Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects


Abstract
:
1. Introduction
2. Data Search and Source
3. Alcohol Absorption
4. Gastric Alcohol Dehydrogenase and First-Pass Metabolism of Alcohol
5. Hepatic Alcohol Metabolism
Ethanol Acetaldehyde
Ethanol Acetaldehyde
Ethanol Acetaldehyde
6. Hepatic Alcohol Dehydrogenase
7. Hepatic Microsomal Ethanol-Oxidizing System
8. Hepatic Acetaldehyde Dehydrogenase
9. Cascade of Molecular Mechanisms and Cellular Events
9.1. The 5-Hit Working Hypothesis of ALD
9.2. Hepatocytes versus Non-Parenchymal Cells
9.2.1. Kupffer Cells
9.2.2. Hepatic Stellate Cells
9.2.3. Liver Sinusoidal Endothelial Cells
9.3. Oxidative Stress and Reactive Oxygen Species
9.4. Signaling Mediators
10. Clinical Issues of Alcoholic Liver Disease
10.1. Natural Course
10.2. Questionaires
10.3. Laboratory Approaches
11. Alcoholic Fatty Liver
12. Alcoholic Steatohepatitis and Alcoholic Hepatitis
13. Alcoholic Cirrhosis
14. Alcoholic Hepatocellular Carcinoma
15. Actual Issues and Future Perspectives
16. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABIC | Age, bilirubin, INR, and creatinine score |
| AASLD | American Association of the Study of Liver Diseases |
| AC | Alcoholic cirrhosis |
| ADH | Alcohol dehydrogenase |
| AFL | Alcoholic fatty liver |
| ALD | Alcoholic liver disease |
| ALDH | Acetaldehyde dehydrogenase |
| AH | Alcoholic hepatitis |
| AHCC | Alcoholic hepatocellular carcinoma |
| ALT | Alanine transaminase |
| ASH | Alcoholic steatohepatitis |
| AST | Aspartate transaminase |
| CDT | Carbohydrate-deficient transferrin |
| CLD | Chronic liver diseases |
| CYP | Cytochrome P450 |
| CYP2E1 | Cytochrome P450 2E1 |
| DILI | Drug induced liver injury |
| EASL | European Association for the Study of the Liver |
| FPM | First pass metabolism |
| GAHS | Glasgow Alcoholic Hepatitis Score |
| GDH | Glutamate dehydrogenase |
| GGT | Gamma-glutamyltransferase |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HILI | Herb-induced liver injury |
| Km | Michaelis-Menten constant |
| MCV | Mean corpuscular volume of erythrocytes |
| MDB | Mallory-Denk bodies |
| MDF | Maddrey Discriminant Function scale |
| MELD | Model for End-stage Liver Disease score |
| MEOS | Microsomal ethanol-oxidizing system |
| MAST | Michigan Alcoholism Screening Test |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| PTU | Propylthiouracil |
| ROS | Reactive oxygen species |
| RUCAM | Roussel Uclaf Causality Assessment Method |
References
- Ingólfsson, H.I.; Anderson, O.S. Alcohol’s effects on lipid bilayer properties. Biophys. J. 2011, 101, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Sergent, O.; Djoudi-Aliche, F.; Lagadic-Gossmann, D. Up-to date insight about membrane remodeling as a mechanism of action for ethanol-induced liver toxicity. In Trends in Alcoholic Liver Disease–Clinical and Scientific Aspects; Shimizu, I., Ed.; InTech: London, UK; Available online: https://cdn.intechopen.com/pdfs-wm/25884.pdf (accessed on 26 October 2018).
- Dopico, A.M.; Lovinger, D.M. Acute alcohol action and desensibilization of ligandgated ion channels. Pharmacol. Rev. 2009, 61, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Sevastianos, V.A.; Dourakis, S.P. Alcoholic liver disease: A clinical review. J. Nutr. Food Sci. 2016, 6, 508. [Google Scholar] [CrossRef]
- Mathurin, P.; Lucey, M.R. Management of alcoholic hepatitis. J. Hepatol. 2012, 56, S39–S45. [Google Scholar] [CrossRef]
- EASL. 2010 Guidelines: Management of Alcoholic Liver Disease. Available online: http://www.easl.eu/research/our-contributions/clinical-practice-guidelines/detail/management-of-alcoholic-liver-disease-easl-clinical-practice-guidelines/report/1 (accessed on 28 April 2018).
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J. Alcoholic liver disease. Am. J. Gastroenterol. 2010, 105, 14–32. Available online: https://www.ncbi.nlm.nih.gov/pubmed/19904248 (accessed on 28 April 2018).
- Toniutto, P.; Zanetto, A.; Ferrarese, A.; Burra, P. Current challenges and future directions for liver transplantation. Liver Int. 2017, 37, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Artru, F.; Louvet, A.; Mathurin, P. Liver transplantation for patients with alcoholic hepatitis. Liver Int. 2017, 37, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.Y.Y. Liver transplantation for severe alcoholic hepatitis–The CON view. Liver Int. 2017, 37, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Lucey, M.R. Liver transplantation for severe alcoholic hepatitis—The PRO view. Liver Int. 2017, 37, 343–344. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Status Report on Alcohol and Health; World Health Organization: Geneva, Switzerland, 2011. Available online: http://www.who.int/substance_abuse/publications/global_alcohol_report/msbgsruprofiles.pdf?ua=1 (accessed on 20 April 2018).
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular analysis of photosystems. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. Available online: http://www.ncbi.nlm.nih.gov/books/NBK21484/ (accessed on 26 October 2018).
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. The Calvin cycle synthesizes hexoses from carbon dioxide and water. In Biochemistry, 5th ed.; W. H. Freeman: New York, NY, USA, 2002. Available online: http://www.ncbi.nlm.nih.gov/books/NBK22344/ (accessed on 26 October 2018).
- Salaspuro, M. Epidemiological aspects of alcohol and alcoholic liver disease, ethanol metabolism, and pathogenesis of alcoholic liver injury. In Oxford Textbook of Clinical Hepatology; Bircher, J., Benhamou, J.P., McIntyre, N., Rizzetto, N., Rodes, J., Eds.; Oxford University Press: Oxford, UK, 1999; pp. 791–810. [Google Scholar]
- Mitchell, M.C.; Teigen, E.L.; Ramchandani, V.A. Absorption and peak blood alcohol concentration after drinking beer, wine, or spirits. Alcohol. Clin. Exp. Res. 2014, 38, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Caballeria, J.; Frezza, M.; Hernández-Muñoz, R.; DiPadova, C.; Korsten, M.A.; Baraona, E.; Lieber, C.S. Gastric origin of the first-pass metabolism of ethanol in humans: Effect of gastrectomy. Gastroenterology 1989, 97, 1205–1209. [Google Scholar] [CrossRef]
- Frezza, M.; di Padova, C.; Pozzato, G.; Terpin, M.; Baraona, E.; Lieber, C.S. High blood alcohol levels in women—Role of decreased gastric alcohol dehydrogenase activity and first-pass metabolism. N. Engl. J. Med. 1990, 322, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcohol and the liver: 1994 update. Gastroenterology 1994, 106, 1085–1105. [Google Scholar] [CrossRef]
- Teschke, R.; Gellert, J. Hepatic microsomal ethanol-oxidizing system (MEOS): Metabolic aspects and clinical implications. Alcohol. Clin. Exp. Res. 1986, 10, 20S–32S. [Google Scholar] [CrossRef] [PubMed]
- Dasgupt, A. Genetic polymorphisms of alcohol metabolizing enzymes associated with protection from or increased risk of alcohol. In Alcohol, Drugs, Genes and the Clinical Laboratory; Dasgupt, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 107–116. [Google Scholar]
- Teschke, R. Alcoholic steatohepatitis (ASH) and acute alcoholic hepatitis (AH): Cascade of events, clinical features, and pharmacotherapy options. Exp. Opin. Pharmacother. 2018, 19, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. Ethanol oxidation by hepatic microsomes: Adaptive increase after ethanol feeding. Science 1968, 162, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. Hepatic microsomal ethanol-oxidizing system. In vitro characteristics and adaptive properties in vivo. J. Biol. Chem. 1970, 245, 2505–2512. [Google Scholar] [PubMed]
- Lieber, C.S.; DeCarli, L.M. Reduced nicotinamide-adenine dinucleotide phosphate oxidase: Enhanced by ethanol consumption. Science 1970, 170, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. The role of the hepatic microsomal ethanol-oxidzing system (MEOS) for ethanol metabolism in vivo. J. Pharmacol. Exp. Ther. 1972, 181, 279–287. [Google Scholar] [PubMed]
- Teschke, R.; Hasumura, Y.; Joly, J.G.; Ishii, H.; Lieber, C.S. Microsomal ethanol-oxidizing system (MEOS): Purification and properties of a rat liver system free of catalase and alcohol dehydrogenase. Biochem. Biophys. Res. Commun. 1972, 49, 1187–1193. [Google Scholar] [CrossRef]
- Ishii, H.; Joly, J.G.; Lieber, C.S. Effect of ethanol on the amount and enzyme activities of hepatic rough and smooth microsomal membranes. Biochim. Biophys. Acta 1973, 291, 411–420. [Google Scholar] [CrossRef]
- Joly, J.G.; Ishii, H.; Teschke, R.; Hasumura, Y.; Lieber, C.S. Effect of chronic ethanol feeding on the activities of and submicrosomal distribution of reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 reductase and the demethylases for aminopyrine and ethylmorphine. Biochem. Pharmacol. 1973, 22, 1532–1535. [Google Scholar] [CrossRef]
- Joly, J.G.; Feinman, L.; Ishii, H.; Lieber, C.S. Effect of chronic ethanol feeding on hepatic microsomal glycerophosphate acyltransferase activity. J. Lipid Res. 1973, 14, 337–343. [Google Scholar] [PubMed]
- Ishii, H.; Joly, J.G.; Lieber, C.S. Increase of microsomal glucose-6-phosphatase activity after chronic ethanol administration. Metabolism 1973, 22, 799–806. [Google Scholar] [CrossRef]
- Mezey, E.; Potter, J.J.; Reed, W.D. Ethanol oxidation by a component of liver microsomes rich in cytochrome P-450. J. Biol. Chem. 1973, 248, 1183–1187. [Google Scholar] [PubMed]
- Teschke, R.; Hasumura, Y.; Lieber, C.S. NADPH-dependent oxidation of methanol, ethanol, propanol, and butanol by hepatic microsomes. Biochem. Biophys. Res. Commun. 1974, 60, 851–857. [Google Scholar] [CrossRef]
- Lieber, C.S.; DeCarli, L.M. Oxidation of ethanol by hepatic microsomes of acatalasemic mice. Biochem. Biophys. Res. Commun. 1974, 60, 1187–1192. [Google Scholar] [CrossRef]
- Teschke, R.; Hasumura, Y.; Lieber, C.S. Hepatic microsomal ethanol oxidizing system: Solubilization, isolation and characterization. Arch. Biochem. Biophys. 1974, 163, 404–415. [Google Scholar] [CrossRef]
- Hasumura, Y.; Teschke, R.; Lieber, C.S. Increased carbon tetrachloride hepatotoxicity, and its mechanism, after chronic ethanol consumption. Gastroenterology 1974, 66, 226–234. [Google Scholar]
- Joly, J.G.; Hétu, C. Effects of chronic ethanol administration in the rat. Relative dependency on dietary lipids—I. Induction of hepatic microsomal drug-metabolizing enzymes in vitro. Biochem. Pharmacol. 1975, 24, 1475–1480. [Google Scholar] [CrossRef]
