
Roles of XBP1s in Transcriptional Regulation of Target Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
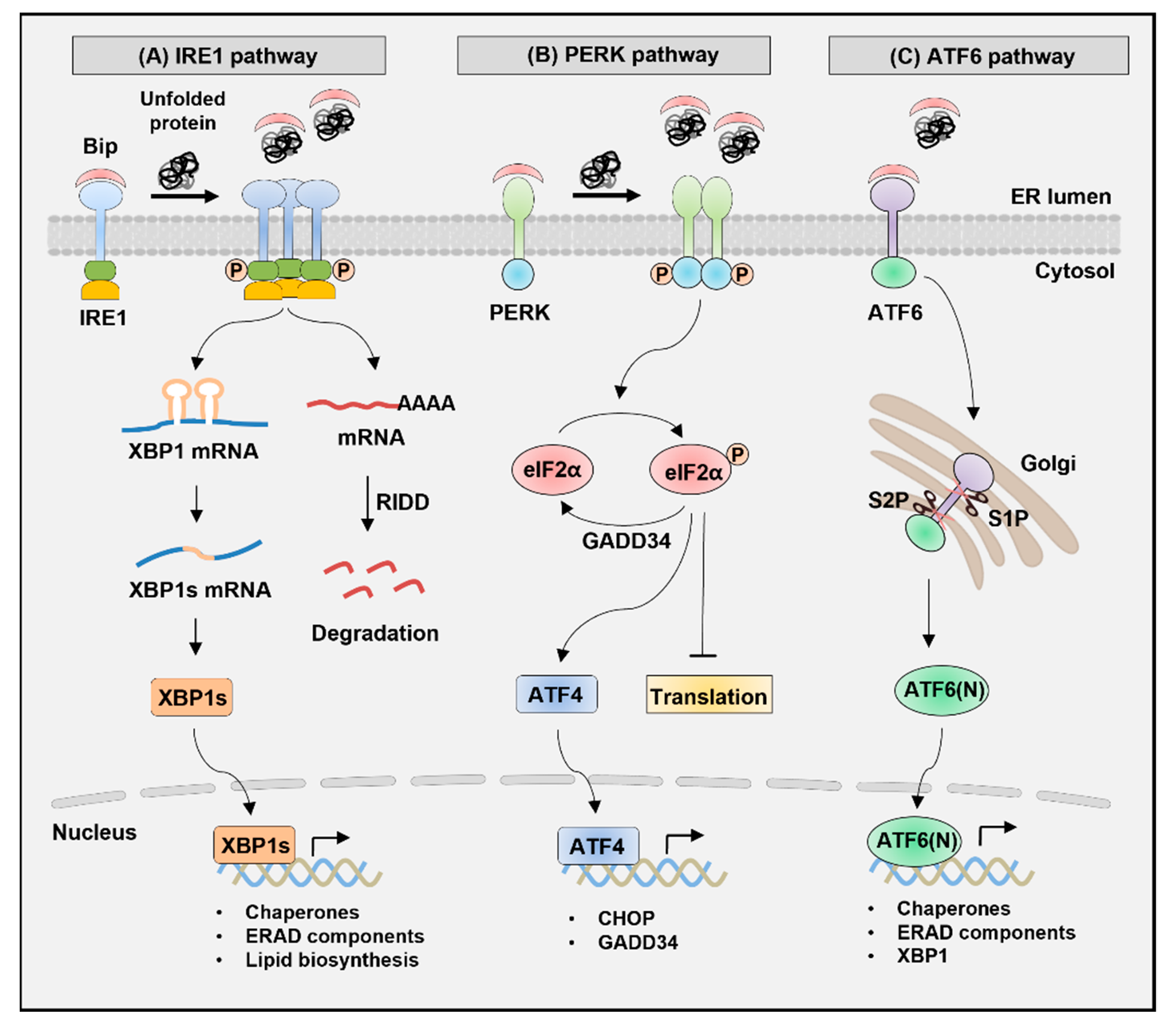
2. Signaling Pathways of UPR
2.1. IRE1 Pathway
2.2. PERK Pathway
2.3. ATF6 Pathway
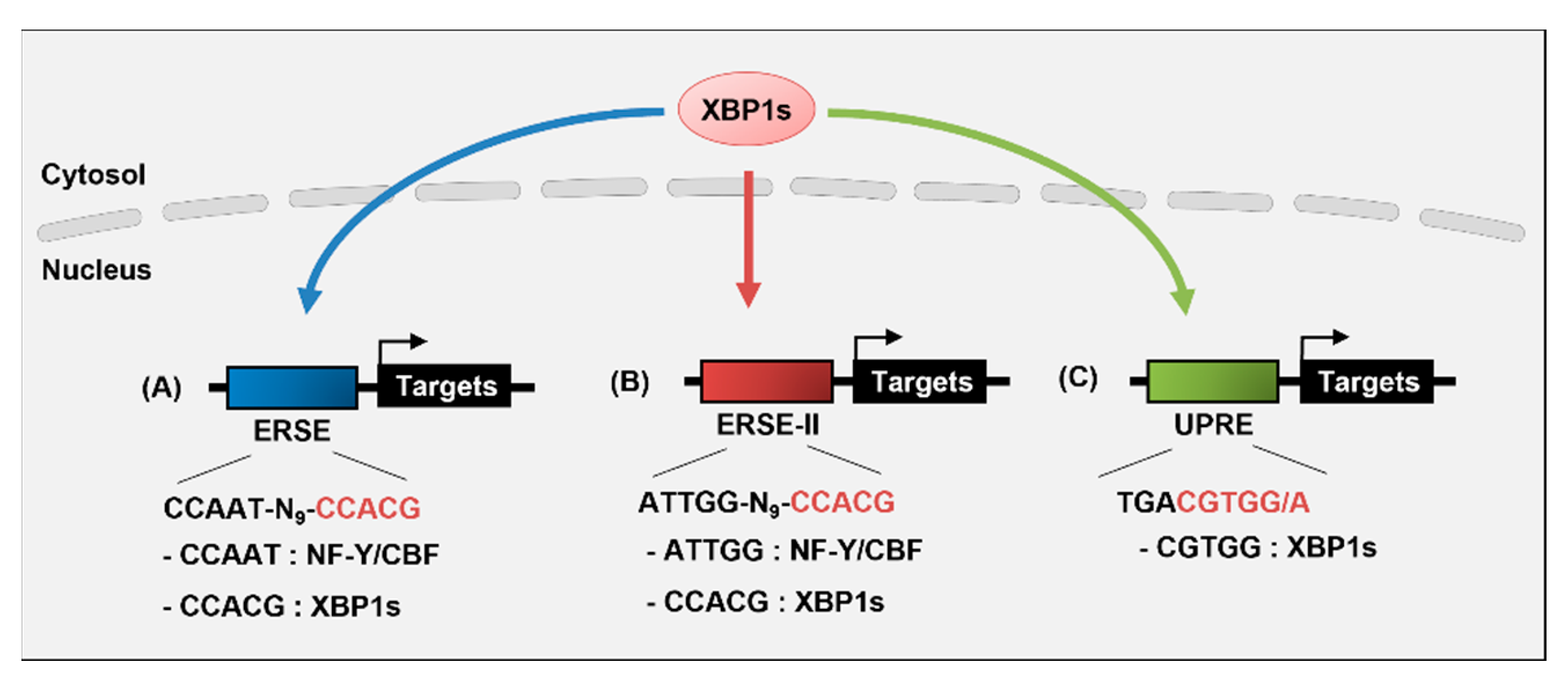
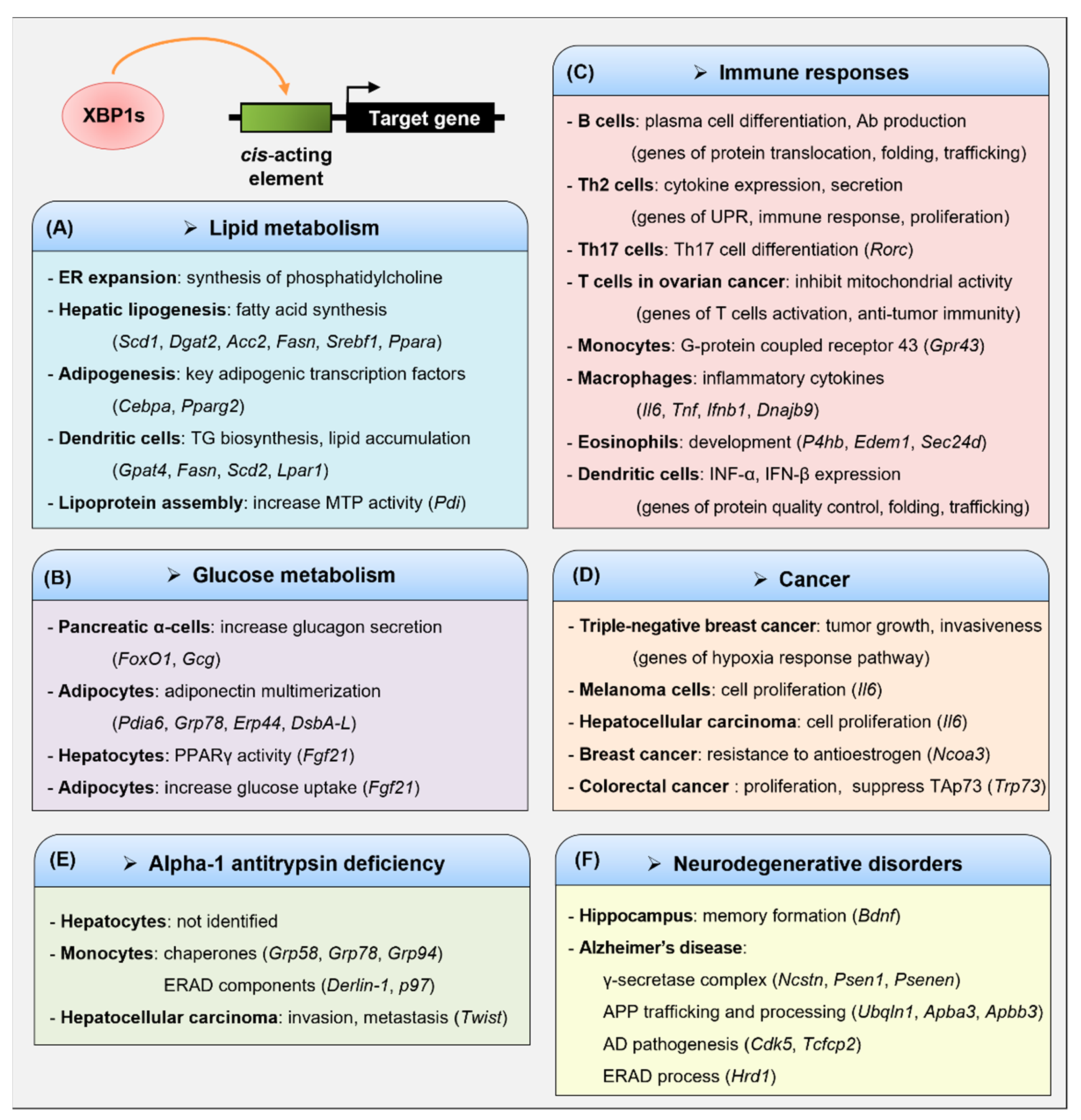
3. Transcriptional Regulation of Target Genes by XBP1s
3.1. Lipid Metabolism
3.2. Glucose Metabolism
3.3. Immune Responses
3.3.1. B Cells
3.3.2. T Cells
3.3.3. Monocytes and Macrophages
3.3.4. Eosinophils and Dendritic Cells (DCs)
3.4. Cancer
3.5. Alpha-1 Antitrypsin Deficiency (AATD)
3.6. Neurodegenerative Disorders
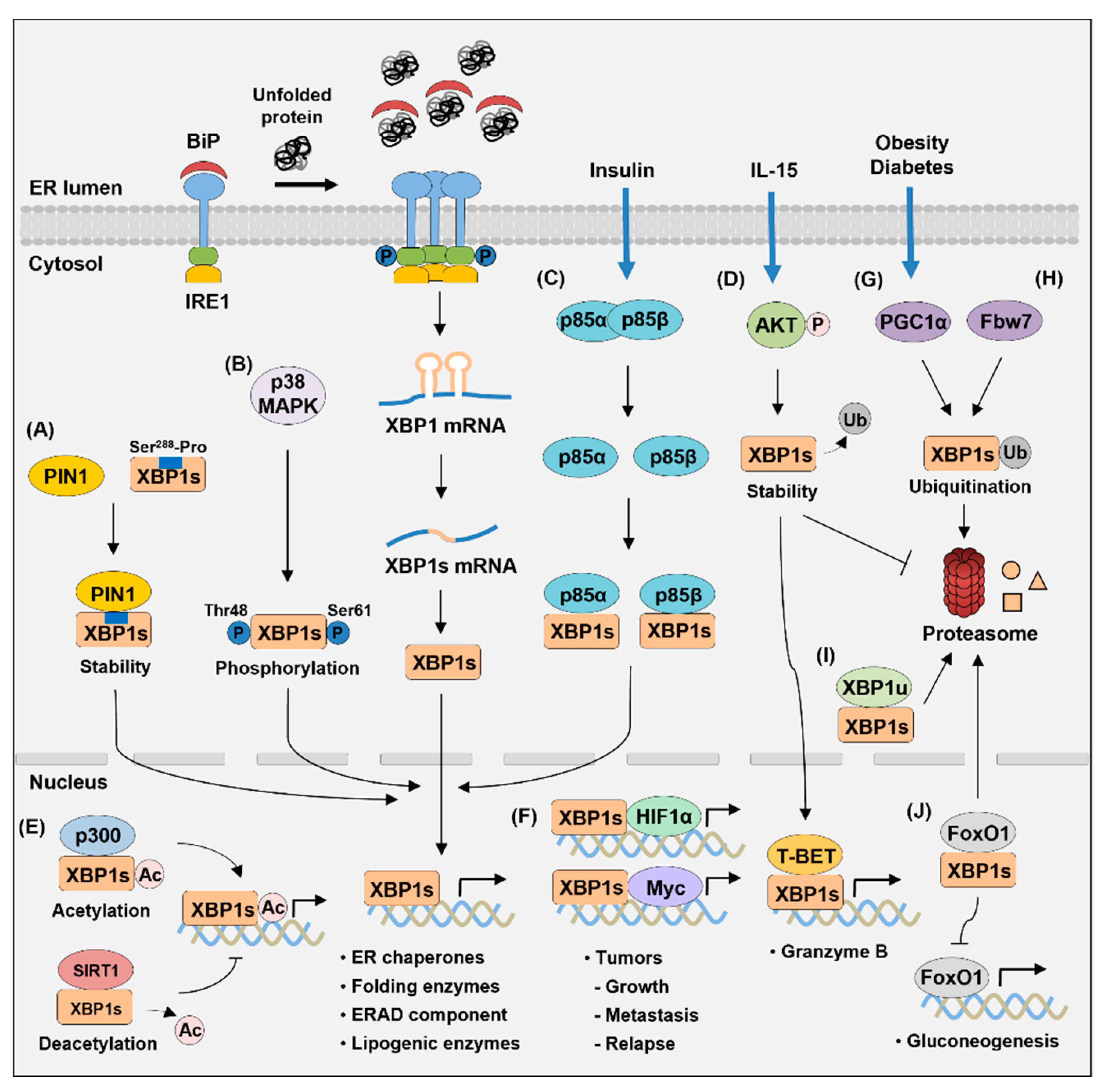
4. Proteins Interacting with XBP1s
5. Transcriptional Regulation of Xbp1 Gene
6. RIDD: Roles of RNase Activity of IRE1α
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribb, A.E.; Peyrou, M.; Muruganandan, S.; Schneider, L. The endoplasmic reticulum in xenobiotic toxicity. Drug Metab. Rev. 2005, 37, 405–442. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Molinari, M.; Helenius, A. Setting the standards: Quality control in the secretory pathway. Science 1999, 286, 1882–1888. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, M. The unfolded protein response triggered by environmental factors. Semin. Immunopathol. 2013, 35, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Calfon, M.; Urano, F.; Novoa, I.; Ron, D. Transcriptional and translational control in the mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 2002, 18, 575–599. [Google Scholar] [CrossRef]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef]
- Hoseki, J.; Ushioda, R.; Nagata, K. Mechanism and components of endoplasmic reticulum-associated degradation. J. Biochem. 2010, 147, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lemus, L.; Goder, V. Regulation of endoplasmic reticulum-associated protein degradation (ERAD) by ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Zhang, Q.-H.; Lu, Y.-J.; Ren, K.; Yi, G.-H. Involvement of the IRE1alpha-XBP1 pathway and XBP1s-dependent transcriptional reprogramming in metabolic diseases. DNA Cell Biol. 2015, 34, 6–18. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.-H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.; Moore, D.D. Endoplasmic reticulum stress and glucose homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 367–373. [Google Scholar] [CrossRef] [PubMed]
- So, J.-S. Roles of endoplasmic reticulum stress in immune responses. Mol. Cells 2018, 41, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of mammalian IRE1α upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 2009, 315, 2496–2504. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [Green Version]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef] [Green Version]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [CrossRef]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a consensus element recognized and cleaved by IRE1 alpha. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Suzuki, N.; Wada, T.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Mori, K. Human HRD1 promoter carries a functional unfolded protein response element to which XBP1 but not ATF6 directly binds. J. Biochem. 2008, 144, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Chen, J.; Hua, X.; Sun, Y.; Cui, R.; Sha, J.; Zhu, X. The emerging role of XBP1 in cancer. Biomed. Pharmacother. 2020, 127, 110069. [Google Scholar] [CrossRef]
