
HIF-α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review


Abstract
:1. Introduction
2. Sensing of Hypoxia and Execution of the Hypoxic Gene Responses
3. Exploration of the Mechanism of Erythropoietin (EPO) Production Induction
4. Molecular Cloning of Hypoxia-Inducible Factor 1
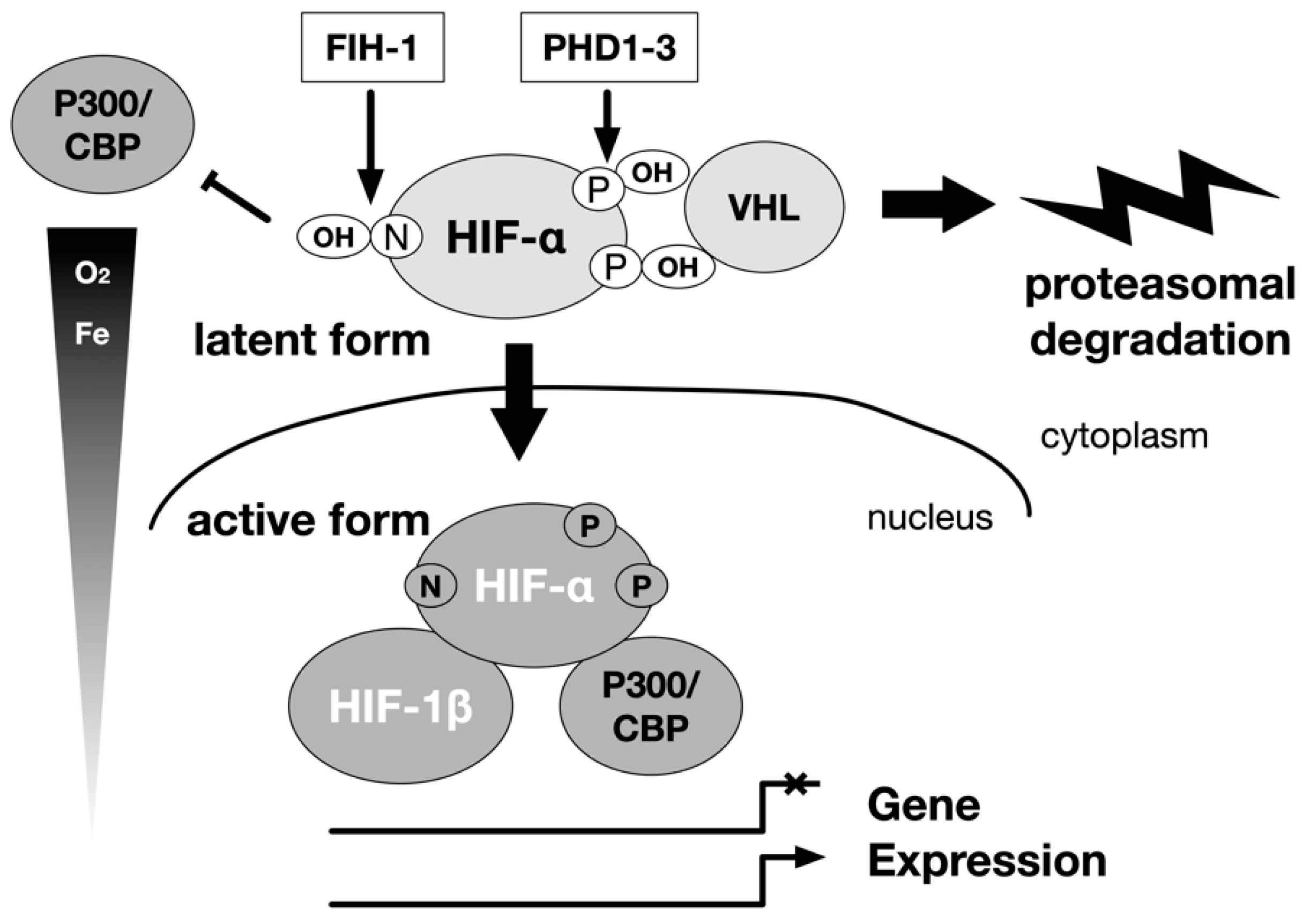
5. Intracellular Signaling Pathways Linking Hypoxia and HIF Activation
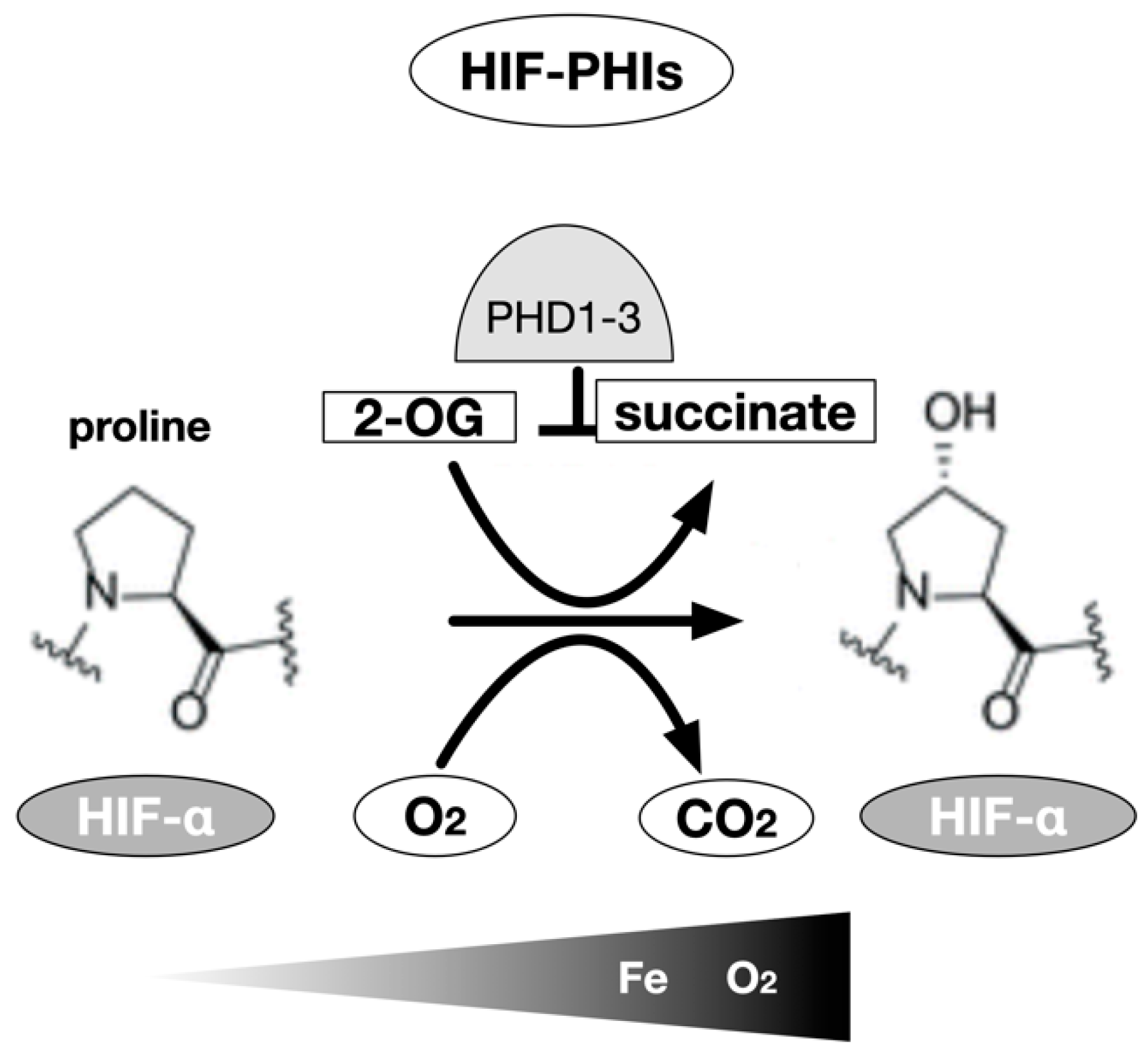
6. Genetic Cloning of Enzymes Modifying HIF-α Hydroxylation (PHDs and FIH-1)
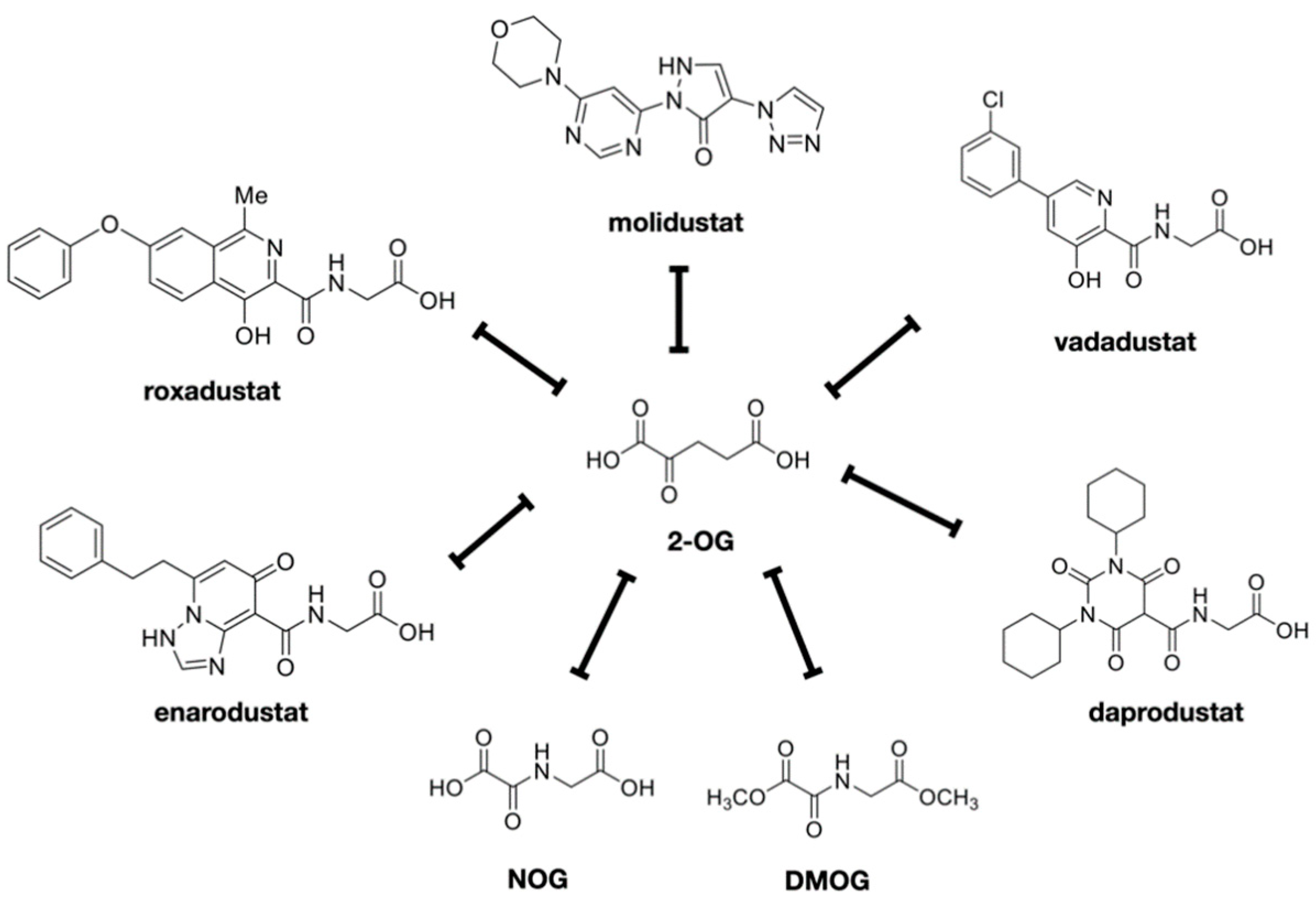
7. Development of HIF-PHIs for Clinical Use
8. Metabolism of HIF-PHIs and Interactions with Other Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Absorption | Excretion Rate of Unchanged Substance in Urine | Half Life (h) | Major Metabolic Pathways | Ref. |
|---|---|---|---|---|---|
| Roxadustat | 40~80% | 1% | 12~15 | CYP2C8, UGT1A9 | [82] |
| Vadadustat | >75% | <1% | 4~7 | UGT1A1/1A9 | [84,91] |
| Dapurodustat | 65% | <0.05% | 1~7 | CYP2C8 | [81,92] |
| Enarodustat | 41.70% | 27~61% | ~11 | Less susceptible to metabolism | [85,93] |
| Molidustat | 59% | 4% | 4~10 | UGT1A1/1A9 | [88,89] |
9. Regulatory Mechanism of Erythropoiesis
10. Renal Anemia Due to CKD
11. HIF-PHIs as a Treatment for Renal Anemia
12. Nephroprotective Effects of HIF-PHIs
13. Diverse Effects of HIF-PHIs
13.1. Ischemia
13.2. Inflammation
14. Adverse Effects of HIF-PHIs
14.1. Iron Deficiency
14.2. Cancers and Malignant Tumors
14.3. Diabetic Retinopathy and Age-Related Macular Degeneration
14.4. Thromboembolism
14.5. Pulmonary Hypertension
14.6. Polycystic Kidney Disease (PCKD)
14.7. Hyperkalemia
15. Establishment of Resistance
16. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nagel, S.; Talbot, N.P.; Mecinovic, J.; Smith, T.G.; Buchan, A.M.; Schofield, C.J. Therapeutic manipulation of the HIF hydroxylases. Antioxid. Redox Signal. 2010, 12, 481–501. [Google Scholar] [CrossRef]
- Thavarajah, S.; Choi, M.J. The Use of Erythropoiesis-Stimulating Agents in Patients With CKD and Cancer: A Clinical Approach. Am. J. Kidney Dis. 2019, 74, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Edmonston, D.; Wolf, M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat. Rev. Nephrol. 2020, 16, 7–19. [Google Scholar] [CrossRef]
- Del Vecchio, L.; Minutolo, R. ESA, Iron Therapy and New Drugs: Are There New Perspectives in the Treatment of Anaemia? J. Clin. Med. 2021, 10, 839. [Google Scholar] [CrossRef]
- Camaschella, C. Iron-deficiency anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T. Anemia of Inflammation. N. Engl. J. Med. 2019, 381, 1148–1157. [Google Scholar] [CrossRef]
- Kaplan, J. Roxadustat and Anemia of Chronic Kidney Disease. N. Engl. J. Med. 2019, 381, 1070–1072. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Life with oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef]
- Semenza, G.L. Perspectives on oxygen sensing. Cell 1999, 98, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 and human disease: One highly involved factor. Genes Dev. 2000, 14, 1983–1991. [Google Scholar]
- Hirota, K. Basic Biology of Hypoxic Responses Mediated by the Transcription Factor HIFs and its Implication for Medicine. Biomedicines 2020, 8, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Semenza, G. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Jiang, B.; Rue, E.; Semenza, G. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Semenza, G. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J. Biol. Chem. 1993, 268, 21513–21518. [Google Scholar] [CrossRef]
- Epstein, A.; Gleadle, J.; McNeill, L.; Hewitson, K.; O’Rourke, J.; Mole, D.; Mukherji, M.; Metzen, E.; Wilson, M.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Fandrey, J.; Schodel, J.; Eckardt, K.U.; Katschinski, D.M.; Wenger, R.H. Now a Nobel gas: Oxygen. Pflugers Arch. 2019. [Google Scholar] [CrossRef]
- West, J.B. Physiological Effects of Chronic Hypoxia. N. Engl. J. Med. 2017, 376, 1965–1971. [Google Scholar] [CrossRef]
- Gilreath, J.A.; Rodgers, G.M. How I treat cancer-associated anemia. Blood 2020, 136, 801–813. [Google Scholar] [CrossRef]
- Bishop, T.; Ratcliffe, P.J. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 2015, 117, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic. Biol. Med. 2019, 133, 118–129. [Google Scholar] [CrossRef]
- Yap, D.Y.H.; McMahon, L.P.; Hao, C.M.; Hu, N.; Okada, H.; Suzuki, Y.; Kim, S.G.; Lim, S.K.; Vareesangthip, K.; Hung, C.C.; et al. Recommendations by the Asian Pacific society of nephrology (APSN) on the appropriate use of HIF-PH inhibitors. Nephrology 2020. [Google Scholar] [CrossRef]
- Gupta, N.; Wish, J.B. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Ho, V.T.; Bunn, H.F. Effects of transition metals on the expression of the erythropoietin gene: Further evidence that the oxygen sensor is a heme protein. Biochem. Biophys. Res. Commun. 1996, 223, 175–180. [Google Scholar] [CrossRef]