- Lieber, C.S.; Hasumura, Y.; Teschke, R.; Matsuzaki, S.; Korsten, M. The effect of chronic ethanol consumption on acetaldehyde metabolism. In The Role of Acetaldehyde in the Actions of Ethanol; Lindros, K.O., Eriksson, C.J.P., Eds.; The Finnish Foundation for Alcohol Studies: Helsinki, Finland, 1975; Volume 23, pp. 83–104. [Google Scholar]
- Teschke, R.; Hasumura, Y.; Lieber, C.S. Hepatic microsomal alcohol-oxidizing system: Affinity for methanol, ethanol, propanol and butanol. J. Biol. Chem. 1975, 250, 7397–7404. [Google Scholar] [PubMed]
- Teschke, R.; Hasumura, Y.; Lieber, C.S. Hepatic microsomal alcohol-oxidizng system in normal and acatalasemic mice: Its dissociation from the peroxidatic activity of catalase-H2O2. Mol. Pharmacol. 1975, 11, 841–849. [Google Scholar] [PubMed]
- Ullrich, V.; Weber, P.; Wollenbeg, P. Tetrahydrofurane—An inhibitor for ethanol-induced liver micosomal cytochrome P-450. Biochem. Biophys. Res. Commun. 1975, 64, 808–813. [Google Scholar] [CrossRef]
- Lieber, C.S.; DeCarli, L.M.; Feinman, L.; Hasumura, Y.; Korsten, M.; Matsuzaki, S.; Teschke, R. Effect of chronic alcohol consumption on ethanol and acetaldehyde metabolism. In Alcohol Intoxication and Withdrawal: Experimental Studies II; Advances in Experimental Medicine and Biology; Gross, M.M., Ed.; Plenum Press: New York, NY, USA, 1975; Volume 59, pp. 185–227. [Google Scholar]
- Hasumura, Y.; Teschke, R.; Lieber, C.S. Acetaldehyde oxidation by hepatic mitochondria: Its decrease after chronic ethanol consumption. Science 1975, 189, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Hasumura, Y.; Teschke, R.; Lieber, C.S. Characteristics of acetaldehyde oxidation in rat liver mitochondria. J. Biol. Chem. 1976, 251, 4908–4913. [Google Scholar] [PubMed]
- Teschke, R.; Hasumura, Y.; Lieber, C.S. Hepatic ethanol metabolism: Respective roles of alcohol dehydrogenase, the microsomal ethanol-oxidizing system, and catalase. Arch. Biochem. Biophys. 1976, 175, 635–643. [Google Scholar] [CrossRef]
- Ohnishi, K.; Lieber, C.S. Reconstitution of the microsomal ethanol-oxidizing system. Qualitative and quantitative changes of cytochrome P-450 after chronic ethanol consumption. J. Biol. Chem. 1977, 252, 7124–7131. [Google Scholar] [PubMed]
- Cederbaum, A.I.; Dicker, E.; Rubin, E.; Lieber, C.S. The effect of dimethylsulfoxide and other radical scavengers on the oxidation of ethanol by rat liver microsomes. Biochem. Biophys. Res. Commun. 1977, 78, 1254–1562. [Google Scholar] [CrossRef]
- Joly, J.G.; Villeneuve, J.P.; Mavier, P. Chronic ethanol administration induces a form of cytochrome P-450 with specific spectral and catalytic properties. Alcohol. Clin. Exp. Res. 1977, 1, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Ohnishi, K.; Hasumura, Y.; Lieber, C.S. Hepatic microsomal ethanol oxidizing system: Isolation and reconstitution. In Microsomes and Drug Oxidations; Ullrich, V., Roots, I., Hildebrandt, A., Estabrook, R.W., Conney, A.H., Eds.; Pergamon Press: Oxford, UK, 1977; pp. 103–110. [Google Scholar]
- Teschke, R.; Matsuzaki, S.; Ohnishi, K.; DeCarli, L.M.; Lieber, C.S. Microsomal ethanol oxidizing system (MEOS): Current status of its characterization and its role. Alcohol. Clin. Exp. Res. 1977, 1, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Matsuzaki, S.; Ohnishi, K.; Hasumura, Y.; Lieber, C.S. Metabolism of alcohol at high concentrations: Role and biochemical nature of the hepatic microsomal ethanol oxidizing system. In Advances in Experimental Medicine and Biology; Vol 85A—Alcohol Intoxication and Withdrawal–IIIa; Gross, M.M., Ed.; Plenum Press: New York, NY, USA, 1977; pp. 257–280. [Google Scholar]
- Lieber, C.S.; DeCarli, L.M.; Matsuzaki, S.; Ohnishi, K.; Teschke, R. The microsomal ethanol-oxidizing system. In Methods in Enzymology; Fleischer, S., Packer, L., Eds.; Academic Press: New York, NY, USA, 1978; pp. 355–368. [Google Scholar]
- Ohnishi, K.; Lieber, C.S. Respective role of superoxide and hydroxyl radical in the activity of the reconstituted microsomal ethanol-oxidizing system. Arch. Biochem. Biophys. 1978, 191, 798–803. [Google Scholar] [CrossRef]
- Miwa, G.T.; Lewin, W.; Thomas, P.E.; Lu, A.Y. The direct oxidation of ethanol by a catalase- free and alcohol dehydrogenase-free reconstituted system containing cytochrome P-450. Arch. Biochem. Biophys. 1978, 187, 464–475. [Google Scholar] [CrossRef]
- Fabry, T.L.; Lieber, C.S. The photochemical action spectrum of the microsomal ethanol oxidizing system. Alcohol. Clin. Exp. Res. 1979, 3, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Korsten, M.A.; Lieber, C.S. Ethanol oxidation by intestinal microsomes: Increased activity after 303 chronic ethanol administration. Life Sci. 1979, 25, 1443–1448. [Google Scholar] [CrossRef]
- Moreno, F.; Teschke, R.; Strohmeyer, G. Effect of thyroid hormones on the activities of hepatic alcohol metabolizing enzymes. Biochem. Biophys. Res. Commun. 1979, 89, 806–812. [Google Scholar] [CrossRef]
- Teschke, R.; Stutz, G.; Strohmeyer, G. Increased paracetamol-induced hepatotoxicity after chronic alcohol consumption. Biochem. Biophys. Res. Commun. 1979, 91, 368–374. [Google Scholar] [CrossRef]
- Teschke, R.; Stutz, G.; Moreno, F. Cholestasis following chronic alcohol consumption: Enhancement after an acute dose of chlorpromazine. Biochem. Biophys. Res. Commun. 1980, 94, 1013–1020. [Google Scholar] [CrossRef]
- Burnett, K.G.; Felder, M.R. Ethanol metabolism in peromyscus genetically deficient in alcohol dehydrogenase. Biochem. Pharmacol. 1980, 29, 125–130. [Google Scholar] [CrossRef]
- Gellert, J.; Moreno, F.; Haydn, M.; Oldiges, H.; Frenzel, H.; Teschke, R.; Strohmeyer, G. Decreased hepatotoxicity of dimethylnitrosamine (DMN) following chronic alcohol consumption. In Alcohol and Aldehyde Metabolizing Systems—IV; Advances in Experimental Medicine and Biology; Thurman, R.G., Ed.; Plenum Press: New York, NY, USA, 1980; Volume 132, pp. 237–243. [Google Scholar]
- Cederbaum, A.I.; Cohen, G. Oxidative demethylation of t-butyl alcohol in rat liver microsomes. Biochem. Biophys. Res. Commun. 1980, 97, 730–736. [Google Scholar] [CrossRef]
- Moreno, F.; Minzlaff, M.; Hauptmeier, K.H.; Teschke, R. Alterations of hepatic alcohol metabolizing enzyme activities due to thyroid hormones. Adv. Exp. Med. Biol. 1980, 132, 109–115. [Google Scholar] [PubMed]
- Cederbaum, A.I.; Qureshi, A.; Messenger, P. Oxidation of isopropanol by rat liver micorosomes. Possible role of hydroxyl radicals. Biochem. Pharmacol. 1981, 30, 825–831. [Google Scholar] [CrossRef]
- Teschke, R.; Moreno, F.; Petrides, A.S. Hepatic microsomal ethanol-oxidizing system (MEOS): Respective roles of ethanol and carbohydrates for the enhanced activity after chronic alcohol consumption. Biochem. Pharmacol. 1981, 30, 1745–1751. [Google Scholar] [CrossRef]
- Seitz, H.K.; Garro, A.J.; Lieber, C.S. Enhanced pulmonary and intestinal activation of procarcinogens and mutagens after chronic ethanol consumption in the rat. Eur. J. Clin. Investig. 1981, 11, 33–38. [Google Scholar] [CrossRef]
- Moreno, F.; Petrides, A.S.; Heinen, E.; Strohmeyer, G.; Teschke, R. Hepatic microsomal ethanol-oxidizing system (MEOS): Increased activity following propylthiouracil administration. Alcohol. Clin. Exp. Res. 1981, 5, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Matsuda, Y.; Lieber, C.S. Increased hepatotoxicity of acetaminophen after chronic ethanol consumption in the rat. Gastroenterology 1981, 80, 140–148. [Google Scholar] [PubMed]
- Teschke, R.; Heymann, K. Effect of sex hormones on the activities of hepatic alcohol-metabolizing enzymes in male rats. Enzyme 1982, 28, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Koop, D.R.; Moran, E.T.; Tarr, G.E.; Coon, M.J. Purification and characterization of a unique isozyme of cytochrome P-450 from liver microsomes of ethanol-treated rabbits. J. Biol. Chem. 1982, 257, 8472–8480. [Google Scholar] [PubMed]
- Morgan, E.T.; Koop, D.R.; Coon, M.J. Catalytic activity of cytochrome P-450 isozyme 3a isolated from liver microsomes of ethanol-treated rabbits. J. Biol. Chem. 1982, 257, 13951–13957. [Google Scholar] [PubMed]
- Ingelman-Sundberg, M.; Hagbjörk, A.L. On the significance of cytochrome P-450-dependent hydroxyl radical-mediated oxygenation mechanism. Xenobiotica 1982, 12, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Petersen, D.R.; Atkinson, N.; Hjelle, J.J. Increase in hepatic microsomal oxidation by a single dose of ethanol. J. Pharmacol. Exp. Ther. 1982, 221, 275–281. [Google Scholar] [PubMed]
- Teschke, R.; Wiese, B. Sex-dependency of hepatic alcohol-metabolizing enzymes. J. Endocrinol. Investig. 1982, 5, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, S.E. The D(V/K) isotope effect of the cytochrome P-450-mediated oxidation of ethanol and its biological applications. Eur. J. Biochem. 1982, 125, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.A.; Lieber, C.S. Hepatic vitamin A depletion in alcoholic liver injury. N. Engl. J. Med. 1982, 307, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bösche, P.; Czygan, P.; Veith, S.; Kommerell, B. Microsomal ethanol oxidation in the colonic mucosa of the rat: Effect of chronic ethanol ingestion. N-S Arch. Pharmacol. 1982, 320, 81–84. [Google Scholar] [CrossRef]
- Teschke, R.; Bolsen, K.; Landmann, H.; Goerz, G. Effect of hexachlorobenzene on the activities of hepatic alcohol-metabolizing enzymes. Biochem. Pharmacol. 1983, 32, 1745–1751. [Google Scholar] [CrossRef]
- Leo, M.A.; Lieber, C.S. Interaction of ethanol with vitamin A. Alcohol. Clin. Exp. Res. 1983, 7, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Minzlaff, M.; Oldiges, H.; Frenzel, H. Effect of chronic alcohol consumption on tumor incidence due to dimethylnitrosamine administration. J. Cancer Res. Clin. Oncol. 1983, 106, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Hauptmeier, K.H.; Frenzel, H. Effect of an acute dose of ethanol on the hepatotoxicity due to carbon tetrachloride. Liver Int. 1983, 3, 100–109. [Google Scholar] [CrossRef]
- Teschke, R. Effect of chronic alcohol pretreatment on the hepatotoxicity elicited by chlorpromazine, paracetamol, and dimethylnitrosamine. Biol. Approach Alcohol. Natl. Inst. Alcohol Abuse Alcohol. Res. Monogr. 1983, 11, 170–189. [Google Scholar]
- Nomura, F.; Pikkarainen, P.; Jauhonen, M.; Arai, E.R.; Gordon, E.; Baraona, E.; Lieber, C.S. Effect of ethanol administration on the metabolism of ethanol in baboons. J. Pharmacol. Exp. Ther. 1983, 227, 78–83. [Google Scholar] [PubMed]
