| 546insCTA | D1152H | G126D | I601F | P5L | R334L | S912L |
| 711 + 3A→G | D1270N | G178E | I618T | P67L | R334Q | S945L |
| 2789 + 5 G→A | E56K | G178R | I807M | P205S | R347H | S977F |
| 3272–26A→G | E60K | G194R | I980K | Q98R | R347L | S1159F |
| 3849 + 10kbC→T | E92K | G194V | I1027T | Q237E | R347P | S1159P |
| A120T | E116K | G314E | I1139V | Q237H | R352Q | S1251N |
| A234D | E193K | G551D | I1269N | Q359R | R352W | S1255P |
| A349V | E403D | G551S | I1366N | Q1291R | R553Q | T338I |
| A455E | E588V | G576A | K1060T | R31L | R668C | T1036N |
| A554E | E822K | G576A/R668C | L15P | R74Q | R751L | T1053I |
| A1006E | E831X | G622D | L206W | R74W | R792G | V201M |
| A1067T | F191V | G970D | L320V | R74W/D1270N | R933G | V232D |
| D110E | F311del | G1069R | L346P | R74W/V201M | R1066H | V562I |
| D110H | F311L | G1244E | L967S | R74W/V201M/D1270N | R1070Q | V754M |
| D192G | F508C | G1249R | L997F | R75Q | R1070W | V1153E |
| D443Y | F508C/S1251N | G1349D | L1324P | R117C | R1162L | V1240G |
| D443Y/G576A/R668C | F508del | H939R | L1335P | R117G | R1283M | V1293G |
| D579G | F575Y | H1054D | L1480P | R117H | R1283S | W1282R |
| D614G | F1016S | H1375P | M152V | R117L | S549N | Y109N |
| D836Y | F1052V | I148T | M265R | R117P | S549R | Y161S |
| D924N | F1074L | I175V | M952I | R170H | S589N | Y1014C |
| D979V | F1099L | I336K | M952T | R258G | S737F | Y1032C |
Table 4.
List of CFTR mutations eligible for the treatment with Ivacaftor/Tezacaftor/Elexacaftor and approved by the FDA. Patients should carry at least one copy of the mutations listed here. Available from https://www.vertexgps.com, accessed on 14 November 2022.
Table 4.
List of CFTR mutations eligible for the treatment with Ivacaftor/Tezacaftor/Elexacaftor and approved by the FDA. Patients should carry at least one copy of the mutations listed here. Available from https://www.vertexgps.com, accessed on 14 November 2022.
| 3141del9 | E193K | G551D | I980K | P574H | R352W | S1255P |
| 546insCTA | E403D | G551S | I1027T | Q98R | R553Q | T338I |
| A46D | E474K | G576A | I1139V | Q237E | R668C | T1036N |
| A120T | E588V | G576A/R668C | I1269N | Q237H | R751L | T1053I |
| A234D | E822K | G622D | I1366N | Q359R | R792G | V201M |
| A349V | F191V | G628R | K1060T | Q1291R | R933G | V232D |
| A455E | F311del | G970D | L15P | R31L | R1066H | V456A |
| A554E | F311L | G1061R | L165S | R74Q | R1070Q | V456F |
| A1006E | F508C | G1069R | L206W | R74W | R1070W | V562I |
| A1067T | F508C/S1251N | G1244E | L320V | R74W/D1270N | R1162L | V754M |
| D110E | F508del | G1249R | L346P | R74W/V201M | R1283M | V1153E |
| D110H | F575Y | G1349D | L453S | R74W/V201M/D1270N | R1283S | V1240G |
| D192G | F1016S | H139R | L967S | R75Q | S13F | V1293G |
| D443Y | F1052V | H199Y | L997F | R117C | S341P | W361R |
| D443Y/G576A/R668C | F1074L | H939R | L1077P | R117G | S364P | W1098C |
| D579G | F1099L | H1054D | L1324P | R117H | S492F | W1282R |
| D614G | G27R | H1085P | L1335P | R117L | S549N | Y109N |
| D836Y | G85E | H1085R | L1480P | R117P | S549R | Y161D |
| D924N | G126D | H1375P | M152V | R170H | S589N | Y161S |
| D979V | G178E | I148T | M265R | R258G | S737F | Y563N |
| D1152H | G178R | I175V | M952I | R334L | S912L | Y1014C |
| D1270N | G194R | I336K | M952T | R334Q | S945L | Y1032C |
| E56K | G194V | I502T | M1101K | R347H | S977F | |
| E60K | G314E | I601F | P5L | R347L | S1159F | |
| E92K | G463V | I618T | P67L | R347P | S1159P | |
| E116K | G480C | I807M | P205S | R352Q | S1251N |
Table 5.
Limitations and possible solutions to challenges faced with organoid cultures and theratyping approach.
Table 5.
Limitations and possible solutions to challenges faced with organoid cultures and theratyping approach.
| Criticisms | Solutions | Refs |
|---|---|---|
|
| [40] |
|
| [68] |
|
| [83,84,85] |
| In progress | |
| In progress | |
| In progress |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Conti, J.; Sorio, C.; Melotti, P. Organoid Technology and Its Role for Theratyping Applications in Cystic Fibrosis. Children 2023, 10, 4. https://doi.org/10.3390/children10010004
AMA Style
Conti J, Sorio C, Melotti P. Organoid Technology and Its Role for Theratyping Applications in Cystic Fibrosis. Children. 2023; 10(1):4. https://doi.org/10.3390/children10010004
Chicago/Turabian StyleConti, Jessica, Claudio Sorio, and Paola Melotti. 2023. "Organoid Technology and Its Role for Theratyping Applications in Cystic Fibrosis" Children 10, no. 1: 4. https://doi.org/10.3390/children10010004
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.