- Shi, W.; Chen, Z.; Li, L.; Liu, H.; Zhang, R.; Cheng, Q.; Xu, D.; Wu, L. Unravel the molecular mechanism of XBP1 in regulating the biology of cancer cells. J. Cancer 2019, 10, 2035–2046. [Google Scholar] [CrossRef] [Green Version]
- Piperi, C.; Adamopoulos, C.; Papavassiliou, A.G. XBP1: A pivotal transcriptional regulator of glucose and lipid metabolism. Trends Endocrinol. Metab. 2016, 27, 119–122. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Lee, A.-H.; Chu, G.C.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005, 24, 4368–4380. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.S.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.-H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Wang, H.-G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zhang, Y.; Scheuner, D.; Chen, J.-J.; Kaufman, R.J.; Ron, D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2α) dephosphorylation in mammalian development. Proc. Natl. Acad. Sci. USA 2009, 106, 1832–1837. [Google Scholar] [CrossRef] [Green Version]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Okada, T.; Haze, K.; Nadanaka, S.; Yoshida, H.; Seidah, N.G.; Hirano, Y.; Sato, R.; Negishi, M.; Mori, K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J. Biol. Chem. 2003, 278, 31024–31032. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins: Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [Green Version]
- Roy, B.; Lee, A.S. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 1999, 27, 1437–1443. [Google Scholar] [CrossRef] [Green Version]
- Kokame, K.; Kato, H.; Miyata, T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 2001, 276, 9199–9205. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6alpha and 6beta that activates the mammalian unfolded protein response. Mol. Cell. Biol. 2001, 21, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Clauss, I.M.; Chu, M.; Zhao, J.-L.; Glimcher, L.H. The basic domain/leucine zipper protein hXBP-1 preferentially binds to and transactivates CRE-like sequences containing an ACGT core. Nucleic Acids Res. 1996, 24, 1855–1864. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef]
- Fink, E.E.; Moparthy, S.; Bagati, A.; Bianchi-Smiraglia, A.; Lipchick, B.C.; Wolff, D.W.; Roll, M.V.; Wang, J.; Liu, S.; Bakin, A.V.; et al. XBP1-KLF9 axis acts as a molecular rheostat to control the transition from adaptive to cytotoxic unfolded protein response. Cell Rep. 2018, 25, 212–223.e4. [Google Scholar] [CrossRef] [Green Version]
- Glimcher, L.H. XBP1: The last two decades. Ann. Rheum. Dis. 2010, 69, i67–i71. [Google Scholar] [CrossRef]
- Lee, A.-H.; Heidtman, K.; Hotamisligil, G.S.; Glimcher, L.H. Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 8885–8890. [Google Scholar] [CrossRef] [Green Version]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar] [CrossRef]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. Available online: https://www.nature.com/articles/ni.1857#supplementary-information (accessed on 29 August 2020). [CrossRef] [PubMed]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Ning, J.; Hong, T.; Ward, A.; Pi, J.; Liu, Z.; Liu, H.Y.; Cao, W. Constitutive role for IRE1alpha-XBP1 signaling pathway in the insulin-mediated hepatic lipogenic program. Endocrinology 2011, 152, 2247–2255. [Google Scholar] [CrossRef] [Green Version]