- Shigeta, Y.; Nomura, F.; Leo, M.A.; Iida, S.; Felder, M.R.; Lieber, C.S. Alcohol dehydrogenase (ADH) independent ethanol metabolism in deermice lacking ADH. Pharmacol. Biochem. Behav. 1983, 18 (Suppl. 1), 195–199. [Google Scholar] [CrossRef]
- Koop, D.R.; Nordblom, G.D.; Coon, M.J. Immunochemical evidence for a role of cytochrome P-450 in liver microsomal ethanol oxidation. Arch. Biochem. Biophys. 1984, 235, 228–238. [Google Scholar] [CrossRef]
- Gadeholt, G. Ethanol and isoniazid induce a hepatic microsomal cytochrome P-450-dependent activity with similar properties towards substrate and inhibitors and different properties from those induced by classical inducers. Biochem. Pharmacol. 1984, 33, 3047–3051. [Google Scholar] [CrossRef]
- Sturtevant, R.P.; Garber, S.L. Circadian rhythms of alcohol dehydrogenase and MEOS in the rat. Proc. Soc. Exp. Biol. Med. 1984, 175, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Jörnvall, H. Induction of the ethanol-inducible form of rabbit liver microsomal cytochrome P-450 by inhibitors of alcohol dehydrogenase. Biochem. Biophys. Res. Commun. 1984, 124, 375–382. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Johansson, I. Mechanisms of hydroxyl radical formation and ethanol oxidation by ethanol-inducible and other forms of rabbit liver microsomal cytochrome P-450. J. Biol. Chem. 1984, 259, 6447–6458. [Google Scholar] [PubMed]
- Leo, M.A.; Lowe, N.; Lieber, C.S. Decreased hepatic vitamin A after drug administration in humans and rats. Am. J. Clin. Nutr. 1984, 40, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Koop, D.R.; Crump, B.L.; Nordblom, G.D.; Coon, M.J. Immunochemical evidence for induction of the alcohol-oxidizing cytochrome P-450 of rabbit liver microsomes by diverse agents: Ethanol, trichloroethylene, acetone, pyrazole, and isoniazid. Proc. Natl. Acad. Sci. USA 1985, 82, 4065–4069. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Koop, D.R.; Wang, T.; Coon, M.J. Immunochemical studies on the metabolism of nitrosamines by ethanol-inducible cytochrome P-450. Biochem. Biophys. Res. Commun. 1985, 128, 1007–1013. [Google Scholar] [CrossRef]
- Johansson, I.; Ingelman-Sundberg, M. Carbon tetrachloride induced lipid peroxidation dependent on an ethanol-inducible form of cytochrome P-450. FEBS Lett. 1985, 183, 265–269. [Google Scholar] [CrossRef]
- Koop, D.R.; Casazza, J.P. Identification of ethanol-inducible P-450 enzyme 3a as the acetone and acetol monooxygenase of rabbit microsomes. J. Biol. Chem. 1985, 260, 13607–13612. [Google Scholar] [PubMed]
- Ryan, D.E.; Ramanthan, L.; Iida, S.; Thomas, P.E.; Haniu, M.; Shively, J.E.; Lieber, C.S.; Levin, W. Characterization of a major form of rat hepatic microsomal cytochrome P-450 induced by isoniazid. J. Biol. Chem. 1985, 260, 6385–6393. [Google Scholar] [PubMed]
- Gellert, J.; Lieber, C.S. Effects of acute ethanol administration and chronic ethanol feeding on mixed function oxidation in Deermice lacking ADH. Alcohol 1985, 2, 13–15. [Google Scholar] [CrossRef]
- Gellert, J.; Alderman, J.; Lieber, C.S. Interaction between ethanol metabolism and mixed-function oxidation in Alcohol Dehydrogenase positive and negative Deermice. Biochem. Pharmacol. 1986, 35, 1037–1041. [Google Scholar] [CrossRef]
- Teschke, R.; Wannagat, F.J.; Löwendorf, F.; Strohmeyer, G. Hepatic alcohol metabolizing enzymes after prolonged administration of sex hormones and alcohol in female rats. Biochem. Pharmacol. 1986, 35, 521–527. [Google Scholar] [CrossRef]
- Teschke, R. Hepatic microsomal ethanol-oxidizing system: Biochemical nature and clinical implications. In Genetics and Alcoholism; Goedde, H.W., Agarwal, D.P., Eds.; Alan R. Liss Inc.: New York, NY, USA, 1986; pp. 173–184. [Google Scholar]
- Leo, M.A.; Lowe, N.; Lieber, C.S. Interaction of drugs and retinol. Biochem. Pharmacol. 1986, 35, 3949–3953. [Google Scholar] [CrossRef]
- Ryan, D.E.; Koop, D.R.; Thomas, P.E.; Coon, M.J.; Levin, W. Evidence that isoniazid and ethanol induce the same microsomal P-450 isozyme 3a. Arch. Biochem. Biophys. 1986, 246, 633–644. [Google Scholar] [CrossRef]
- Wrighton, S.A.; Thomas, P.E.; Molowa, D.T.; Haniu, M.; Shively, J.E.; Maines, S.L.; Watkins, P.B.; Parker, G.; Mendez-Picon Levin, W.; Guzelman, P.S. Characterization of ethanol-inducible human liver N-nitrosodimethylamine demethylase. Biochemistry 1986, 25, 6731–6735. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Koop, D.R.; Crump, B.I.; Coon, M.J. Immunochemical identification of cytochrome P-450 isozyme 3a [P-450ALC] in rabbit nasal and kidney microsomes and evidence for differential induction by alcohol. Mol. Pharmacol. 1986, 30, 370–378. [Google Scholar] [PubMed]
- Johansson, I.J.; Eliasson, E.; Norsten, C.; Ingelman-Sundberg, M. Hydroxylation of acetone by ethanol- and acetone-inducible cytochrome P-450 in liver microsomes and reconstituted membranes. FEBS Lett. 1986, 196, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Song, B.J.; Gelboin, H.V.; Park, S.S.; Yang, C.S.; Gonzalez, F.J. Complementary DNA and protein sequences of ethanol-inducible rat and human cytochrome P-450s: Transcriptional and posttranscriptional regulation of the rat enzyme. J. Biol. Chem. 1986, 261, 16689–16697. [Google Scholar] [PubMed]
- Bellward, G.D.; Chang, T.; Rodrigues, J.H.; McNeil, J.H.; Maines, S.; Ryan, D.E.; Levin, W.; Thomas, B.E. Hepatic cytochrome P-450j induction in the spontaneously diabetic BB rat. Mol. Pharmacol. 1987, 33, 140–143. [Google Scholar]
- Lasker, J.M.; Raucy, J.; Kubota, S.; Bloswick, B.P.; Black, M.; Lieber, C.S. Purification and characterization of human liver cytochrome P-450-ALC. Biochem. Biophys. Res. Commun. 1987, 148, 232–238. [Google Scholar] [CrossRef]
- Khani, S.C.; Zaphiropoulos, P.G.; Fujita, V.S.; Porter, T.D.; Koop, D.R.; Coon, M.J. cDNA and derived amino acid sequence of ethanol inducible rabbit liver cytochrome P-450 isozyme 3a (P-450ALC). Proc. Natl. Acad. Sci. USA 1987, 84, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Lasker, J.M.; Alderman, J.; Leo, M.A. The microsomal ethanol oxidizing system and its interaction with other drugs, carcinogens, and vitamins. Ann. N. Y. Acad. Sci. 1987, 492, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Alderman, J.; Takagi, T.; Lieber, C.S. Ethanol-metabolizing pathways in Deermice: Estimation of flux calculated from isotope effects. J. Biol. Chem. 1987, 262, 7497–7503. [Google Scholar] [PubMed]
- Ingelman-Sundberg, M.; Johansson, I.; Penttila, K.E.; Glaumann, H.; Lindros, K.O. Centrilobular expression of ethanol-inducible cytochrome P-450 (IIE1) in rat liver. Biochem. Biophys. Res. Commun. 1988, 157, 55–60. [Google Scholar] [CrossRef]
- Salazar, D.E.; Sorge, C.L.; Cocoran, G.B. Obesity as a risk factor for drug-induced organ injury. VI. Increased hepatic P450 concentration and microsomal ethanol oxidizing activity in the obese overfed rat. Biochem. Biophys. Res. Commun. 1988, 157, 315–320. [Google Scholar] [CrossRef]
- Behrens, U.H.; Hoerner, M.; Lasker, J.M.; Lieber, C.S. Formation of acetaldehyde adducts with ethanol-inducible P-450IIE1 in vivo. Biochem. Biophys. Res. Commun. 1988, 154, 584–590. [Google Scholar] [CrossRef]
- Ding, X.; Coon, M.J. Induction of cytochrome P-450 isozyme 3a (P-450IIE1) in rabbit olfactory mucosa by ethanol and acetone. Drug Metab. Dispos. 1988, 18, 742–745. [Google Scholar]
- Kubota, S.; Lasker, J.M.; Lieber, C.S. Molecular regulation of ethanol inducible cytochrome P-450IIE1 in hamsters. Biochem. Biophys. Res. Commun. 1988, 150, 304–310. [Google Scholar] [CrossRef]
- Johansson, I.J.; Ingelman-Sundberg, M. Benzene metabolism by ethanol-, acetone-, and benzene-inducuble cytochrome P-450 (IIE1) in rat and rabbit liver microsomes. Cancer Res. 1988, 48, 5387–5390. [Google Scholar] [PubMed]
- Eliasson, E.; Johansson, I.; Ingelman-Sundberg, M. Ligand-dependent maintenance of ethanol-inducible cytochrome P-450 in primary rat hepatocyte cell cultures. Biochem. Biophys. Res. Commun. 1988, 150, 436–443. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Shimizu, M.; Lasker, J.M.; Lieber, C.S. Intralobular distribution of ethanol-inducible cytochrome P-450IIE1 in liver. Hepatology 1988, 8, 1237A. [Google Scholar]
- Dicker, E.; Cederbaum, A.I. Increased oxygen radical dependent inactivation of metabolic enzymes by liver microsomes after chronic ethanol consumption. FASEB J. 1988, 2, 2901–2906. [Google Scholar] [CrossRef] [PubMed]
- Porter, T.D.; Khani, S.C.; Coon, M.J. Induction and tissue-specific expression of rabbit cytochrome P-450IIE1 and II2 genes. Mol. Pharmacol. 1989, 36, 61–65. [Google Scholar] [PubMed]
- Tsutsumi, M.; Lasker, J.M.; Shimizu, M.; Rosman, A.S.; Lieber, C.S. The intralobular distribution of ethanol-inducible P-450IIE1 in rat and human liver. Hepatology 1989, 10, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Tinberg, N.; Ingelman-Sundberg, M.; Köhler, C. Regional distribution of ethanol-inducible cytochrome P-450 2E1 in the rat central nervous system. Neuroscience 1990, 34, 451–463. [Google Scholar] [CrossRef]
- Hong, J.Y.; Ning, S.M.; Ma, B.L.; Lee, M.J.; Pan, J.M.; Yang, C.S. Roles of pituitary hormones in the regulation of hepatic cytochrome P-450IIE1in rats and mice. Arch. Biochem. Biophys. 1990, 281, 132–138. [Google Scholar] [CrossRef]
- Koop, D.R.; Tierney, D.J. Multiple mechanisms in the regulation of ethanol-inducible cytochrome P-450IIE1. Bioassays 1990, 9, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Veech, R.L.; Saenger, P. Cytochrome P-450IIE1 is elevated in lymphocytes from poorly controlled insulin-dependent diabetics. J. Clin. Endorinol. Metab. 1990, 71, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Novak, R.F. Induction of rat hepatic cytochrome P-450 IIE1 (CYP 2E1) by pyridine: Evidence for a role of protein synthesis in the absence of transcriptional activation. Biochem. Biophys. Res. Commun. 1990, 166, 1072–1079. [Google Scholar] [CrossRef]