- Weir, E.K.; Lopez-Barneo, J.; Buckler, K.J.; Archer, S.L. Acute oxygen-sensing mechanisms. N. Engl. J. Med. 2005, 353, 2042–2055. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Peng, Y.J. Oxygen Sensing by the Carotid Body: Past and Present. Adv. Exp. Med. Biol. 2017, 977, 3–8. [Google Scholar] [CrossRef]
- Simon, M.C. The Hypoxia Response Pathways—Hats Off! N. Engl. J. Med. 2016, 375, 1687–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reissmann, K.R. Studies on the mechanism of erythropoietic stimulation in parabiotic rats during hypoxia. Blood 1950, 5, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Fried, W.; Goldwasser, E.; Jacobson, L.O.; Plzak, L.F. Studies on erythropoiesis. III. Factors controlling erythropoietin production. Proc. Soc. Exp. Biol. Med. 1957, 94, 237–241. [Google Scholar] [CrossRef]
- Jacobson, L.O.; Goldwasser, E.; Fried, W.; Plzak, L.F. Studies on erythropoiesis. VII. The role of the kidney in the production of erythropoietin. Trans. Soc. Assoc. Am. Physicians 1957, 70, 305–317. [Google Scholar]
- Goldwasser, E.; Fried, W.; Jacobson, L.O. Studies on erythropoiesis. VIII. The effect of nephrectomy on response to hypoxic anoxia. J. Lab. Clin. Med. 1958, 52, 375–378. [Google Scholar]
- Miyake, T.; Kung, C.K.; Goldwasser, E. Purification of human erythropoietin. J. Biol. Chem. 1977, 252, 5558–5564. [Google Scholar] [CrossRef]
- Jacobs, K.; Shoemaker, C.; Rudersdorf, R.; Neill, S.D.; Kaufman, R.J.; Mufson, A.; Seehra, J.; Jones, S.S.; Hewick, R.; Fritsch, E.F.; et al. Isolation and characterization of genomic and cDNA clones of human erythropoietin. Nature 1985, 313, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Yamamoto, M. Roles of renal erythropoietin-producing (REP) cells in the maintenance of systemic oxygen homeostasis. Pflug. Arch. 2016, 468, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.A.; Lundby, C.; Cotter, J.D.; Burke, L.M. Maximizing Cellular Adaptation to Endurance Exercise in Skeletal Muscle. Metab. Cell Metab. 2018, 27, 962–976. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.A.; Glass, G.A.; Cunningham, J.M.; Bunn, H.F. The regulated expression of erythropoietin by two human hepatoma cell lines. Proc. Natl. Acad. Sci. USA 1987, 84, 7972–7976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldwasser, E.; Jacobson, L.O.; Fried, W.; Plzak, L.F. Studies on erythropoiesis. V. The effect of cobalt on the production of erythropoietin. Blood 1958, 13, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.A.; Dunning, S.P.; Bunn, H.F. Regulation of the erythropoietin gene: Evidence that the oxygen sensor is a heme protein. Science 1988, 242, 1412–1415. [Google Scholar] [CrossRef]
- Jiang, B.H.; Semenza, G.L.; Bauer, C.; Marti, H.H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996, 271, C1172–C1180. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N. Erythropoietin gene expression: Developmental-stage specificity, cell-type specificity, and hypoxia inducibility. Tohoku J. Exp. Med. 2015, 235, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Gradin, K.; Poellinger, L.; Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp. Cell Res. 2017, 356, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallio, P.J.; Wilson, W.J.; O’Brien, S.; Makino, Y.; Poellinger, L. Regulation of the hypoxia-inducible transcription factor 1alpha by the ubiquitin-proteasome pathway. J. Biol. Chem. 1999, 274, 6519–6525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Srinivas, V.; Zhang, L.P.; Zhu, X.H.; Caro, J. Characterization of an oxygen/redox-dependent degradation domain of hypoxia-inducible factor alpha (HIF-alpha) proteins. Biochem. Biophys. Res. Commun. 1999, 260, 557–561. [Google Scholar] [CrossRef]
- Srinivas, V.; Zhu, X.; Salceda, S.; Nakamura, R.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) is a non-heme iron protein. Implications for oxygen sensing. J. Biol. Chem. 1999, 274, 1180. [Google Scholar] [CrossRef]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Krek, W. VHL takes HIF’s breath away. Nat. Cell Biol. 2000, 2, E121–E123. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Wilkins, S.E.; Abboud, M.I.; Hancock, R.L.; Schofield, C.J. Targeting Protein-Protein Interactions in the HIF System. ChemMedChem 2016, 11, 773–786. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 610–616. [Google Scholar] [CrossRef]
- Yeh, T.L.; Leissing, T.M.; Abboud, M.I.; Thinnes, C.C.; Atasoylu, O.; Holt-Martyn, J.P.; Zhang, D.; Tumber, A.; Lippl, K.; Lohans, C.T.; et al. Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem. Sci. 2017, 8, 7651–7668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, K.; Yamanaka, K.; Kamura, T.; Minato, N.; Conaway, R.C.; Conaway, J.W.; Klausner, R.D.; Pause, A. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 12436–12441. [Google Scholar] [CrossRef] [Green Version]
- Kamura, T.; Sato, S.; Iwai, K.; Czyzyk-Krzeska, M.; Conaway, R.C.; Conaway, J.W. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc. Natl. Acad. Sci. USA 2000, 97, 10430–10435. [Google Scholar] [CrossRef] [Green Version]