- Sha, H.; He, Y.; Chen, H.; Wang, C.; Zenno, A.; Shi, H.; Yang, X.; Zhang, X.; Qi, L. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009, 9, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Shao, M.; Shan, B.; Liu, Y.; Deng, Y.; Yan, C.; Wu, Y.; Mao, T.; Qiu, Y.; Zhou, Y.; Jiang, S.; et al. Hepatic IRE1α regulates fasting-induced metabolic adaptive programs through the XBP1s–PPARα axis signalling. Nat. Commun. 2014, 5, 3528. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.M.; Kwak, S.N.; Joo, N.S.; Kim, D.H.; Lee, A.H.; Kim, K.S.; Seo, J.B.; Jeong, S.W.; Kwon, O.J. X-box binding protein 1 is a novel key regulator of peroxisome proliferator-activated receptor gamma2. FEBS J. 2014, 281, 5132–5146. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Ginsberg, H.N. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol. Metab. 2011, 22, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chen, Z.; Lam, V.; Han, J.; Hassler, J.; Finck, B.N.; Davidson, N.O.; Kaufman, R.J. IRE1alpha-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012, 16, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res. 2003, 44, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Wetterau, J.R.; Combs, K.A.; McLean, L.R.; Spinner, S.N.; Aggerbeck, L.P. Protein disulfide isomerase appears necessary to maintain the catalytically active structure of the microsomal triglyceride transfer protein. Biochemistry 1991, 30, 9728–9735. [Google Scholar] [CrossRef]
- Akiyama, M.; Liew, C.W.; Lu, S.; Hu, J.; Martinez, R.; Hambro, B.; Kennedy, R.T.; Kulkarni, R.N. X-box binding protein 1 is essential for insulin regulation of pancreatic alpha-cell function. Diabetes 2013, 62, 2439–2449. [Google Scholar] [CrossRef] [Green Version]
- Sha, H.; Yang, L.; Liu, M.; Xia, S.; Liu, Y.; Liu, F.; Kersten, S.; Qi, L. Adipocyte spliced form of X-box-binding protein 1 promotes adiponectin multimerization and systemic glucose homeostasis. Diabetes 2014, 63, 867–879. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.M.; Kim, D.H.; Lee, K.H.; Jeong, S.W.; Kwon, O.J. The IRE1alpha-XBP1s pathway promotes insulin-stimulated glucose uptake in adipocytes by increasing PPARgamma activity. Exp. Mol. Med. 2018, 50, 102. [Google Scholar] [CrossRef] [Green Version]
- Turer, A.T.; Scherer, P.E. Adiponectin: Mechanistic insights and clinical implications. Diabetologia 2012, 55, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Yan, C.; Fang, Q.-c.; Shao, M.-l.; Zhang, Y.-l.; Liu, Y.; Deng, Y.-p.; Shan, B.; Liu, J.-q.; Li, H.-t.; et al. Fibroblast growth factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J. Biol. Chem. 2014, 289, 29751–29765. [Google Scholar] [CrossRef] [Green Version]
- Rahmati, M.; Moosavi, M.A.; McDermott, M.F. ER Stress: A therapeutic target in rheumatoid arthritis? Trends Pharmacol. Sci. 2018, 39, 610–623. [Google Scholar] [CrossRef]
- Qiu, Q.; Zheng, Z.; Chang, L.; Zhao, Y.-S.; Tan, C.; Dandekar, A.; Zhang, Z.; Lin, Z.; Gui, M.; Li, X.; et al. Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013, 32, 2477–2490. [Google Scholar] [CrossRef] [Green Version]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [Green Version]
- Vattemi, G.; Engel, W.K.; McFerrin, J.; Askanas, V. Endoplasmic reticulum dtress and unfolded protein response in inclusion body myositis muscle. Am. J. Pathol. 2004, 164, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 2010, 16, 396–399. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.-H.; Qian, S.-B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Iwakoshi, N.N.; Lee, A.-H.; Glimcher, L.H. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 2003, 194, 29–38. [Google Scholar] [CrossRef]