- Kim, S.G.; Shehin, S.E.; States, J.C.; Novak, R.F. Evidence for increased translational efficiency in the induction of P-450IIE1 by solvents: Analysis of P-450IIE1 mRNA polyribosomal distribution. Biochem. Biophys. Res. Commun. 1990, 172, 767–774. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Leo, A.M.; Kim, C.; Tsutsumi, M.; Lasker, J.M.; Lowe, N.; Lieber, C.S. Interaction of ethanol with enflurane metabolism and toxicity: Role of P-450IIE1. Alcohol. Clin. Exp. Res. 1990, 14, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Yoo, J.S.; Ishizaki, H.; Hong, J. Cytochrome P-450IIE1: Roles of nitrosamine metabolism and mechanisms of regulation. Drug Metab. Rev. 1990, 22, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Peter, R.; Bocker, R.; Beaune, P.H.; Iwasaki, M.; Guengerich, F.P.; Yang, C.S. Hydroxylation of chlorzoxazone as a specific probe for human liver cytochrome P-450IIE1. Chem. Res. Toxicol. 1990, 3, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Lasker, J.M.; Tsutsumi, M.; Lieber, C.S. Immunohistochemical localization of ethanol-inducible P-450IIE1 in the rat alimentary tract. Gastroenterology 1990, 99, 1044–1053. [Google Scholar] [CrossRef]
- Sohn, D.H.; Yun, Y.P.; Park, S.S.; Veech, R.L.; Song, B.J. Post-translational reduction of cytochrome P450 2E1 by CCl4, its substrate. Biochem. Biophys. Res. Commun. 1991, 179, 449–454. [Google Scholar] [CrossRef]
- Johansson, E.; Eliasson, E.; Ingelman-Sundberg, M. Hormone controlled phosphorylation and degradation of CYP2B1 and 2E1 in isolated rat hepatocytes. Biochem. Biophys. Res. Commun. 1991, 174, 37–42. [Google Scholar] [CrossRef]
- Yoo, J.; Ning, S.M.; Pantuck, E.J.; Yang, C.S. Regulation of hepatic microsomal cytochrome P-450 2E1 level by dietary lipids and carbohydrates in rats. J. Nutr. 1991, 121, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Raucy, J.L.; Lasker, J.M.; Kramer, J.C.; Salazer, D.E.; Lieber, C.S.; Corcoran, G.B. Induction of P-450IIE1 in the obese rat. Mol. Pharmacol. 1991, 39, 275–280. [Google Scholar] [PubMed]
- Terelius, Y.; Norsten-Hoog, C.; Cronholm, T.; Ingelman-Sundberg, M. Acetaldehyde as a substrate for ethanol-inducible cytochrome P-450 (CYP2E1). Biochem. Biophys. Res. Commun. 1991, 179, 689–694. [Google Scholar] [CrossRef]
- Koop, D.R.; Chernosky, A.; Brass, E.P. Identification and induction of cytochrome P-450 2E1 in rat Kupffer cells. J. Pharmacol. Exp. Ther. 1991, 251, 1072–1076. [Google Scholar]
- Hayashi, S.; Watanabe, J.; Kaname, K. Genetic polymorphism in the 5’-flanking region change transcriptional regulation of the human cytochrome P-450IIE1 gene. J. Biochem. 1991, 110, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.A.; Kim, C.I.; Lowe, N.; Lieber, C.S. Interaction of ethanol with β-carotene: Delayed blood clearance and enhanced hepatotoxicity. Hepatology 1992, 15, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, E.; Mkrtchian, S.; Ingelman-Sundberg, M. Hormone- and substrate-regulated intracellular degradation of cytochrome P450 (2E1) involving MgATP-activated rapid proteolysis in the endoplasmic reticulum membranes. J. Biol. Chem. 1992, 267, 15765–15769. [Google Scholar] [PubMed]
- Sohda, T.; Shimizu, M.; Okumura, M. Distribution in pancreas of ethanol-inducible P-4502E1 in rats fed ethanol plus high fat or low fat diet. Alcohol Metab. Liver 1992, 1, 110–113. [Google Scholar]
- Koop, D.R. Oxidative and reductive metabolism by cytochrome P-450 2E1. FASEB J. 1992, 6, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Inui, Y.; Yun, C.H.; Guengrich, F.P.; Shimada, T. Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis 1992, 13, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Kharasch, E.D.; Thummel, K.E.; Mhyre, J.; Lillibridge, J.H. Single-dose disulfiram inhibition of chlorzoxazone metabolism: A clinical probe for P450 2E1. Clin. Pharmacol. Ther. 1993, 53, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Thummel, K.E.; Kharasch, E.D.; Podoll, T.; Kunze, K. Human liver microsomal enflurane defluorination catalyzed by cytochrome P-450 2E1. Drug Metab. Dispos. 1993, 21, 350–357. [Google Scholar] [PubMed]
- Day, Y.; Rashba-Step, J.; Cederbaum, A.I. Stable expression of human cytochrome P-4502E1 in HepG2 cells. Characterization of catalytic activities and production of reactive intermediates. Biochemistry 1993, 32, 6928–6937. [Google Scholar] [CrossRef]
- Hirvonen, A.; Husgafvel-Pursiainen, K.; Anttilla, S.; Karjalainen, A.; Vainio, H. The human CYP2E1 gene and lung cancer: Dra I and Rsa I restriction fragment length polymorphisms in a Finish study population. Carcinogensis 1993, 1, 85–88. [Google Scholar] [CrossRef]
- Morimoto, M.; Hagbjork, A.L.; Nanji, A.A.; Ingelman-Sundberg, M.; Lindros, K.O.; Fu, P.C.; Albano, E.; French, S.W. Role of cytochrome P-450 2E1 in alcoholic liver disease pathogenesis. Alcohol 1993, 10, 459–464. [Google Scholar] [CrossRef]
- Rashba-Step, J.; Turro, N.J.; Cederbaum, A.I. Increased NADPH- and NADH-dependent production of superoxide and hydroxyl radical by microsomes after chronic ethanol consumption. Arch. Biochem. Biophys. 1993, 300, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, M.; Lasker, J.M.; Takahashi, T.; Lieber, C.S. The in vivo induction of hepatic P-4502E1: Role of increased enzyme synthesis. Arch. Biochem. Biophys. 1993, 304, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Laethem, R.M.; Balaxy, M.; Falck, J.R.; Laethem, C.L.; Koop, D.R. Formation of 19(S)-, 19(R)-, and 18(R)-hydroxyeicosatetraenoic acids by alcohol-inducible cytochrome P-4502E1. J. Biol. Chem. 1993, 268, 12912–12918. [Google Scholar] [PubMed]
- Badger, T.M.; Huang, J.; Ronis, M.; Lumpkin, C.K. Induction of cytochrome P-450 2E1 during chronic ethanol exposure occurs via transcription of the CYP 2E1 gene when blood alcohol concentrations are high. Biochem. Biophys. Res. Commun. 1993, 190, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Kunitoh, S.; Tanaka, T.; Imaoka, S.; Funae, Y.; Monna, Y. Contribution of cytochrome P-450s to MEOS (microsomal ethanol-oxidizing system): A specific and sensitive assay of MEOS activity by HPLC with fluorescence labeling. Alcohol Alcohol. 1993, 28, 63–68. [Google Scholar] [CrossRef]
- Lucas, D.; Berthou, F.; Dreano, Y.; Lozach, P.; Volant, A.; Menez, J.F. Comparison of levels of cytochrome P-450, CYP1A2, CYP2E1, and their related monooxygenase activities in human surgical samples. Alcohol. Clin. Exp. Res. 1993, 17, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.; Huang, J.; Crouch, J.; Mercado, C.; Irby, D.; Valentine, C.R.; Lumpkin, C.K.; Ingelman-Sundberg, M.; Badger, T.M. Cytochrome P-450 CYP2E1 induction during chronic ethanol exposure occurs by a two-step mechanism associated with blood alcohol concentration in rats. J. Pharmacol. Exp. Ther. 1993, 264, 944–950. [Google Scholar] [PubMed]
- Kharasch, E.D.; Thummel, K.E. Identification of cytochrome P-450 2E1 as the predominant enzyme catalyzing human liver microsomal defluorination of sevoflurane, isoflurane, and methoxyflurane. Anesthesiology 1993, 79, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Lasker, J.M.; Rosman, A.S.; Lieber, C.S. Inhibition of cytochrome P-4502E1 in the human liver by ethanol is caused by corresponding increase in encoding messenger RNA. Hepatology 1993, 17, 236–245. [Google Scholar] [PubMed]
- Tassaneeyakul, W.; Veronese, M.E.; Birkett, D.J.; Gonzalez, F.J.; Miners, O. Validation of 4-nitrophenol as an in vitro substrate probe for human liver CYP2E1 using CDNA expression and microsomal kinetic techniques. Biochem. Pharmacol. 1993, 46, 1975–1981. [Google Scholar] [CrossRef]
- Albano, E.; Tomasi, A.; Ingelman-Sundberg, M. Spin trapping of alcohol derived radicals in microsomes and reconstituted systems by electron spin resonance. Methods Enzymol. 1994, 223, 117–127. [Google Scholar]
- Carrocio, A.; Wu, D.; Cederbaum, A.I. Ethanol increases content and activity of human cytochrome P-4502E1 in a transduced HEPG2 cell line. Biochem. Biophys. Res. Commun. 1994, 203, 727–733. [Google Scholar] [CrossRef]
- Botto, F.; Seree, E.; Khyari, S.E.; DeSoussa, G.; Massacrier, A.; Placidi, M.; Cau, P.; Pellet, W.; Rahmani, R.; Barra, Y. Tissue-specific expression and methylation of the human CYP2E1 gene. Biochem. Pharmacol. 1994, 48, 1095–1103. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Takata, A.; Wang, J.S. Genetic polymorphism of cytochrome P-4502E1 related to the development of alcoholic liver disease. Gastroenterology 1994, 107, 1430–1435. [Google Scholar] [CrossRef]
- Stephens, E.A.; Taylor, J.A.; Kaplan, N.; Yang, C.H.; Hsieh, L.L.; Lucier, G.W.; Bell, D.A. Ethnic variation in the CYP2E1 gene: Polymorphism analysis of 695 African-Americans, European-Americans and Taiwanese. Pharmacogenetics 1994, 4, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lee, M.; Hong, J.Y.; Huang, W.; Wang, E.; Yang, C.S. Relationship between cytochrome P-450 2E1 and acetone catabolism in rats as studied with diallyl sulfide as an inhibitor. Biochem. Pharmacol. 1994, 48, 2199–2205. [Google Scholar] [CrossRef]
- Maezawa, Y.; Yamauchi, M.; Toda, G. Association between restriction fragment length polymorphism of the human cytochrome P-450IIE1 gene and susceptibility to alcoholic liver cirrhosis. Am. J. Gastroenterol. 1994, 89, 561–565. [Google Scholar] [PubMed]
- Amet, Y.; Berthou, F.; Goasduff, T.; Schaun, J.P.; Le Breton, L.; Menez, J.F. Evidence that cytochrome P-450 2E1 is involved in the (omega-1)-hydroxylation of lauric acid in rat liver microsomes. Biochem. Biophys. Res. Commun. 1994, 203, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- McGehee, R.E.; Ronis, M.J.J.; Cowherd, R.M.; Ingelman-Sunderberg, M.; Badger, T.M. Characterization of cytochrome P-450 2E1 induction in a rat hepatoma FGC-4 cell model by ethanol. Biochem. Pharmacol. 1994, 48, 1823–1833. [Google Scholar] [CrossRef]
- Kang, M.H.; Won, S.M.; Park, S.S.; Kim, S.G.; Novak, R.F.; Kim, N.D. Piperine effects on the expression of P4502E1, P4502B and P4501A in rat. Xenobiotica 1994, 24, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, F.; Ikawa, S.; Kikuchi, H.; Sagami, I.; Kanamara, R.; Abe, T.; Satoh, K.; Motomiya, M.; Watanabe, M. Restriction fragment length polymorphism of the human CYP2E1 (cytochrome P-450IIE1) gene and susceptibility to lung cancer: Possible relevance to low smoking exposure. Pharmacogenetics 1994, 4, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Hayashi, S.; Kawajiri, K. Different regulations and expression of the human CYP2E1 gene due to the Rsal polymorphism in the 5′ flanking region. J. Biochem. 1994, 116, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Yang, J.P.; Eguchi, H.; Hayashi, S.; Imai, K.; Nakachi, K.; Kawajiri, K. An Rsa I polymorphism in the CYP2E1 gene does not affect lung cancer risk in a Japanese population. Jpn. Cancer Res. 1995, 86, 245–248. [Google Scholar] [CrossRef]
- Roberts, B.J.; Song, B.J.; Soh, Y.; Park, S.S.; Shoaf, S.E. Ethanol induces CYP2E1 by protein stabilization. J. Biol. Chem. 1995, 270, 29632–29635. [Google Scholar] [PubMed]