- Hirsila, M.; Koivunen, P.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef] [Green Version]
- Koivunen, P.; Hirsila, M.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joharapurkar, A.A.; Pandya, V.B.; Patel, V.J.; Desai, R.C.; Jain, M.R. Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases. J. Med. Chem. 2018, 61, 6964–6982. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Linde, N.S.; Wynter, A.; Metzger, M.; Wong, C.; Langsetmo, I.; Lin, A.; Smith, R.; Rodgers, G.P.; Donahue, R.E.; et al. HIF prolyl hydroxylase inhibition results in endogenous erythropoietin induction, erythrocytosis, and modest fetal hemoglobin expression in rhesus macaques. Blood 2007, 110, 2140–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cases, A. The latest advances in kidney diseases and related disorders. Drug News Perspect. 2007, 20, 647–654. [Google Scholar]
- Chan, M.C.; Holt-Martyn, J.P.; Schofield, C.J.; Ratcliffe, P.J. Pharmacological targeting of the HIF hydroxylases—A new field in medicine development. Mol. Aspects Med. 2016, 47–48, 54–75. [Google Scholar] [CrossRef]
- Hewitson, K.S.; Schofield, C.J.; Ratcliffe, P.J. Hypoxia-inducible factor prolyl-hydroxylase: Purification and assays of PHD2. Methods Enzymol. 2007, 435, 25–42. [Google Scholar] [CrossRef]
- Rose, N.R.; McDonough, M.A.; King, O.N.; Kawamura, A.; Schofield, C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev. 2011, 40, 4364–4397. [Google Scholar] [CrossRef]
- Ehrismann, D.; Flashman, E.; Genn, D.N.; Mathioudakis, N.; Hewitson, K.S.; Ratcliffe, P.J.; Schofield, C.J. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem. J. 2007, 401, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Kaule, G.; Gunzler, V. Assay for 2-oxoglutarate decarboxylating enzymes based on the determination of [1-14C]succinate: Application to prolyl 4-hydroxylase. Anal. Biochem. 1990, 184, 291–297. [Google Scholar] [CrossRef]
- McNeill, L.A.; Hewitson, K.S.; Claridge, T.D.; Seibel, J.F.; Horsfall, L.E.; Schofield, C.J. Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the beta-carbon of asparagine-803. Biochem. J. 2002, 367, 571–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, N.A.; Rakhman, I.; Moroz, N.; Basso, M.; Payappilly, J.; Kazakov, S.; Hernandez-Guzman, F.; Gaisina, I.N.; Kozikowski, A.P.; Ratan, R.R.; et al. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem. Biol. 2010, 17, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.C.; Atasoylu, O.; Hodson, E.; Tumber, A.; Leung, I.K.; Chowdhury, R.; Gomez-Perez, V.; Demetriades, M.; Rydzik, A.M.; Holt-Martyn, J.; et al. Potent and Selective Triazole-Based Inhibitors of the Hypoxia-Inducible Factor Prolyl-Hydroxylases with Activity in the Murine Brain. PLoS ONE 2015, 10, e0132004. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jeong Hee, M.; Eun Ah, C.; Ryu, S.E.; Myung Kyu, L. Monoclonal antibody-based screening assay for factor inhibiting hypoxia-inducible factor inhibitors. J. Biomol. Screen. 2008, 13, 494–503. [Google Scholar] [CrossRef]
- Snell, C.E.; Turley, H.; McIntyre, A.; Li, D.; Masiero, M.; Schofield, C.J.; Gatter, K.C.; Harris, A.L.; Pezzella, F. Proline-hydroxylated hypoxia-inducible factor 1alpha (HIF-1alpha) upregulation in human tumours. PLoS ONE 2014, 9, e88955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.M.; Yeoh, K.K.; Lee, M.K.; Eriksson, T.; Kessler, B.M.; Kramer, H.B.; Edelmann, M.J.; Willam, C.; Pugh, C.W.; Schofield, C.J.; et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J. Biol. Chem. 2011, 286, 13041–13051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, R.; Candela-Lena, J.I.; Chan, M.C.; Greenald, D.J.; Yeoh, K.K.; Tian, Y.M.; McDonough, M.A.; Tumber, A.; Rose, N.R.; Conejo-Garcia, A.; et al. Selective small molecule probes for the hypoxia inducible factor (HIF) prolyl hydroxylases. ACS Chem. Biol. 2013, 8, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; Lienard, B.M.; McDonough, M.A.; Clifton, I.J.; Butler, D.; Soares, A.S.; Oldham, N.J.; McNeill, L.A.; Schofield, C.J. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J. Biol. Chem. 2007, 282, 3293–3301. [Google Scholar] [CrossRef] [Green Version]
- Oehme, F.; Jonghaus, W.; Narouz-Ott, L.; Huetter, J.; Flamme, I. A nonradioactive 96-well plate assay for the detection of hypoxia-inducible factor prolyl hydroxylase activity. Anal. Biochem. 2004, 330, 74–80. [Google Scholar] [CrossRef]
- Zheng, Q.; Yang, H.; Sun, L.; Wei, R.; Fu, X.; Wang, Y.; Huang, Y.; Liu, Y.N.; Liu, W.J. Efficacy and safety of HIF prolyl-hydroxylase inhibitor vs epoetin and darbepoetin for anemia in chronic kidney disease patients not undergoing dialysis: A network meta-analysis. Pharmacol. Res. 2020, 159, 105020. [Google Scholar] [CrossRef]
- Dhillon, S. Daprodustat: First Approval. Drugs 2020, 80, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Roxadustat: First Global Approval. Drugs 2019, 79, 563–572. [Google Scholar] [CrossRef]