- Gass, J.N.; Gifford, N.M.; Brewer, J.W. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J. Biol. Chem. 2002, 277, 49047–49054. [Google Scholar] [CrossRef] [Green Version]
- Zygmunt, B.; Veldhoen, M. T helper cell differentiation: More than just cytokines. Adv. Immunol. 2011, 109, 159–196. [Google Scholar] [CrossRef]
- Saravia, J.; Chapman, N.M.; Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 2019, 16, 634–643. [Google Scholar] [CrossRef]
- Pramanik, J.; Chen, X.; Kar, G.; Henriksson, J.; Gomes, T.; Park, J.-E.; Natarajan, K.; Meyer, K.B.; Miao, Z.; McKenzie, A.N.J.; et al. Genome-wide analyses reveal the IRE1α-XBP1 pathway promotes T helper cell differentiation by resolving secretory stress and accelerating proliferation. Genome Med. 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.A.; McKenzie, A.N.J. TH2 cell development and function. Nat. Rev. Immunol. 2018, 18, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Brucklacher-Waldert, V.; Ferreira, C.; Stebegg, M.; Fesneau, O.; Innocentin, S.; Marie, J.C.; Veldhoen, M. Cellular stress in the context of an inflammatory environment supports TGF-β-independent T helper-17 differentiation. Cell Rep. 2017, 19, 2357–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Sandoval, T.A.; Chae, C.-S.; Chopra, S.; Tan, C.; Rutkowski, M.R.; Raundhal, M.; Chaurio, R.A.; Payne, K.K.; Konrad, C.; et al. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 2018, 562, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Ang, Z.; Er, J.Z.; Ding, J.L. The short-chain fatty acid receptor GPR43 is transcriptionally regulated by XBP1 in human monocytes. Sci. Rep. 2015, 5, 8134. [Google Scholar] [CrossRef] [Green Version]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in obesity and inflammation – protective or causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Chen, S.; Deng, B.; Tan, C.; Deng, J.; Zhu, G.; Yin, Y.; Ren, W. Implication of G protein-coupled receptor 43 in intestinal inflammation: A mini-review. Front. Immunol. 2018, 9, 1434. [Google Scholar] [CrossRef]
- Senga, T.; Iwamoto, S.; Yoshida, T.; Yokota, T.; Adachi, K.; Azuma, E.; Hamaguchi, M.; Iwamoto, T. LSSIG is a novel murine leukocyte-specific GPCR that is induced by the activation of STAT3. Blood 2003, 101, 1185–1187. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Taylor, P.R. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Turner, M.J.; DeLay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Liu, Y.-P.; Sha, H.; Chen, H.; Qi, L.; Smith, J.A. XBP-1 couples endoplasmic reticulum stress to augmented IFN-β induction via a cis-acting enhancer in macrophages. J. Immunol. 2010, 185, 2324–2330. [Google Scholar] [CrossRef] [Green Version]
- Bettigole, S.E.; Lis, R.; Adoro, S.; Lee, A.-H.; Spencer, L.A.; Weller, P.F.; Glimcher, L.H. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat. Immunol. 2015, 16, 829–837. [Google Scholar] [CrossRef]
- Furuta, G.T.; Atkins, F.D.; Lee, N.A.; Lee, J.J. Changing roles of eosinophils in health and disease. Ann. Allergy Asthma Immunol. 2014, 113, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Yu, X.; Wang, H.; Zuo, D.; Guo, C.; Yi, H.; Tirosh, B.; Subjeck, J.R.; Qiu, X.; Wang, X.-Y. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur. J. Immunol. 2011, 41, 1086–1097. [Google Scholar] [CrossRef]