- Speerschneider, P.; Dekant, W. Renal tumorigenicity of 1,1-dichloroethene in mice: The role of male-specific expression of cytochrome P-450 2E1 in the renal bioactivation of 1,1-dichloroethene. Toxicol. Appl. Pharmacol. 1995, 130, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, U.; Ahmed, A.E. Intestinal toxicity of acrylonitrile: In vitro metabolism by intestinal P-450 2E1. Toxicol. Appl. Pharmacol. 1995, 135, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schmalix, W.A.; Barrenscheen, N.; Landsiedel, R.; Janzowski, C.; Eisenbrand, G.; Gonzalez, F.; Eliasson, B.; Ingelman-Sundberg, M.; Perchermeier, M.; Greim, H. Stable expression of human cytochrome P-450 2E1 in V79 Chinese hamster cells. Eur. J. Pharmacol. 1995, 293, 123–131. [Google Scholar] [PubMed]
- Carr, L.G.; Hartleroad, J.Y.; Liang, Y.B.; Mendenhall, C.; Moritz, T.; Thomasson, H. Polymorphism at the P-450IIE1 locus is not associated with alcoholic liver disease in Caucasian men. Alcohol. Clin. Exp. Res. 1995, 19, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, M.; Hagbjork, A.L.; Wan, Y.J.Y.; Fu, P.C.; Clot, P.; Albano, E.; Ingelman-Sundberg, M.; French, S.W. Modulation of experimental alcohol-induced liver disease by cytochrome P-450 2E1 inhibitors. Hepatology 1995, 21, 1610–1617. [Google Scholar] [PubMed]
- Rannug, A.; Alexandrie, A.K.; Persson, I.; Ingelman-Sundberg, M. Genetic polymorphism of cytochrome P-450 1A1, 2D6 and 2E1: Regulation and toxicological significance. J. Occup. Environ. Med. 1995, 37, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; Kitteringham, N.R.; Quest, L.I.; Allott, R.L.; Green, V.J.; Gilmore, I.T.; Park, B.K. Genetic polymorphism of cytochrome P-4502E1 and risk of alcoholic liver disease in Caucasians. Pharmacogenetics 1995, 5, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.; Menez, C.; Girre, C.; Berthou, F.; Bodenez, P.; Joannet, I.; Hispard, E.; Bardon, L.G.; Menez, J.F. Cytochrome P-450 2E1 genotype and chlorzoxazone metabolism in healthy and alcoholic Caucasian subjects. Pharmacogenetics 1995, 5, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.; Menez, C.; Girre, C.; Bodenez, P.; Hispard, E.; Menez, J.F. Decrease in cytochrome P-4502E1 as assessed by the rate of chlorzoxazone hydroxylation in alcoholics during the withdrawal phase. Alcohol. Clin. Exp. Res. 1995, 19, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Mishin, V.; Koivisto, T.; Lieber, C.S. Respective roles of CYP2E1 and CYPA2 in chlorzoxazone and ethanol metabolism in mammalian liver microsomes. Alcohol. Clin. Exp. Res. 1995, 19, 78A. [Google Scholar]
- Jayyosi, Z.; Knoble, D.; Muc, M.; Erick, J.; Thomas, P.E.; Kelley, N. Cytochrome P-450 2E1 is not the sole catalyst of chlorzoxazone hydroxylation in rat liver microsomes. J. Pharmacol. Exp. Ther. 1995, 273, 1156–1161. [Google Scholar] [PubMed]
- Yamazaki, H.; Guo, Z.; Guengerich, F.P. Selectivity of cytochrome P4502E1 in chlorzoxazone 6-hydroxylation. Drug Metab. Dispos. 1995, 23, 438–440. [Google Scholar] [PubMed]
- De Waziers, I.; Garlatti, M.; Bouguet, J.; Beaune, P.H.; Barouki, R. Insulin down-regulates cytochrome P-450 2B and 2E expression at the posttranscriptional level in the rat hepatoma cell line. Mol. Pharmacol. 1995, 47, 474–479. [Google Scholar] [PubMed]
- Sampol, E.; Mirrione, A.; Villard, P.H.; Picerelle, P.; Scoma, H.; Berbis, P.; Barra, Y.; Durand, A.; Lacarelle, B. Evidence for a tissue-specific induction of cutaneous CYP2E1 by dexamethasone. Biochem. Biophys. Res. Commun. 1995, 235, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Crabb, D.W. Ethanol oxidizing enzyme: Roles in alcohol metabolism and alcoholic liver disease. Prog. Liver Dis. 1995, 13, 151–172. [Google Scholar] [PubMed]
- Zerilli, A.; Lucas, D.; Amet, Y.; Beauge, F.; Volant, A.; Floch, H.H.; Berthou, F.; Menez, J.F. Cytochrome P-450 2E1 in rat liver, kidney and lung microsomes after chronic administration of ethanol either orally or by inhalation. Alcohol Alcohol. 1995, 30, 357–365. [Google Scholar] [PubMed]
- Asai, H.; Imaoka, S.; Kuroki, T.; Monna, T.; Funnae, Y. Microsomal ethanol oxidizing system activity by 2 DNA single strand breaks. Biochem. Biophys. Res. Commun. 1996, 219, 429–434. [Google Scholar]
- Seree, E.M.; Villard, P.H.; Re, J.L.; De Meo, M.; Lacarelle, B.; Attolini, L.; Dumenil, G.; Catalin, J.; Durand, A.; Barra, Y. High inducibility of mouse renal CYP2E1 gene by tobacco smoke and its possible effect on DNA single strand breaks. Biochem. Biophys. Res. Commun. 1996, 219, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Hakkak, R.; Korourian, S.; Ronis, M.J.; Ingelman-Sundberg, M.; Badger, T.M. Effects of diet and ethanol on the expression and localization of cytochromes P-450 2E1 and P-450 2C7 in the colon of male rats. Biochem. Pharmacol. 1996, 51, 61–69. [Google Scholar] [CrossRef]
- Koivisto, T.; Mishin, V.M.; Mak, K.M.; Cohen, P.A.; Lieber, C.S. Induction of cytochrome P-4502E1 by ethanol in rat Kuppfer cells. Alcohol. Clin. Res. Exp. Res. 1996, 20, 207–212. [Google Scholar] [CrossRef]
- Carriere, V.; Berthou, F.; Baird, S.; Belloc, C.; Beaune, P.; De Waziers, I. Human cytochrome P-450 2E1 (CYP2E1): From genotype to phenotype. Pharmacogenetics 1996, 6, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Tindberg, N.; Ingelman-Sundberg, M. Expression, catalytic activity, and inducibility of cytochrome P-450 2E1 (CYP2E1) in the rat central nervous system. J. Neurochem. 1996, 67, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Von Moltke, L.L.; Shader, R.I.; Greenblatt, D.J. Biotransformation of chlorzoxazone by hepatic microsomes from humans and ten other mammalian species. Biopharm. Drug Dispos. 1997, 18, 213–226. [Google Scholar] [CrossRef]
- McGehee, R.E.; Ronis, M.J.J.; Badger, T.M. Regulation of the hepatic CYP2E1 gene during chronic alcohol exposure: Lack of an ethanol response element in the proximal 5′-flanking sequence. DNA Cell Biol. 1997, 16, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.A.; Mak, K.M.; Rosman, A.S.; Kessova, I.; Mishin, V.M.; Kovisto, T.; Lieber, C.S. Immunohistochemical determination of hepatic cytochrome P-4502E1 in formalin-fixed, paraffin-embedded sections. Alcohol. Clin. Exp. Res. 1997, 21, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Zangar, R.C.; Novak, R.F. Effect of fatty acids and ketone bodies on cytochromes P-450 2B, 4A, and 2E1 expression in primary cultured rat hepatocytes. Arch. Biochem. Biophys. 1997, 337, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Zimatkin, S.M.; Deitrich, R.A. Ethanol metabolism in the brain. Addict. Biol. 1997, 2, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.D.; Kwat, M.K.; Kim, S.G. Inhibition of cytochrome P-450 2E1 expression by 2-(allylthio) pyrazine, a potential chemoprotective agent: Hepatoprotective effects. Biochem. Pharmacol. 1997, 53, 261–269. [Google Scholar] [CrossRef]
- Woodcroft, K.J.; Novak, R.F. Insulin effects on CYP2E1, 2B, 3A, and 4A expression in primary cultured rat hepatocytes. Chem. Biol. Interact. 1997, 107, 75–91. [Google Scholar] [CrossRef]
- French, S.; Morimoto, M.; Reitz, R.; Koop, D.; Klopfenstein, B.; Estes, K.; Clot, B.; Ingelman-Sundberg, M.; Albano, E. Lipidperoxidation, CYP2E1 and arachidonoid acid metabolism in alcoholic liver disease in rats. J. Nutr. 1997, 127 (Suppl. 5), 907S–911S. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, A.C.; Lucas, D.; Menez, J.F.; Seitz, H.K. Chlormethiazole inhibition of cytochrome P450 2E1 as assessed by chlorzoxazone hydroxylation in humans. Hepatology 1997, 26, 957–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, D.; Farez, C.; Bardou, L.G.; Vaisse, J.; Attali, J.R.; Valensi, P. Cytochrome P-450 2E1 activity in diabetic and obese patients as assessed by chlorzoxazone hydroxylation. Fundam. Clin. Pharmacol. 1998, 12, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Powell, H.; Kitteringham, N.R.; Pirmohamed, M.; Smith, D.A.; Park, B.K. Expression of cytochrome P-4502E1 in human liver: Assessment by MRNA, genotype and phenotype. Pharmacogenetics 1998, 8, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Weltman, M.D.; Farrell, G.C.; Hall, P.; Ingelman-Sundberg, M.; Liddle, C. Hepatic cytochrome P4502E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 1998, 27, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Salmela, K.S.; Kessova, I.G.; Tsyrlov, I.B.; Lieber, C.S. Respective roles of human cytochrome P-4502E1 and 3A4 in the hepatic microsomal ethanol oxidizing system. Alcohol. Clin. Exp. Res. 1998, 22, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Salmela, K.; Lieber, C.S. Microsomal acetaldehyde oxidation is negible in the presence of ethanol. Alcohol. Clin. Exp. Res. 1998, 22, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.M.; Etheridge, A.S.; Raymer, J.H.; Black, S.R.; Pulliam, D.W.; Bucher, J.M. Selective inhibition of cytochrome P-450 2E1 in vivo and in vitro with trans-1, 2-dichloroethylene. Chem. Res. Toxicol. 1998, 11, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Kuo, C.L.; Pernecky, S.J.; Piper, W.N. The detection of cytochrome P-450 2E1 and its catalytic activity in rat testis. Biochem. Biophys. Res. Commun. 1998, 246, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Kessova, I.G.; DeCarli, L.M.; Lieber, C.S. Inducibility of cytochrome P-450 2E1 & P-450 1A1 in rat pancreas. Alcohol. Clin. Exp. Res. 1998, 22, 501–504. [Google Scholar] [PubMed]
- Norton, I.D.; Apte, M.V.; Haber, P.S.; McCaughan, G.W.; Pirola, R.C.; Wilson, J.S. Cytochrome P-450 2E1 is present in rat pancreas is induced by chronic ethanol administration. Gut 1998, 42, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Aleynik, M.K.; Leo, M.A.; Aleynik, S.I.; Lieber, C.S. Polyenylphosphatidylcholine opposes the increase of cytochrome P-4502E1 by ethanol and corrects the iron-induced decrease. Alcohol. Clin. Exp. Res. 1998, 23, 96–100. [Google Scholar] [CrossRef]
- Adas, F.; Betthou, F.; Picart, D.; Lozach, P.; Beauge, F.; Amet, Y. Involvement of cytochrome P-450 2E1 in the (omega-1)-hydroxylation of oleic acid in human and rat liver microsomes. J. Lipid Res. 1998, 39, 1210–1219. [Google Scholar] [PubMed]
- Mishin, V.M.; Rosma, A.S.; Basu, P.; Kessova, I.; Oneta, C.M.; Lieber, C.S. Chlorzoxazone pharmacogenetics as a marker of hepatic cytochrome P-4502E1 in humans. Am. J. Gastroenterol. 1998, 93, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Eap, C.B.; Schnyder, C.; Besson, J.; Savary, L.; Buclin, T. Inhibition of CYP2E1 by chlormethiazole as measured by chlorzoxazone pharmacokinetics in patients with alcoholism and in healthy volunteers. Clin. Pharmacol. Ther. 1998, 64, 52–57. [Google Scholar] [CrossRef]