- Backman, J.T.; Filppula, A.M.; Niemi, M.; Neuvonen, P.J. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol. Rev. 2016, 68, 168–241. [Google Scholar] [CrossRef]
- Markham, A. Vadadustat: First Approval. Drugs 2020, 80, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Enarodustat: First Approval. Drugs 2020. [Google Scholar] [CrossRef]
- Pai, S.M.; Connaire, J.; Yamada, H.; Enya, S.; Gerhardt, B.; Maekawa, M.; Tanaka, H.; Koretomo, R.; Ishikawa, T. A Mass Balance Study of (14) C-Labeled JTZ-951 (Enarodustat), a Novel Orally Available Erythropoiesis-Stimulating Agent, in Patients With End-Stage Renal Disease on Hemodialysis. Clin. Pharmacol. Drug Dev. 2020, 9, 728–741. [Google Scholar] [CrossRef] [PubMed]
- van der Mey, D.; Gerisch, M.; Jungmann, N.A.; Kaiser, A.; Yoshikawa, K.; Schulz, S.; Radtke, M.; Lentini, S. Drug-drug interaction of atazanavir on UGT1A1-mediated glucuronidation of molidustat in human. Basic Clin. Pharmacol. Toxicol. 2020. [Google Scholar] [CrossRef]
- Lentini, S.; van der Mey, D.; Kern, A.; Thuss, U.; Kaiser, A.; Matsuno, K.; Gerisch, M. Absorption, distribution, metabolism and excretion of molidustat in healthy participants. Basic Clin. Pharmacol. Toxicol. 2020, 127, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Lentini, S.; Kaiser, A.; Kapsa, S.; Matsuno, K.; van der Mey, D. Effects of oral iron and calcium supplement on the pharmacokinetics and pharmacodynamics of molidustat: An oral HIF-PH inhibitor for the treatment of renal anaemia. Eur. J. Clin. Pharmacol. 2020, 76, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Akizawa, T.; Taguchi, M.; Matsuda, Y.; Iekushi, K.; Yamada, T.; Yamamoto, H. Molidustat for the treatment of renal anaemia in patients with dialysis-dependent chronic kidney disease: Design and rationale of three phase III studies. BMJ Open 2019, 9, e026602. [Google Scholar] [CrossRef]
- Haase, V.H. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial. Int. 2017, 21 (Suppl. S1), S110–S124. [Google Scholar] [CrossRef] [Green Version]
- Caltabiano, S.; Mahar, K.M.; Lister, K.; Tenero, D.; Ravindranath, R.; Cizman, B.; Cobitz, A.R. The drug interaction potential of daprodustat when coadministered with pioglitazone, rosuvastatin, or trimethoprim in healthy subjects. Pharmacol. Res. Perspect. 2018, 6, e00327. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Shinozaki, Y.; Kobayashi, H.; Deai, K.; Yoshiuchi, H.; Matsui, T.; Matsuo, A.; Matsushita, M.; Tanaka, T.; Nangaku, M. JTZ-951 (enarodustat), a hypoxia-inducibe factor prolyl hydroxylase inhibitor, stabilizes HIF-alpha protein and induces erythropoiesis without effects on the function of vascular endothelial growth factor. Eur. J. Pharmacol. 2019, 859, 172532. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; Segel, G.B. Developmental biology of erythropoiesis. Blood Rev. 1998, 12, 106–114. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- de la Chapelle, A.; Sistonen, P.; Lehvaslaiho, H.; Ikkala, E.; Juvonen, E. Familial erythrocytosis genetically linked to erythropoietin receptor gene. Lancet 1993, 341, 82–84. [Google Scholar] [CrossRef]
- Ang, S.O.; Stockton, D.W.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Sergueeva, A.I.; Maxwell, P.H.; Semenza, G.L.; Prchal, J.T. Oxygen sensing and Chuvash Polycythemia. Exp. Hematol. 2002, 30, 43. [Google Scholar]
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621. [Google Scholar] [CrossRef]
- Percy, M.J.; Zhao, Q.; Flores, A.; Harrison, C.; Lappin, T.R.; Maxwell, P.H.; McMullin, M.F.; Lee, F.S. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; Furlow, P.W.; Beer, P.A.; Lappin, T.R.; McMullin, M.F.; Lee, F.S. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood 2007, 110, 2193–2196. [Google Scholar] [CrossRef] [Green Version]
- Percy, M.J.; Furlow, P.W.; Lucas, G.S.; Li, X.; Lappin, T.R.; McMullin, M.F.; Lee, F.S. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N. Engl. J. Med. 2008, 358, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Nangaku, M.; Eckardt, K.U. Pathogenesis of renal anemia. Semin. Nephrol. 2006, 26, 261–268. [Google Scholar] [CrossRef]
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of Diabetic Kidney Disease: Current and Future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Tanaka, T.; Nangaku, M. How the Target Hemoglobin of Renal Anemia Should Be. Nephron 2015, 131, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souma, T.; Suzuki, N.; Yamamoto, M. Renal erythropoietin-producing cells in health and disease. Front. Physiol. 2015, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Suzuki, N.; Hirano, I.; Yamazaki, S.; Minegishi, N.; Yamamoto, M. Isolation and characterization of renal erythropoietin-producing cells from genetically produced anemia mice. PLoS ONE 2011, 6, e25839. [Google Scholar] [CrossRef] [Green Version]