- Osorio, F.; Tavernier, S.J.; Hoffmann, E.; Saeys, Y.; Martens, L.; Vetters, J.; Delrue, I.; De Rycke, R.; Parthoens, E.; Pouliot, P.; et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat. Immunol. 2014, 15, 248–257. [Google Scholar] [CrossRef]
- Sykes, E.K.; Mactier, S.; Christopherson, R.I. Melanoma and the unfolded protein response. Cancers 2016, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, X. IRE1α-XBP1 pathway promotes melanoma progression by regulating IL-6/STAT3 signaling. J. Transl. Med. 2017, 15, 42. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Xiang, L.; Huang, S.; Jin, L.; Zhou, G.; Zhuge, L.; Li, J.; Fan, H.; Zhou, L.; Pan, C.; et al. IRE1α-XBP1 signaling pathway regulates IL-6 expression and promotes progression of hepatocellular carcinoma. Oncol. Lett. 2018, 16, 4729–4736. [Google Scholar] [CrossRef]
- Gupta, A.; Hossain, M.M.; Miller, N.; Kerin, M.; Callagy, G.; Gupta, S. NCOA3 coactivator is a transcriptional target of XBP1 and regulates PERK-eIF2α-ATF4 signalling in breast cancer. Oncogene 2016, 35, 5860–5871. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Huang, C.; Wu, S.; Kasim, V. XBP1-s promotes colorectal cancer cell proliferation by inhibiting TAp73 transcriptional activity. Biochem. Biophys. Res. Commun. 2019, 508, 203–209. [Google Scholar] [CrossRef]
- Xu, J.; Wu, R.-C.; O’Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Yasui, K.; Lee, C.J.; Kurioka, H.; Hosokawa, Y.; Oka, T.; Inazawa, J. Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer 2003, 98, 18–23. [Google Scholar] [CrossRef]
- Stantic, M.; Sakil, H.A.M.; Zirath, H.; Fang, T.; Sanz, G.; Fernandez-Woodbridge, A.; Marin, A.; Susanto, E.; Mak, T.W.; Arsenian Henriksson, M.; et al. TAp73 suppresses tumor angiogenesis through repression of proangiogenic cytokines and HIF-1α activity. Proc. Natl. Acad. Sci. USA 2015, 112, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Bae, W.-K.; Hong, C.-S.; Park, M.-R.; Sun, E.-G.; Lee, J.-H.; Kang, K.; Ryu, K.-H.; Shim, H.-J.; Hwang, J.-E.; Cho, S.-H.; et al. TAp73 inhibits cell invasion and migration by directly activating KAI1 expression in colorectal carcinoma. Cancer Lett. 2018, 415, 106–116. [Google Scholar] [CrossRef]
- Fra, A.; Cosmi, F.; Ordoñez, A.; Berardelli, R.; Perez, J.; Guadagno, N.A.; Corda, L.; Marciniak, S.J.; Lomas, D.A.; Miranda, E. Polymers of Z α1-antitrypsin are secreted in cell models of disease. Eur. Respir. J. 2016, 47, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, C.; Saldova, R.; Wormald, M.R.; Rudd, P.M.; McElvaney, N.G.; Reeves, E.P. The role and importance of glycosylation of acute phase proteins with focus on alpha-1 antitrypsin in acute and chronic inflammatory conditions. J. Proteome Res. 2014, 13, 3131–3143. [Google Scholar] [CrossRef]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. α1-Antitrypsin deficiency. Nat. Rev. Dis. Primers 2016, 2, 16051. [Google Scholar] [CrossRef]
- Greene, C.M.; McElvaney, N.G. Z α-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J. Gastrointest. Pharmacol. Ther. 2010, 1, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Fang, D. Endoplasmic reticulum stress signaling and the pathogenesis of hepatocarcinoma. Int. J. Mol. Sci. 2021, 22, 1799. [Google Scholar] [CrossRef] [PubMed]