- Peng, H.M.; Coon, M.J. Regulation of rabbit cytochrome P-450 2E1 expression in HepG2 cells by insulin and thyroid hormones. Mol. Pharmacol. 1998, 54, 740–747. [Google Scholar] [PubMed]
- Dupont, I.; Lucas, D.; Clot, P.; Menez, C.; Albano, E. Cytochrome P-4502E1 inducibility and hydroxyethyl radical formation among alcoholics. J. Hepatol. 1998, 28, 564–571. [Google Scholar] [CrossRef]
- Leo, M.A.; Lieber, C.S. Alcohol, vitamin, A.; and beta-carotene: Adverse interactions, including hepatotoxicity and carcinogenicity. Am. J. Clin. Nutr. 1999, 69, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Hutson, J.L.; Wickramasinghe, S.N. Expression of CYP2E1 by human monocyte-derived macrophages. J. Pathol. 1999, 188, 197–200. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Jacob, P.; Saunders, S.; Gourlay, S. Carbon monoxide, cigarette smoking and CYP2E1 activity. Clin. Pharmacol. 1999, 65, 154. [Google Scholar] [CrossRef]
- Lucas, D.; Ferrara, R.; Gonzalez, E.; Bodenez, P.; Albores, A.; Manno, M.; Berthou, F. Chlorzoxazone, a selective probe for phenotyping CYP2E1 in humans. Pharmacogenetics 1999, 9, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Loizou, G.D.; Cocker, J. The effects of alcohol and diallylsulphide on CYP2E1 activity in humans: A phenotyping study using chlorzoxazone. Hum. Exp. Toxicol. 2001, 20, 321–327. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F. Inhibition of CYP2E1 with natural agents may be a feasible strategy for minimizing the hepatotoxicity of ethanol. Med. Hypotheses 2001, 56, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.Y.; Magnus Ingelman-Sundberg, M.; Neve, E.; Matsumoto, H.; Nishitani, Y.; Minowa, Y.; Fukui, Y.; Bailey, S.M.; Patel, V.B.; Cunningham, C.C.; et al. Ethanol and oxidative stress. Alcohol. Clin. Exp. Res. 2001, 25, 237S–243S. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.F.; Branch, R.A. Effect of chronic disulfiram administration on the activities of CYP1A2, CYP2C19, CYP2D6, CYP2E1, and N-acetyltransferase in healthy human subjects. Br. J. Clin. Pharmacol. 2002, 53, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosal, A.; Hapangama, N.; Yuan, Y.; Lu, X.; Horne, D.; Patrick, J.E.; Zbaida, S. Rapid determination of enzyme activities of recombinant human cytochromes P450, human liver microsomes and hepatocytes. Pharm. Drug Dispos. 2003, 24, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Kessova, I.; Cederbaum, A.I. CYP2E1: Biochemistry, toxicology, regulation and function in ethanol-induced liver injury. Curr. Mol. Med. 2003, 3, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Ernstgard, L.; Warholm, M.; Johanson, G. Robustness of chlorzoxazone as an in vivo measure of cytochrome P450 2E1 activity. Br. J. Clin. Pharmacol. 2004, 58, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Kim, K.A.; Park, P.W.; Ha, J.M. Effect of high-dose aspirin on CYP2E1 activity in healthy subjects measured using chlorzoxazone as a probe. J. Clin. Pharmacol. 2006, 46, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellman, M.; Siest, G. Cytochrome P-450-mediated differential oxidative modification of proteins: Albumin, apolipoprotein, E.; and CYP2E1 as targets. J. Toxicol. Environ. Health 2010, 67, 2061–2071. [Google Scholar] [CrossRef]
- Kim, S.K.; Park, H.J.; Seok, H.; Jeon, H.S.; Lee, T.W.; Lee, S.H.; Moon, S.Y.; Ihm, C.G.; Kim, T.H.; Kim, Y.H.; et al. Association studies of cytochrome P450, family 2, subfamily, E.; polypeptide 1 (CYP2E1) gene polymorphisms with acute rejection in kidney transplantation recipients. Clin. Transplant. 2014, 28, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedada, S.K.; Neerati, P. Resveratrol pretreatment affects CYP2E1 activity of chlorzoxazone in healthy human. Phytother. Res. 2016, 30, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bedada, S.K.; Boga, P.K. Effect of piperine on CYP2E1 enzyme activity of chlorzoxazone in healthy volunteers. Xenobiotica 2017, 47, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ma, H.Y.; Liang, S.; Sun, M.; Karin, G.; Koyama, Y.; Hu, R.; Quehenberger, O.; Davidson, N.O.; Dennis, E.A.; et al. The role of human cytochrome P450 2E1 in liver inflammation and fibrosis. Hepatol. Commun. 2017, 1, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmegeed, M.A.; Choi, Y.; Ha, S.K.; Song, B.J. Cytochrome P450-2E1 is involved in aging-related kidney damage in mice through increased nitroxidative stress. Food Chem. Toxicol. 2017, 109, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, R. Exposure to sub-parts per million levels of vinyl chloride can increase the risk of developing liver injury. Hepatol. Commun. 2018, 2, 227–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.L.; Chen, L.; Poff, G.D.; Ding, W.X.; Barnett, R.A.; Arteel, G.E.; Beier, J.I. Vinylchloride dysregulates metabolic homeostasis and enhances diet-induced liver injury in mice. Hepatol. Commun. 2018, 2, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Teschke, R.; Ohnishi, K.; Lieber, C.S. Acceleration of ethanol metabolism by high ethanol concentrations and chronic alcohol consumption: Role of the microsomal ethanol oxidizing system (MEOS). In Alcohol and the Liver; Fisher, M.M., Rankin, J.G., Eds.; Plenum Press: New York, NY, USA, 1977; Volume III, pp. 119–143. [Google Scholar]
- Lieber, C.S. Mechanism of ethanol induced hepatic injury. Pharmacol. Ther. 1990, 46, 1–41. [Google Scholar] [CrossRef]
- Lieber, C.S. Cytochrome P-4502E1: Its physiological and pathological role. Physiol. Rev. 1997, 77, 517–544. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Microsomal ethanol-oxidizing system (MEOS): The first 30 years (1968-1998)—A review. Alcohol. Clin. Exp. Res. 1999, 2, 991–1007. [Google Scholar] [CrossRef]
- Tanaka, E.; Terada, M.; Misawa, S. Cytochrome P450 2E1: Its clinical and toxicological role. Clin. Pharm. Ther. 2000, 25, 165–175. [Google Scholar] [CrossRef]
- Lieber, C.S. The discovery of the microsomal ethanol oxidizing system and its physiologic and pathologic role. Drug Metab. Rev. 2004, 36, 511–529. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Milestones in liver disease, a commentary. The unexpected outcomes of medical research: Serendipidy and the microsomal ethanol oxidizing system. J. Hepatol. 2004, 40, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Iseri, O.A.; Lieber, C.S.; Gottlieb, I.S. The ultrastructure of fatty liver induced by prolonged ethanol ingestion. Am. J. Pathol. 1966, 48, 535–555. [Google Scholar] [PubMed]
- Lane, B.P.; Lieber, C.S. Ultrastructural alterations in human hepatocytes following ingestion of ethanol with adequate diets. Am. J. Pathol. 1966, 49, 593–603. [Google Scholar] [PubMed]
- Joly, J.G.; Ishii, H.; Lieber, C.S. Microsomal cyanide-binding cytochrome: Its role in hepatic ethanol oxidation. Gastroenterology 1972, 62, 174A. [Google Scholar]
- Villeneuve, J.P.; Mavier, P.; Joly, J.G. Ethanol-induced cytochro 257me P-450: Catalytic activity after prtial purification. Biochem. Biophys. Res. Commun. 1976, 70, 723–728. [Google Scholar] [CrossRef]
- Teschke, R. Liver injury by carbon tetrachloride intoxication in 16 patients treated with forced ventilation to accelerate toxin removal via the lungs: A clinical report. Toxics 2018, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, M.P.; Lieber, C.S. Nonuniformaty of blood ethanol elimination: Its exaggeration after chronic consumption. Ann. Clin. Res. 1978, 10, 294–297. [Google Scholar] [PubMed]
- Feinman, L.; Baraona, E.; Matsuzaki, S.; Korsten, M.; Lieber, C.S. Concentration dependence of ethanol metabolism in vivo in rats and man. Alcohol. Clin. Exp. Res. 1978, 2, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I.; Dicker, E.; Lieber, C.S.; Rubin, E. Ethanol oxidation by isolated hepatocytes from ethanol-treated and control rats: Factors contributing to the metabolic adaptation after chronic ethanol consumption. Biochem. Pharmacol. 1978, 27, 7–15. [Google Scholar] [CrossRef]
- Pikkarainen, P.H.; Lieber, C.S. Concentration dependence of ethanol elimination rates on baboons: Effect of chronic alcohol consumption. Alcohol. Clin. Exp. Res. 1980, 4, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Matzsuzaki, S.; Gordon, E.; Lieber, C.S. Increased alcohol dehydrogenase independent ethanol oxidation at high ethanol concentrations in isolated rat hepatocytes: The effect of chronic ethanol feeding. J. Pharmacol. Exp. Ther. 1981, 217, 133–137. [Google Scholar]
- Shigeta, Y.; Nomura, F.; Iida, S.; Leo, M.A.; Felder, M.R.; Lieber, C.S. Ethanol metabolism in vivo by the microsomal ethanol-oxidizing system in Deermice lacking alcohol dehydrogenase (ADH). Biochem. Pharmacol. 1984, 33, 807–814. [Google Scholar] [CrossRef]
- Korsten, M.A.; Matsuzaki, S.; Feinman, L.; Lieber, C.S. High blood acetaldehyde levels after ethanol administration—Difference between alcoholic and nonalcoholic subjects. N. Engl. J. Med. 1975, 292, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Davis, V.E.; Walsh, M.J. Alcohol, amines, and alkaloids: A possible biochemical basis for alcohol addition. Science 1970, 167, 1005–1007. [Google Scholar]
- Tsukamoto, H.; Machida, K.; Dynnyk, A.; Mkrtchyan, H. “Second hit” models for alcoholic liver disease. Semin. Liver Dis. 2009, 29, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Dara, L.; Kaplowitz, N. Drug-induced liver injury: Cascade of events leading to cell death, apoptosis or necrosis. Int. J. Mol. Sci. 2017, 18, 1018. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, S.; Farrell, G.C. Etiopathogenesis of nonalcoholic steatohepatitis. Semin. Liver Dis. 2001, 21, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Emery, M.G.; Fisher, J.M.; Chien, J.Y.; Kharasch, E.D.; Dellinger, E.P.; Kowdley, K.V.; Thummel, K.E. CYP2E1 activity before and after weight loss in morbidly obese subjects with nonalcoholic fatty liver disease. Hepatology 2003, 38, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschke, R.; Xuan, T.D. Viewpoint: A contributory role of Shell ginger (Alpinia zerumbet) for human longevity of Okinawa in Japan? Nutrients 2018, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-induced hepatotoxicity: A comprehensive update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Intoxications by aliphatic halogenated hydrocarbons: Hepatotoxic risks for patients and clinical issues including role of CO2-induced hyperventilation as therapy option. J. Clin. Exp. Tox. 2018, 6, 1–21. [Google Scholar] [CrossRef]