- Obara, N.; Suzuki, N.; Kim, K.; Nagasawa, T.; Imagawa, S.; Yamamoto, M. Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood 2008, 111, 5223–5232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Souma, T.; Nezu, M.; Nakano, D.; Yamazaki, S.; Hirano, I.; Sekine, H.; Dan, T.; Takeda, K.; Fong, G.H.; Nishiyama, A.; et al. Erythropoietin Synthesis in Renal Myofibroblasts Is Restored by Activation of Hypoxia Signaling. J. Am. Soc. Nephrol. 2016, 27, 428–438. [Google Scholar] [CrossRef]
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamamura, K.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Investig. 2011, 121, 3981–3990. [Google Scholar] [CrossRef] [Green Version]
- Souza, E.; Cho, K.H.; Harris, S.T.; Flindt, N.R.; Watt, R.K.; Pai, A.B. Hypoxia-inducible factor prolyl hydroxylase inhibitors: A paradigm shift for treatment of anemia in chronic kidney disease? Expert Opin. Investig. Drugs 2020, 29, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Aapro, M.; Gascon, P.; Patel, K.; Rodgers, G.M.; Fung, S.; Arantes, L.H., Jr.; Wish, J. Erythropoiesis-Stimulating Agents in the Management of Anemia in Chronic Kidney Disease or Cancer: A Historical Perspective. Front. Pharmacol. 2018, 9, 1498. [Google Scholar] [CrossRef] [PubMed]
- Bazeley, J.; Wish, J.B. The Evolution of Target Hemoglobin Levels in Anemia of Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2019, 26, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Bohlius, J.; Bohlke, K.; Castelli, R.; Djulbegovic, B.; Lustberg, M.B.; Martino, M.; Mountzios, G.; Peswani, N.; Porter, L.; Tanaka, T.N.; et al. Management of cancer-associated anemia with erythropoiesis-stimulating agents: ASCO/ASH clinical practice guideline update. Blood Adv. 2019, 3, 1197–1210. [Google Scholar] [CrossRef] [Green Version]
- KDIGO. KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int. Suppl. 2012, 2, 279. [Google Scholar]
- Drueke, T.B.; Parfrey, P.S. Summary of the KDIGO guideline on anemia and comment: Reading between the (guide)line(s). Kidney Int. 2012, 82, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Liu, B.C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008, 7, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Hirano, I.; Pan, X.; Minegishi, N.; Yamamoto, M. Erythropoietin production in neuroepithelial and neural crest cells during primitive erythropoiesis. Nat. Commun. 2013, 4, 2902. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.L.; Ang, S.O.; Weigent, D.A.; Prchal, J.T.; Bloomer, J.R. Regulation of ferrochelatase gene expression by hypoxia. Life Sci. 2004, 75, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Riby, J.E.; Firestone, G.L.; Bjeldanes, L.F. 3,3’-diindolylmethane reduces levels of HIF-1alpha and HIF-1 activity in hypoxic cultured human cancer cells. Biochem. Pharmacol. 2008, 75, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.; Grondin, F.; McDonald, P.P.; Richard, D.E.; Dubois, C.M. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: Impact on the bioactivation of proproteins. J. Biol. Chem. 2005, 280, 6561–6569. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Evrard, S.; Badiola, I.; Siegfried, G.; Khatib, A.M. Regulation of the proprotein convertases expression and activity during regenerative angiogenesis: Role of hypoxia-inducible factor (HIF). Eur. J. Cell Biol. 2017, 96, 457–468. [Google Scholar] [CrossRef]
- Shah, Y.M.; Matsubara, T.; Ito, S.; Yim, S.H.; Gonzalez, F.J. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Metab. Cell Metab. 2009, 9, 152–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J. Clin. Investig. 2009, 119, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.Q.; Qian, Z.M.; Zhou, Y.F.; Zhang, M.W.; Wang, D.; Zhu, L.; Ke, Y. Expression of Iron Regulatory Protein 1 Is Regulated not only by HIF-1 but also pCREB under Hypoxia. Int. J. Biol. Sci. 2016, 12, 1191–1202. [Google Scholar] [CrossRef]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Akizawa, T.; Iwasaki, M.; Yamaguchi, Y.; Majikawa, Y.; Reusch, M. Phase 3, Randomized, Double-Blind, Active-Comparator (Darbepoetin Alfa) Study of Oral Roxadustat in CKD Patients with Anemia on Hemodialysis in Japan. J. Am. Soc. Nephrol. 2020, 31, 1628–1639. [Google Scholar] [CrossRef]
- Akizawa, T.; Nangaku, M.; Yamaguchi, T.; Arai, M.; Koretomo, R.; Matsui, A.; Hirakata, H. A Placebo-Controlled, Randomized Trial of Enarodustat in Patients with Chronic Kidney Disease Followed by Long-Term Trial. Am. J. Nephrol. 2019, 49, 165–174. [Google Scholar] [CrossRef]
- Akizawa, T.; Nangaku, M.; Yonekawa, T.; Okuda, N.; Kawamatsu, S.; Onoue, T.; Endo, Y.; Hara, K.; Cobitz, A.R. Efficacy and Safety of Daprodustat Compared with Darbepoetin Alfa in Japanese Hemodialysis Patients with Anemia: A Randomized, Double-Blind, Phase 3 Trial. Clin. J. Am. Soc. Nephrol. 2020, 15, 1155–1165. [Google Scholar] [CrossRef]
- Nangaku, M.; Hamano, T.; Akizawa, T.; Tsubakihara, Y.; Nagai, R.; Okuda, N.; Kurata, K.; Nagakubo, T.; Jones, N.P.; Endo, Y.; et al. Daprodustat Compared with Epoetin Beta Pegol for Anemia in Japanese Patients Not on Dialysis: A 52-Week Randomized Open-Label Phase 3 Trial. Am. J. Nephrol. 2021, 1–10. [Google Scholar] [CrossRef]