- Gooptu, B.; Dickens, J.A.; Lomas, D.A. The molecular and cellular pathology of α1-antitrypsin deficiency. Trends Mol. Med. 2014, 20, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.; Carlson, J.; Velez, R. Risk of cirrhosis and primary liver cancer in alpha1-antitrypsin deficiency. N. Engl. J. Med. 1986, 314, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Paraboschi, E.M.; Duga, S. Hereditary hypofibrinogenemia with hepatic storage. Int. J. Mol. Sci. 2020, 21, 7830. [Google Scholar] [CrossRef]
- Lawless, M.W.; Greene, C.M.; Mulgrew, A.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z α1-antitrypsin deficiency. J. Immunol. 2004, 172, 5722–5726. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.; Greene, C.M.; Carroll, T.P.; McElvaney, N.G.; O’Neill, S.J. Selenoprotein S/SEPS1 modifies endoplasmic reticulum stress in Z variant α1-antitrypsin deficiency. J. Biol. Chem. 2009, 284, 16891–16897. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.D.W.; Greene, C.M.; McLean, C.; Lawless, M.W.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of bad. Hepatology 2007, 46, 496–503. [Google Scholar] [CrossRef]
- Greene, C.M.; Miller, S.D.W.; Carroll, T.P.; Oglesby, I.K.; Ahmed, F.; O’Mahony, M.; Taggart, C.C.; McElvaney, N.G.; O’Neill, S.J. Anti-apoptotic effects of Z α1-antitrypsin in human bronchial epithelial cells. Eur. Respir. J. 2010, 35, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- Carroll, T.P.; Greene, C.M.; O’Connor, C.A.; Nolan, Á.M.; O’Neill, S.J.; McElvaney, N.G. Evidence for unfolded protein response activation in monocytes from individuals with α-1 antitrypsin deficiency. J. Immunol. 2010, 184, 4538–4546. [Google Scholar] [CrossRef] [Green Version]
- Molmenti, E.P.; Perlmutter, D.H.; Rubin, D.C. Cell-specific expression of alpha 1-antitrypsin in human intestinal epithelium. J. Clin. Investig. 1993, 92, 2022–2034. [Google Scholar] [CrossRef] [Green Version]
- Hidvegi, T.; Schmidt, B.Z.; Hale, P.; Perlmutter, D.H. Accumulation of mutant α1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFκB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005, 280, 39002–39015. [Google Scholar] [CrossRef] [Green Version]
- Hidvegi, T.; Mirnics, K.; Hale, P.; Ewing, M.; Beckett, C.; Perlmutter, D.H. Regulator of G Signaling 16 Is a marker for the distinct endoplasmic reticulum stress state associated with aggregated mutant α1-antitrypsin Z in the classical form of α1-antitrypsin deficiency. J. Biol. Chem. 2007, 282, 27769–27780. [Google Scholar] [CrossRef] [Green Version]
- Kamimoto, T.; Shoji, S.; Hidvegi, T.; Mizushima, N.; Umebayashi, K.; Perlmutter, D.H.; Yoshimori, T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. 2006, 281, 4467–4476. [Google Scholar] [CrossRef] [Green Version]
- Ordóñez, A.; Snapp, E.L.; Tan, L.; Miranda, E.; Marciniak, S.J.; Lomas, D.A. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology 2013, 57, 2049–2060. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Du, R.; Gao, C.; Kang, J.; Wen, J.; Sun, T. The role of XBP1s in the metastasis and prognosis of hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 500, 530–537. [Google Scholar] [CrossRef]
- Pavlović, N.; Calitz, C.; Thanapirom, K.; Mazza, G.; Rombouts, K.; Gerwins, P.; Heindryckx, F. Inhibiting IRE1α-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma. eLife 2020, 9, e55865. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Saito, A.; Imaizumi, K. Unfolded protein response-dependent communication and contact among endoplasmic reticulum, mitochondria, and plasma membrane. Int. J. Mol. Sci. 2018, 19, 3215. [Google Scholar] [CrossRef] [Green Version]