- Zeng, T.; Zhang, C.L.; Xiao, M.; Yang, R.; Xie, K.Q. Critical roles of Kupffer cells in the pathogenesis of alcoholic liver disease: From basic science to clinical trials. Front. Immunol. 2016, 7, 538. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.G.; Jeong, W.I. Hepatic stellate cells and innate immunity in alcoholic liver disease. World J. Gastroenterol. 2011, 17, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Reeves, H.L.; Burt, A.D.; Wood, S.; Day, C.P. Hepatic stellate cell activation occurs in the absence of hepatitis in alcoholic liver disease and correlates with the severity of steatosis. J. Hepatol. 1996, 25, 677–683. [Google Scholar] [CrossRef]
- DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Ann. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Eickhoff, A.; Wolff, A.; Xuan, T.D. Liver injury from herbs and “dietary supplements”: Highlights of a literature review from 2015 to 2017. Curr. Pharmacol. Rep. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- Teschke, R.; Melchart, D.; Xuan, T.D. Editorial: Hormesis and dose-responses in herbal traditional Chinese medicine (TCM) alone are insufficient solving real clinical TCM challenges and associated herbal quality issues. Longhua Chin Med. 2018, 19, 779–793. [Google Scholar] [CrossRef]
- Wang, X.; Fang, H.; Huang, Z.; Shang, W.; Hou, T.; Cheng, A.; Cheng, H. Imaging ROS signaling in cells and animals. J. Mol. Med. (Berl) 2013, 91, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschke, R.; Wolff, A.; Eickhoff, A.; Danan, G. Is obesity rather than dietary supplements used for weight reduction the cause of liver injury? J. Gastroenterol. Hepatol. Open 2018, 2, 152–157. [Google Scholar] [CrossRef]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Cederbaum, A.I. Alcohol and oxidative liver injury. Hepatology 2006, 43, S63–S74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewing, J.A. Detecting alcoholism: The CAGE questionnaire. JAMA 1984, 252, 1905–1907. [Google Scholar] [CrossRef] [PubMed]
- Selzer, M.L. The Michigan Alcohol Screening Test: The quest for a new diagnostic instrument. Am. J. Psychiatr. 1971, 127, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.B.; Aasland, O.G.; Babor, T.F.; De La Fuente, J.R.; Grant, M. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO Collaborative Project on early detection of persons with harmful alcohol consumption—II. Addiction 1993, 88, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Aertgeerts, B.; Buntinx, F.; Kester, A. Review: Sensitivity of the CAGE questionnaire for the 5DSM diagnosis of alcohol abuse and dependence in general clinical populations was 71% at cut points ≥2. J. Clin. Epidemiol. 2004, 57, 30–39. [Google Scholar] [CrossRef]
- Teschke, R.; Danan, G. Review article: Diagnosis and management of drug-induced liver injury (DILI) in patients with pre-existing liver disease. Drug Saf. 2016, 39, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Danan, G. Drug-induced liver injury: Is chronic liver disease a risk factor and a clinical issue? Exp. Opin. Drug Metab. Toxicol. 2017, 13, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Amadi, C.N.; Orisakwe, O.E. Review. Herb-induced liver injuries in developing nations: An update. Toxics 2018, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Teschke, R. RUCAM in drug and herb induced liver injury: The update. Int. J. Mol. Sci. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Danan, G. Systematic review: Drug induced liver injury with analysis of alternative causes as confounding variables. Br. J. Clin. Pharmacol. 2018, 84, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Brand, A.; Strohmeyer, G. Induction of hepatic microsomal gamma-glutamyltransferase following chronic alcohol consumption. Biochem. Biophys. Res. Commun. 1977, 75, 718–724. [Google Scholar] [CrossRef]
- Lieber, C.S.; DeCarli, L.M. The feeding of alcohol in liquid diets: Two decades of applications and 1982 update. Alcohol. Clin. Exp. Res. 1982, 6, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Neuefeind, M.; Nishimura, N.; Strohmeyer, G. Hepatic gamma-glutamyltransferase activity in alcoholic fatty liver: Comparison with other liver enzymes in man and rats. Gut 1983, 24, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Stein, H.; Berges, W.; Teschke, R. Gamma-glutamyltransferase activity of liver plasma membrane: Induction following chronic alcohol consumption. Biochem. Biophys. Res. Commun. 1981, 99, 142–148. [Google Scholar] [CrossRef]
- Nishimura, M.; Teschke, R. Effect of chronic alcohol consumption on the activities of liver plasma membrane enzymes: Gamma-glutamyltransferase, alkaline phosphatase and 5’-nucleotidase. Biochem. Pharmacol. 1982, 31, 377–381. [Google Scholar]
- Teschke, R.; Krukenberg, S.; Stremmel, W.; Nishimura, M. Enhanced biliary gamma-glutamyltransferase excretion following prolonged alcohol consumption in rats. Eur. J. Clin. Investig. 1987, 17, 347–353. [Google Scholar] [CrossRef]
- Milstein, H.J.; Bloomer, J.R.; Klatskin, G. Serum bile acids in alcoholic liver disease. Am. J. Dig. Dis. 1976, 21, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Koch, T. Biliary excretion of gamma-glutamyltransferase: Selective enhancement by acute ethanol administration. Biochem. Pharmacol. 1986, 35, 2521–2525. [Google Scholar] [CrossRef]
- Nishimura, M.; Teschke, R. Alcohol and gamma-glutamyltransferase. Klin. Wochenschr. 1983, 61, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Chang, B.; Sun, Y.; Lin, H.; Li, B.; Teng, G.; Zou, Z.S. Disease spectrum of alcoholic liver disease in Beijing 302 Hospital from 2002 to 2013. Medicine 2017, 96, e6163. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Kaplan, M.M. The SGOT/SGPT ratio an indicator of alcoholic liver disease. Dig. Dis. Sci. 1979, 24, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Koskinas, J.; Kenna, J.G.; Bird, G.L.; Alexander, G.J.M.; Williams, R. Immunglobulin A antibody to a 200-Kilodalton cytosolic acetaldehyde adduct in alcoholic hepatitis. Gastroenterology 1992, 103, 1860–1867. [Google Scholar] [CrossRef]
- Van Waes, L.; Lieber, C.S. Glutamate dehydrogenase: A reliable marker of liver cell necrosis in the alcoholic. Br. Med. J. 1977, 2, 1508–1510. [Google Scholar] [CrossRef] [PubMed]
- Blaya, D.; Coll, M.; Rodrigo-Torres, D.; Vila-Casadesús, M.; Altamirano, J.; Llopis, M.; Graupera, I.; Perea, L.; Aguilar-Bravo, B.; Díaz, A.; et al. Integrative microRNA profiling in alcoholic hepatitis reveals a role for microRNA-182 in liver injury and inflammation. Gut 2016, 65, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Schulze, J.; Eickhoff, A.; Danan, G. Drug induced liver injury: Can biomarkers assist RUCAM in causality assessment? Special issue: Molecular Research on Drug Induced Liver Injury. Int. J. Mol. Sci. 2017, 18, 803. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Larrey, D.; Melchart, D.; Danan, G. Traditional Chinese Medicine (TCM) and herbal hepatotoxicity: RUCAM and the role of novel diagnostic biomarkers such as microRNAs. Medicines 2016, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Celli, R.; Zhang, X. Pathology of alcoholic liver disease. J. Clin. Transl. Hepatol. 2014, 2, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003, 289, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Basra, S.; Anand, B.S. Definition, epidemiology and magnitude of alcoholic hepatitis. World J. Hepatol. 2011, 3, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Theise, N.D. Histopathology of alcoholic liver disease. Clin. Liver Dis. 2013, 2, 64–67. [Google Scholar] [CrossRef]
- Sakhuja, P. Pathology of alcoholic liver disease, can it be differentiated from nonalcoholic steatohepatitis? World J. Gastroenterol. 2014, 20, 16474–16479. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C. Histopathology of alcoholic liver disease. Clin. Liver Dis. 1998, 2, 753–763. [Google Scholar] [CrossRef]
- Toivola, D.M.; Ku, N.O.; Resurreccion, E.Z.; Nelson, D.R.; Wright, T.L.; Omary, M.B. Keratin 8 and 18 hyperphosphorylation is a marker of progression of human liver disease. Hepatology 2004, 40, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.E.; Snyder, H.S.; Twilla, J.D.; Satapathy, S.K. Pharmacologic treatment of alcoholic hepatitis: Examining outcomes based on disease severity stratification. J. Clin. Exp. Hepatol. 2016, 6, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Vuittonet, C.L.; Halse, M.; Leggio, L.; Frichione, S.B.; Brickley, M.; Haass-Koffler, C.L.; Tavares, T.; Swift, R.M.; Kenna, G.A. Pharmacotherapy for alcoholic patients with alcoholic liver disease. Am. J. Health Syst. Pharm. 2014, 71, 1265–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, H.P.; Simon, F.R.; Pope, C.E.; Volwiler, W.; Fenster, L.F. Corticosteroid therapy in severe alcoholic hepatitis. A double-blind drug trial. N. Engl. J. Med. 1971, 284, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Helman, R.A.; Temko, M.H.; Nye, S.W.; Fallon, H.J. Alcoholic hepatitis. Natural history and evaluation of prednisolone therapy. Ann. Intern. Med. 1971, 74, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Campra, J.L.; Hamlin, E.M.; Kirshbaum, R.J.; Olivier, M.; Redeker, A.G.; Reynolds, T.B. Prednisone therapy in acute alcoholic hepatitis. Report of a controlled trial. Ann. Intern. Med. 1973, 79, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Blitzer, B.L.; Mutchnick, M.G.; Joshi, P.H.; Phillips, M.M.; Fessel, J.M.; Conn, H.O. Adrenocorticosteroid therapy in alcoholic hepatitis. A prospective, double-blind randomized study. Am. J. Dig. Dis. 1977, 22, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, J.B.; Resnick, R.H.; Galambos, J.T.; Makopour, H.; Iber, F.L. A controlled trial of 6-methylprednisolone in acute alcoholic hepatitis. With a note on published results in encephalopathic patients. Am. J. Gastroenterol. 1978, 69, 443–449. [Google Scholar] [PubMed]
- Lesesne, H.R.; Bozymski, E.M.; Fallon, H.J. Treatment of alcoholic hepatitis with encephalopathy: Comparison with caloric supplements. Gastroenterology 1978, 74, 169–173. [Google Scholar] [PubMed]