- Del Vecchio, L.; Locatelli, F. Hypoxia response and acute lung and kidney injury: Possible implications for therapy of COVID-19. Clin. Kidney J. 2020, 13, 494–499. [Google Scholar] [CrossRef]
- Spath, M.R.; Koehler, F.C.; Hoyer-Allo, K.J.R.; Grundmann, F.; Burst, V.; Muller, R.U. Preconditioning strategies to prevent acute kidney injury. F1000Research 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Bullen, A.; Liu, Z.Z.; Hepokoski, M.; Li, Y.; Singh, P. Renal Oxygenation and Hemodynamics in Kidney Injury. Nephron 2017, 137, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Mimura, I.; Shoji, K.; Tanaka, T.; Nangaku, M. Hypoxia and fibrosis in chronic kidney disease: Crossing at pericytes. Kidney Int. Suppl. 2014, 4, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangaku, M.; Rosenberger, C.; Heyman, S.N.; Eckardt, K.U. Regulation of hypoxia-inducible factor in kidney disease. Clin. Exp. Pharmacol. Physiol. 2013, 40, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.G.; Smith, J.A.; Wright, C.; Gardiner, B.S.; Smith, D.W.; Cochrane, A.D. Urinary oxygen tension: A clinical window on the health of the renal medulla? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R45–R50. [Google Scholar] [CrossRef] [Green Version]
- Lankadeva, Y.R.; Okazaki, N.; Evans, R.G.; Bellomo, R.; May, C.N. Renal Medullary Hypoxia: A New Therapeutic Target for Septic Acute Kidney Injury? Semin. Nephrol. 2019, 39, 543–553. [Google Scholar] [CrossRef]
- Heyman, S.N.; Gorelik, Y.; Zorbavel, D.; Rosenberger, C.; Abassi, Z.; Rosen, S.; Khamaisi, M. Near-drowning: New perspectives for human hypoxic acute kidney injury. Nephrol. Dial. Transplant. 2020, 35, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Nangaku, M. The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 2010, 6, 667–678. [Google Scholar] [CrossRef]
- Manotham, K.; Tanaka, T.; Matsumoto, M.; Ohse, T.; Miyata, T.; Inagi, R.; Kurokawa, K.; Fujita, T.; Nangaku, M. Evidence of tubular hypoxia in the early phase in the remnant kidney model. J. Am. Soc. Nephrol. 2004, 15, 1277–1288. [Google Scholar] [CrossRef]
- Nangaku, M.; Eckardt, K.U. Hypoxia and the HIF system in kidney disease. J. Mol. Med. 2007, 85, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Nangaku, M. Founding papers of current nephrology: From acute kidney injury to diabetic kidney disease. Kidney Int. 2020, 98, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Hirakawa, Y.; Nangaku, M. The role of oxidative stress and hypoxia in renal disease. Kidney Res. Clin. Pract. 2019, 38, 414–426. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Tanaka, T.; Ishii, T.; Wakashima, T.; Fukui, K.; Nangaku, M. Prolyl hydroxylase inhibition protects the kidneys from ischemia via upregulation of glycogen storage. Kidney Int. 2020, 97, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Jaffe, J.; Michael, M.; Swan, C.E.; Duffy, K.J.; Erickson-Miller, C.L.; Haase, V.H. Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am. J. Physiol. Ren. Physiol. 2012, 302, F1172–F1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Investig. 2014, 124, 2396–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wen, L.; Hu, X.; Wei, Q.; Dong, Z. HIF in Nephrotoxicity during Cisplatin Chemotherapy: Regulation, Function and Therapeutic Potential. Cancers 2021, 13, 180. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, X.; Zhang, Y.; Ding, G.; Zhu, C.; Huang, S.; Jia, Z.; Zhang, A. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, W.M.; Gottmann, U.; Doyon, F.; Buchholz, B.; Campean, V.; Schodel, J.; Reisenbuechler, A.; Klaus, S.; Arend, M.; Flippin, L.; et al. Donor treatment with a PHD-inhibitor activating HIFs prevents graft injury and prolongs survival in an allogenic kidney transplant model. Proc. Natl. Acad. Sci. USA 2009, 106, 21276–21281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraisl, P.; Aragones, J.; Carmeliet, P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 2009, 8, 139–152. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Hyvarinen, J.; Hassinen, I.E.; Sormunen, R.; Maki, J.M.; Kivirikko, K.I.; Koivunen, P.; Myllyharju, J. Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J. Biol. Chem. 2010, 285, 13646–13657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.; Keely, S.; Karhausen, J.; Gerich, M.E.; Furuta, G.T.; Colgan, S.P. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 2008, 134, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Vogler, M.; Zieseniss, A.; Hesse, A.R.; Levent, E.; Tiburcy, M.; Heinze, E.; Burzlaff, N.; Schley, G.; Eckardt, K.U.; Willam, C.; et al. Pre- and post-conditional inhibition of prolyl-4-hydroxylase domain enzymes protects the heart from an ischemic insult. Pflugers Arch. 2015, 467, 2141–2149. [Google Scholar] [CrossRef]