- Sprenkle, N.T.; Sims, S.G.; Sanchez, C.L.; Meares, G.P. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Martinez, G.; Vidal, R.L.; Mardones, P.; Serrano, F.G.; Ardiles, A.O.; Wirth, C.; Valdes, P.; Thielen, P.; Schneider, B.L.; Kerr, B.; et al. Regulation of memory formation by the transcription factor XBP1. Cell Rep. 2016, 14, 1382–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017, 134, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Gerakis, Y.; Dunys, J.; Bauer, C.; Checler, F. Aβ42 oligomers modulate β-secretase through an XBP-1s-dependent pathway involving HRD1. Sci. Rep. 2016, 6, 37436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [Green Version]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [Green Version]
- Chu, B.; Li, M.; Cao, X.; Li, R.; Jin, S.; Yang, H.; Xu, L.; Wang, P.; Bi, J. IRE1α-XBP1 affects the mitochondrial function of Aβ25–35-treated SH-SY5Y cells by regulating mitochondria-associated endoplasmic reticulum membranes. Front. Cell. Neurosci. 2021, 15, 614556. [Google Scholar] [CrossRef]
- Rusiñol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef]
- Hetz, C.; Lee, A.-H.; Gonzalez-Romero, D.; Thielen, P.; Castilla, J.; Soto, C.; Glimcher, L.H. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.-M.; Chen, Y.-J.; Ouyang, H.-J. Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem. J. 2011, 433, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.E., Jr.; Pruitt, W.M.; Pruitt, K. Diverse roles of SIRT1 in cancer biology and lipid metabolism. Int. J. Mol. Sci. 2015, 16, 950–965. [Google Scholar] [CrossRef]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Kishino, A.; Hayashi, K.; Hidai, C.; Masuda, T.; Nomura, Y.; Oshima, T. XBP1-FoxO1 interaction regulates ER stress-induced autophagy in auditory cells. Sci. Rep. 2017, 7, 4442. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Lin, E.A.; Liu, P.; Lin, J.; Liu, C. XBP1U inhibits the XBP1S-mediated upregulation of the iNOS gene expression in mammalian ER stress response. Cell. Signal. 2010, 22, 1818–1828. [Google Scholar] [CrossRef]
- Lee, J.; Salazar Hernandez, M.A.; Auen, T.; Mucka, P.; Lee, J.; Ozcan, U. PGC-1α functions as a co-suppressor of XBP1s to regulate glucose metabolism. Mol. Metab. 2018, 7, 119–131. [Google Scholar] [CrossRef]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of XBP1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Lee, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B.; et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [Green Version]
- Chae, U.; Park, S.-J.; Kim, B.; Wei, S.; Min, J.-S.; Lee, J.-H.; Park, S.H.; Lee, A.-H.; Lu, K.P.; Lee, D.-S.; et al. Critical role of XBP1 in cancer signalling is regulated by PIN1. Biochem. J. 2016, 473, 2603–2610. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Liou, Y.-C.; Zhou, X.Z.; Lu, K.P. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 2011, 36, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Hunter, T. Prolyl isomerase Pin1 in cancer. Cell Res. 2014, 24, 1033–1049. [Google Scholar] [CrossRef] [Green Version]
- Chae, U.; Lee, H.; Kim, B.; Jung, H.; Kim, B.M.; Lee, A.-H.; Lee, D.-S.; Min, S.-H. A negative feedback loop between XBP1 and Fbw7 regulates cancer development. Oncogenesis 2019, 8, 12. [Google Scholar] [CrossRef]