- Philipp, S.; Jurgensen, J.S.; Fielitz, J.; Bernhardt, W.M.; Weidemann, A.; Schiche, A.; Pilz, B.; Dietz, R.; Regitz-Zagrosek, V.; Eckardt, K.U.; et al. Stabilization of hypoxia inducible factor rather than modulation of collagen metabolism improves cardiac function after acute myocardial infarction in rats. Eur. J. Heart Fail. 2006, 8, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.G.; Lee, W.H.; Theodorou, L.; Kodo, K.; Lim, S.Y.; Shukla, D.H.; Briston, T.; Kiriakidis, S.; Ashcroft, M.; Davidson, S.M.; et al. HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc. Res. 2014, 104, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Furuta, G.T.; Turner, J.R.; Taylor, C.T.; Hershberg, R.M.; Comerford, K.; Narravula, S.; Podolsky, D.K.; Colgan, S.P. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J. Exp. Med. 2001, 193, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Keely, S.; Campbell, E.L.; Baird, A.W.; Hansbro, P.M.; Shalwitz, R.A.; Kotsakis, A.; McNamee, E.N.; Eltzschig, H.K.; Kominsky, D.J.; Colgan, S.P. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol. 2014, 7, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, E.; Goggins, B.J.; Cardona, J.; Cole, S.; Minahan, K.; Mateer, S.; Walker, M.M.; Shalwitz, R.; Keely, S. Oral delivery of prolyl hydroxylase inhibitor: AKB-4924 promotes localized mucosal healing in a mouse model of colitis. Inflamm. Bowel Dis. 2015, 21, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Rehman, J.; Tang, H.; Wary, K.; Mittal, M.; Chaturvedi, P.; Zhao, Y.Y.; Komarova, Y.A.; Vogel, S.M.; Malik, A.B. HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Investig. 2015, 125, 652–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhara, T.; Hishiki, T.; Kasahara, M.; Hayakawa, N.; Oyaizu, T.; Nakanishi, T.; Kubo, A.; Morisaki, H.; Kaelin, W.G., Jr.; Suematsu, M.; et al. Inhibition of the oxygen sensor PHD2 in the liver improves survival in lactic acidosis by activating the Cori cycle. Proc. Natl. Acad. Sci. USA 2015, 112, 11642–11647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeley, T.W.; Sternlicht, M.D.; Klaus, S.J.; Neff, T.B.; Liu, D.Y. Induction of erythropoiesis by hypoxia-inducible factor prolyl hydroxylase inhibitors without promotion of tumor initiation, progression, or metastasis in a VEGF-sensitive model of spontaneous breast cancer. Hypoxia 2017, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Nishide, S.; Uchida, J.; Matsunaga, S.; Tokudome, K.; Yamaguchi, T.; Kabei, K.; Moriya, T.; Miura, K.; Nakatani, T.; Tomita, S. Prolyl-hydroxylase inhibitors reconstitute tumor blood vessels in mice. J. Pharmacol. Sci. 2020, 143, 122–126. [Google Scholar] [CrossRef]
- Nishide, S.; Matsunaga, S.; Shiota, M.; Yamaguchi, T.; Kitajima, S.; Maekawa, Y.; Takeda, N.; Tomura, M.; Uchida, J.; Miura, K.; et al. Controlling the Phenotype of Tumor-Infiltrating Macrophages via the PHD-HIF Axis Inhibits Tumor Growth in a Mouse Model. iScience 2019, 19, 940–954. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Taguchi, M.; Matsuda, Y.; Iekushi, K.; Yamada, T.; Akizawa, T. Molidustat for the treatment of renal anaemia in patients with non-dialysis-dependent chronic kidney disease: Design and rationale of two phase III studies. BMJ Open 2019, 9, e026704. [Google Scholar] [CrossRef] [Green Version]
- Biguzzi, E.; Siboni, S.M.; Peyvandi, F. How I treat gastrointestinal bleeding in congenital and acquired von Willebrand disease. Blood 2020, 136, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Reveiz, L.; Gyte, G.M.; Cuervo, L.G.; Casasbuenas, A. Treatments for iron-deficiency anaemia in pregnancy. Cochrane Database Syst. Rev. 2011, CD003094. [Google Scholar] [CrossRef]
- Mokas, S.; Lariviere, R.; Lamalice, L.; Gobeil, S.; Cornfield, D.N.; Agharazii, M.; Richard, D.E. Hypoxia-inducible factor-1 plays a role in phosphate-induced vascular smooth muscle cell calcification. Kidney Int. 2016, 90, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaco, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef] [Green Version]
- Gale, D.P.; Harten, S.K.; Reid, C.D.; Tuddenham, E.G.; Maxwell, P.H. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Mouradian, G.; Li, M.; Riddle, S.; et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- Smith, K.A.; Waypa, G.B.; Dudley, V.J.; Budinger, G.R.S.; Abdala-Valencia, H.; Bartom, E.; Schumacker, P.T. Role of Hypoxia-Inducible Factors in Regulating Right Ventricular Function and Remodeling during Chronic Hypoxia-induced Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 63, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Macias, D.; Moore, S.; Crosby, A.; Southwood, M.; Du, X.; Tan, H.; Xie, S.; Vassallo, A.; Wood, A.J.T.; Wallace, E.M.; et al. Targeting HIF2alpha-ARNT hetero-dimerisation as a novel therapeutic strategy for pulmonary arterial hypertension. Eur. Respir. J. 2021, 57. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Investig. 2020, 130, 5638–5651. [Google Scholar] [CrossRef]
- Siques, P.; Brito, J.; Pena, E. Reactive Oxygen Species and Pulmonary Vasculature During Hypobaric Hypoxia. Front. Physiol. 2018, 9, 865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, A.A.; Aragones, J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines 2018, 6, 68. [Google Scholar] [CrossRef] [Green Version]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, A.; Klanke, B.; Wagner, M.; Volk, T.; Willam, C.; Wiesener, M.S.; Eckardt, K.U.; Warnecke, C. Hypoxia, via stabilization of the hypoxia-inducible factor HIF-1alpha, is a direct and sufficient stimulus for brain-type natriuretic peptide induction. Biochem. J. 2008, 409, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, B.; Schley, G.; Faria, D.; Kroening, S.; Willam, C.; Schreiber, R.; Klanke, B.; Burzlaff, N.; Jantsch, J.; Kunzelmann, K.; et al. Hypoxia-inducible factor-1alpha causes renal cyst expansion through calcium-activated chloride secretion. J. Am. Soc. Nephrol. 2014, 25, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Raptis, V.; Bakogiannis, C.; Loutradis, C.; Boutou, A.K.; Lampropoulou, I.; Intzevidou, E.; Sioulis, A.; Balaskas, E.; Sarafidis, P.A. Levels of Endocan, Angiopoietin-2, and Hypoxia-Inducible Factor-1a in Patients with Autosomal Dominant Polycystic Kidney Disease and Different Levels of Renal Function. Am. J. Nephrol. 2018, 47, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.; Fu, H.; Li, Y.; Wang, L.; Luo, S.; Lu, H. Hypoxia exacerbates nonalcoholic fatty liver disease via the HIF-2alpha/PPARalpha pathway. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E710–E722. [Google Scholar] [CrossRef]
- He, Y.; Yang, W.; Gan, L.; Liu, S.; Ni, Q.; Bi, Y.; Han, T.; Liu, Q.; Chen, H.; Hu, Y.; et al. Silencing HIF-1alpha aggravates non-alcoholic fatty liver disease in vitro through inhibiting PPAR-alpha/ANGPTL4 singling pathway. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; He, Y.; Zhao, H.; Xu, X. Hypoxia inducible factor-1 promotes liver fibrosis in nonalcoholic fatty liver disease by activating PTEN/p65 signaling pathway. J. Cell. Biochem. 2019, 120, 14735–14744. [Google Scholar] [CrossRef]
- Meadowcroft, A.M.; Cizman, B.; Holdstock, L.; Biswas, N.; Johnson, B.M.; Jones, D.; Nossuli, A.K.; Lepore, J.J.; Aarup, M.; Cobitz, A.R. Daprodustat for anemia: A 24-week, open-label, randomized controlled trial in participants on hemodialysis. Clin. Kidney J. 2019, 12, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Oguro, A.; Imaoka, S. Feedback of hypoxia-inducible factor-1alpha (HIF-1alpha) transcriptional activity via redox factor-1 (Ref-1) induction by reactive oxygen species (ROS). Free Radic. Res. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aprelikova, O.; Chandramouli, G.V.; Wood, M.; Vasselli, J.R.; Riss, J.; Maranchie, J.K.; Linehan, W.M.; Barrett, J.C. Regulation of HIF prolyl hydroxylases by hypoxia-inducible factors. J. Cell. Biochem. 2004, 92, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Markova, D.; Anderson, D.G.; Chiba, K.; Toyama, Y.; Shapiro, I.M.; Risbud, M.V. Expression of prolyl hydroxylases (PHDs) is selectively controlled by HIF-1 and HIF-2 proteins in nucleus pulposus cells of the intervertebral disc: Distinct roles of PHD2 and PHD3 proteins in controlling HIF-1alpha activity in hypoxia. J. Biol. Chem. 2012, 287, 16975–16986. [Google Scholar] [CrossRef] [Green Version]
- Henze, A.T.; Riedel, J.; Diem, T.; Wenner, J.; Flamme, I.; Pouyseggur, J.; Plate, K.H.; Acker, T. Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer Res. 2010, 70, 357–366. [Google Scholar] [CrossRef] [Green Version]




| Substrate | Km (µM) | ||||||
|---|---|---|---|---|---|---|---|
| Gene Name | Enzyme | Pro-402 | Pro-564 | O2 | 2-OG | Ascorbate | Fe (II) |
| EGLN1 | PHD2 | + | + | 230 | 60 | 170 | 0.03 |
| EGLN2 | PHD1 | + | + | 250 | 60 | 180 | 0.1 |
| EGLN3 | PHD3 | − | + | 230 | 55 | 140 | 0.03 |
| HIF1AN | FIH-1 | Asp-803 | 90 | 25 | 260 | 0.5 | |
| Protein | Gene | HIF1/HIF2 | Function | Ref. |
|---|---|---|---|---|
| Ceruloplasmin | CP | HIF-1 | ferroxidase | [121] |
| Duodenal cytochrome b | CYBRD1 | HIF-2 | Ferric reductase | [122] |
| Erythropoietin | EPO | HIF-2 | promote red blood cell production | [36,123] |
| Ferrochelatase | FECH | HIF-1 | Heme synthesis | [124] |
| Furin | FURIN | HIF-1 | subtilisin-like proprotein convertase | [125,126,127] |
| Hepcidin | HAMP | HIF-2 | maintenance of iron homeostasis | [128,129] |
| Heme oxygenase-1 | HMOX1 | HIF-1 | Heme degradation | [121] |
| Aconitase | IRP1 | HIF-1 | Cellular iron sensing | [130] |
| Transferrin | TF | HIF-1 | Serum iron transfporter | [131,132] |
| Transferrin receptor | TFRC | HIF-1 | Cellular iron uptake | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirota, K. HIF-α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review. Biomedicines 2021, 9, 468. https://doi.org/10.3390/biomedicines9050468
Hirota K. HIF-α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review. Biomedicines. 2021; 9(5):468. https://doi.org/10.3390/biomedicines9050468
Chicago/Turabian StyleHirota, Kiichi. 2021. "HIF-α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review" Biomedicines 9, no. 5: 468. https://doi.org/10.3390/biomedicines9050468