- Xu, W.; Taranets, L.; Popov, N. Regulating Fbw7 on the road to cancer. Semin. Cancer Biol. 2016, 36, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Li, G. Role of the ubiquitin ligase Fbw7 in cancer progression. Cancer Metastasis Rev. 2012, 31, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Yi, P.; Dong, W.; Nalin, A.P.; Zhang, J.; Zhu, Z.; Chen, L.; Benson, D.M.; Mundy-Bosse, B.L.; et al. The IL-15-AKT-XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat. Immunol. 2019, 20, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.-J.; Xiong, Z.; Lu, X.; Ye, M.; Han, X.; Jiang, R. ATF6 upregulates XBP1s and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cell. Signal. 2014, 26, 332–342. [Google Scholar] [CrossRef]
- Moore, B.D.; Jin, R.U.; Lo, H.; Jung, M.; Wang, H.; Battle, M.A.; Wollheim, C.B.; Urano, F.; Mills, J.C. Transcriptional regulation of X-box-binding protein one (XBP1) by hepatocyte nuclear factor 4α (HNF4A) is vital to beta-cell function. J. Biol. Chem. 2016, 291, 6146–6157. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Coelho, D.S.; Domingos, P.M. Physiological roles of regulated Ire1 dependent decay. Front. Genet. 2014, 5, 76. [Google Scholar] [CrossRef] [Green Version]
- Lipson, K.L.; Ghosh, R.; Urano, F. The role of IRE1α in the degradation of insulin mRNA in pancreatic β-cells. PLoS ONE 2008, 3, e1648. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.-P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [Green Version]
- So, J.-S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Hur, K.Y.; So, J.-S.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Iwawaki, T.; Glimcher, L.H.; Lee, A.-H. IRE1α activation protects mice against acetaminophen-induced hepatotoxicity. J. Exp. Med. 2012, 209, 307–318. [Google Scholar] [CrossRef] [Green Version]
- So, J.-S.; Cho, S.; Min, S.-H.; Kimball, S.R.; Lee, A.-H. IRE1α-dependent decay of CReP/Ppp1r15b mRNA increases eukaryotic initiation factor 2α phosphorylation and suppresses protein synthesis. Mol. Cell. Biol. 2015, 35, 2761–2770. [Google Scholar] [CrossRef] [Green Version]
- Benhamron, S.; Hadar, R.; Iwawaky, T.; So, J.-S.; Lee, A.-H.; Tirosh, B. Regulated IRE1-dependent decay participates in curtailing immunoglobulin secretion from plasma cells. Eur. J. Immunol. 2014, 44, 867–876. [Google Scholar] [CrossRef]
- Tang, C.-H.A.; Chang, S.; Paton, A.W.; Paton, J.C.; Gabrilovich, D.I.; Ploegh, H.L.; Del Valle, J.R.; Hu, C.-C.A. Phosphorylation of IRE1 at S729 regulates RIDD in B cells and antibody production after immunization. J. Cell Biol. 2018, 217, 1739–1755. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.S.; Koh, J.M.; So, J.-S.; Jeon, Y.K.; Kim, H.Y.; Chung, D.H. Palmitate inhibits arthritis by inducing t-bet and gata-3 mRNA degradation in iNKT cells via IRE1α-dependent decay. Sci. Rep. 2017, 7, 14940. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.-P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.-P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell. Biol. 2008, 28, 3943–3951. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote pogrammed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-M.; Kang, T.-I.; So, J.-S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. https://doi.org/10.3390/biomedicines9070791
Park S-M, Kang T-I, So J-S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines. 2021; 9(7):791. https://doi.org/10.3390/biomedicines9070791
Chicago/Turabian StylePark, Sung-Min, Tae-Il Kang, and Jae-Seon So. 2021. "Roles of XBP1s in Transcriptional Regulation of Target Genes" Biomedicines 9, no. 7: 791. https://doi.org/10.3390/biomedicines9070791