- Maddrey, W.C.; Boitnott, J.K.; Bedine, M.S.; Weber, F.L.; Mezey, E.; White, R.I. Corticosteroid therapy of alcoholic hepatitis. Gastroenterology 1978, 75, 193–199. [Google Scholar] [PubMed]
- Depew, W.; Boyer, T.; Omata, M.; Redeker, A.; Reynolds, T. Double-blind controlled trial of prednisolone therapy in patients with severe acute alcoholic hepatitis and spontaneous encephalopathy. Gastroenterology 1980, 78, 524–529. [Google Scholar] [PubMed]
- Theodossi, A.; Eddleston, A.L.; Williams, R. Controlled trial of methylprednisolone therapy in severe acute alcoholic hepatitis. Gut 1982, 23, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, C.L.; Anderson, S.; Garcia-Pont, P.; Goldberg, S.; Kiernan, T.; Seeff, L.B.; Sorell, M.; Tamburro, C.; Weesner, R.; Zetterman, R.; et al. The Veterans Administration Cooperative Study on Alcoholic Hepatitis. Short-term and long-term survival in patients with alcoholic hepatitis treated with oxandrolone and prednisolone. N. Engl. J. Med. 1984, 311, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Bories, P.; Guedj, J.Y.; Mirouze, D.; Yousfi, A.; Michel, H. Treatment of acute alcoholic hepatitis with prednisolone. 45 patients. Presse Med. 1987, 16, 769–772. [Google Scholar] [PubMed]
- Carithers, R.L.; Herlong, H.F.; Diehl, A.M.; Shaw, E.W.; Combes, B.; Fallon, H.J.; Maddrey, W.C. Methylprednisolone therapy in patients with severe alcoholic hepatitis. A randomized multicenter trial. Ann. Intern. Med. 1989, 110, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Ramond, M.J.; Poynard, T.; Rueff, B.; Mathurin, P.; Theodore, C.; Chaput, J.C.; Benhamou, J.P. A randomized trial of prednisolone in patients with severe alcoholic hepatitis. N. Engl. J. Med. 1992, 326, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; OGrady, J.; Carithers, R.L.; Phillips, M.; Louvet, A.; Mendenhall, C.L.; Ramond, M.J.; Naveau, S.; Maddrey, W.C.; Morgan, T.R. Corticosteroids improve short-term survival in patients with severe alcoholic hepatitis: Meta-analysis of individual patient data. Gut 2011, 60, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Thursz, M.R.; Richardson, P.; Allison, M.; Austin, A.; Bowers, M.; Day, C.P.; Downs, N.; Gleeson, D.; MacGilchrist, A.; Grant, A.; et al. Prednisolone or Pentoxifylline for alcoholic hepatitis. N. Engl. J. Med. 2015, 372, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Akriviadis, E.; Botla, R.; Briggs, W.; Han, S.; Reynolds, T.; Shakil, O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: A double-blind, placebo-controlled trial. Gastroenterology 2000, 119, 16371648. [Google Scholar] [CrossRef]
- Naveau, S.; Chollet-Martin, S.; Dharancy, S.; Mathurin, P.; Jouet, P.; Piquet, M.A.; Davion, T.; Oberti, F.; Broët, P.; Emilie, F. A double blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 2004, 39, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.F.; Lischner, M.W.; Galambos, J.T. Natural history of alcoholic hepatitis. II. The long-term prognosis. Am. J. Gastroenterol. 1971, 56, 515–525. [Google Scholar] [PubMed]
- Jepsen, P.; Ott, P.; Andersen, P.K.; Srensen, H.T.; Vilstrup, H. The clinical course of alcoholic liver cirrhosis: A Danish population-based cohort study. Hepatology 2010, 51, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.B.; Walters, J.R.F.; Davie, P.; Paton, A. A 20-year prospective study of cirrhosis. BMJ 1981, 282, 263266. [Google Scholar] [CrossRef]
- Fleming, K.M.; Aithal, G.P.; Card, T.R.; West, J. The rate of decompensation and clinical progression of disease in people with cirrhosis: A cohort study. Aliment. Pharmacol. Ther. 2010, 32, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.J., Jr.; Klatskin, G. Duration of survival in patients with Laennec’s cirrhosis. Influence of alcohol withdrawal, and possible effects of recent changes in general management of the disease. Am. J. Med. 1968, 44, 406–420. [Google Scholar] [CrossRef]
- Martini, G.A.; Teschke, R. Alcohol abstinence in alcoholic liver disease. Acta Med. Scand. 1985, 703, 185–194. [Google Scholar] [CrossRef]
- O’Grady, J.G. Liver transplantation alcohol related liver disease: (Deliberately) stirring a hornet’s nest! Gut 2006, 55, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; Naveau, S.; Doffoel, M.; Boudjema, K.; Vanlemmens, C.; Mantion, G.; Messner, M.; Launois, B.; Samuel, D.; Cherqui, D.; et al. Evaluation of efficacy of liver transplantation in alcoholic cirrhosis using matched and simulated controls: 5-year survival. Multi-centre group. J. Hepatol. 1999, 30, 1130–1137. [Google Scholar] [CrossRef]
- Worner, T.M.; Lieber, C.S. Perivenular fibrosis as precursor lesion of cirrhosis. JAMA 1985, 254, 627–630. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.J.; O’Connor, J.F.B. Alcoholic liver disease: Proposed recommendations for the American College of Gastroenterology. Am. J. Gastroenterol. 1998, 93, 2022–2036. [Google Scholar] [CrossRef] [PubMed]
- Testino, G.; Leone, S.; Borro, P. Alcohol and hepatocellular carcinoma: A review and a point of view. World J. Gastroenterol. 2014, 20, 15943–15954. [Google Scholar] [CrossRef] [PubMed]
- Burra, P.; Zanetto, A.; Germani, G. Liver transplantation for alcoholic liver disease and hepatocellular carcinoma. Cancers 2018, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–844. [Google Scholar] [CrossRef]
- Robin, M.A.; Anandatheerthavarada, H.K.; Biswas, G.; Sepuri, N.B.; Gordon, D.M.; Pain, D.; Avadhani, N.G. Bimodal targeting of microsomal CYP2E1 to mitochondria through activation of an N-terminal chimeric signal by AMP-mediated phosphorylation. J. Biol. Chem. 2002, 277, 40583–40593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, D.; Dong, W.; Zhang, L.; Zhang, X.; Quan, X.; Ma, C.; Lian, H.; Zhang, L. Expression of CYP2E1 increases oxidative stress and induces apoptosis of cardiomyocytes in transgenic mice. FEBS J. 2011, 278, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, S.; Liu, C.P.; Sepuri, N.B.V.; Anandatheerthavarada, H.K.; Selvaraj, V.; Hoek, J.; Milne, G.L.; Guengerich, F.P.; Avadhani, N.G. Mitochondria-targeted cytochrome P450 2E1 induced oxidative damage and augments alcohol-mediated stress. J. Biol. Chem. 2010, 285, 24609–24619. Available online: http://www.jbc.org/cgi/doi/10.1074/jbc.M110.121822 (accessed on 19 September 2018). [CrossRef] [PubMed]
- Raza, H.; Prabu, S.K.; Robin, M.A.; Avadhani, N.G. Elevated mitochondrial cytochrome P450 2E1 and glutathione S-transferase A4-4 in streptomycin-induced diabetic rats. Tissue-specific variations and roles in oxidative stress. Diabetes 2004, 53, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.A.; Sauvage, I.; Grandperret, T.; Descatoire, V.; Pessayre, D.; Fromenty, B. Ethanol increases mitochondrial cytochrome 2E1 in mouse liver and rat hepatocytes. FEBS Lett. 2005, 579, 6895–6902. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Suzuki, H.; Sakaguchi, S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J. Toxicol. Sci. 2007, 32, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Role of CYP 2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig. Dis. 2010, 28, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, D.; Wang, X.; Cederbaum, A.I. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radic Biol. Med. 2012, 53, 1170–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.H.; Yun, J.W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederbaum, A.I. Methodology to assay CYP2E1 mixed function oxidase catalytic activity and its function. Redox Biol. 2014, 2, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Gaviria, M.M.; Arango, G.C.; Navas, M.C. Alcohol, cirrhosis, and genetic predisposition. Rev. Colomb. Gastroenterol. 2016, 31, 26–33. [Google Scholar] [CrossRef]
- Cho, Y.E.; Mezey, E.; Hardwick, J.P.; Salem, N.; Clemens, D.L.; Song, B.J. Increased ethanol-inducible cytochrome P450-2E1 and cytochrome P450 isoforms in exosomes of alcohol-exposed rodents and patients with alcoholism through oxidative and endoplasmic reticulum stress. Hepatol. Commun. 2017, 1, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolbright, B.L.; Jaeschke, H. Alcoholic hepatitis: Lost in translation. J. Clin. Transl. Hepatol. 2018, 6, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Zhu, Y. Paracetamol (acetaminophen), alcohol, and liver injury: Biomarkers, clinical issues, and experimental aspects. SL Pharmacol. Toxicol. 2018, 5, 11. [Google Scholar]
- Ramachandran, A.; Jaeschke, H. Acetaminophen toxicity: novel insights into mechanisms and future perspectives. Gene Expr. 2018, 18, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Leo, M.A.; Wang, X.; DeCarli, L.M. Effect of chronic alcohol consumption on hepatic SIRT1 and PGC- 1alpha in rats. Biochem. Biophys. Res. Commun. 2008, 370, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.J.; Humphries, J.R.; Niemeyer, D.J.; Sindram, D.; McKillop, I.H. The effect of alcohol on Sirt1 expression and function in animal and human models of hepatocellular carcinoma (HCC). Adv. Exp. Med. Biol. 2015, 815, 361–373. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Jogasuria, A.; Taylor, C.; Wu, J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.A.; Rosman, A.S.; Lieber, C.S. Differential depletion of carotenoids and tocopherol in liver disease. Hepatology 1993, 17, 977–986. [Google Scholar] [PubMed]
- Albano, E.; Vidali, M. Immune mechanisms in alcoholic liver disease. Genes Nutr. 2010, 5, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Mandrekar, P.; Petrasek, J.; Catalano, D. The unfolding web of innate immune dysregulation in alcoholic liver injury. Alcohol. Clin. Exp. Res. 2011, 35, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Duryee, M.J.; Klassen, L.W.; Thiele, G.M. Immunological response in alcoholic liver disease. World J. Gastroenterol. 2007, 13, 4938–4946. [Google Scholar] [CrossRef] [PubMed]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The gastrointestinal microbiome: Alcohol effects on the composition of intestinal microbiota. Alcohol. Res. Curr. Rev. 2015, 37, 223–236. [Google Scholar]
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal. 2011, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Udoh, U.S.; Valcin, J.A.; Gamble, K.L.; Bailey, S.M. The molecular circadian clock and alcohol-induced liver injury. Biomolecules 2015, 5, 2504–2537. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Aliphatic halogenated hydrocarbons: Liver injury in 60 patients. J. Clin. Transl. Hepatol. 2018, 6, 1–12. [Google Scholar]























{kind=link}
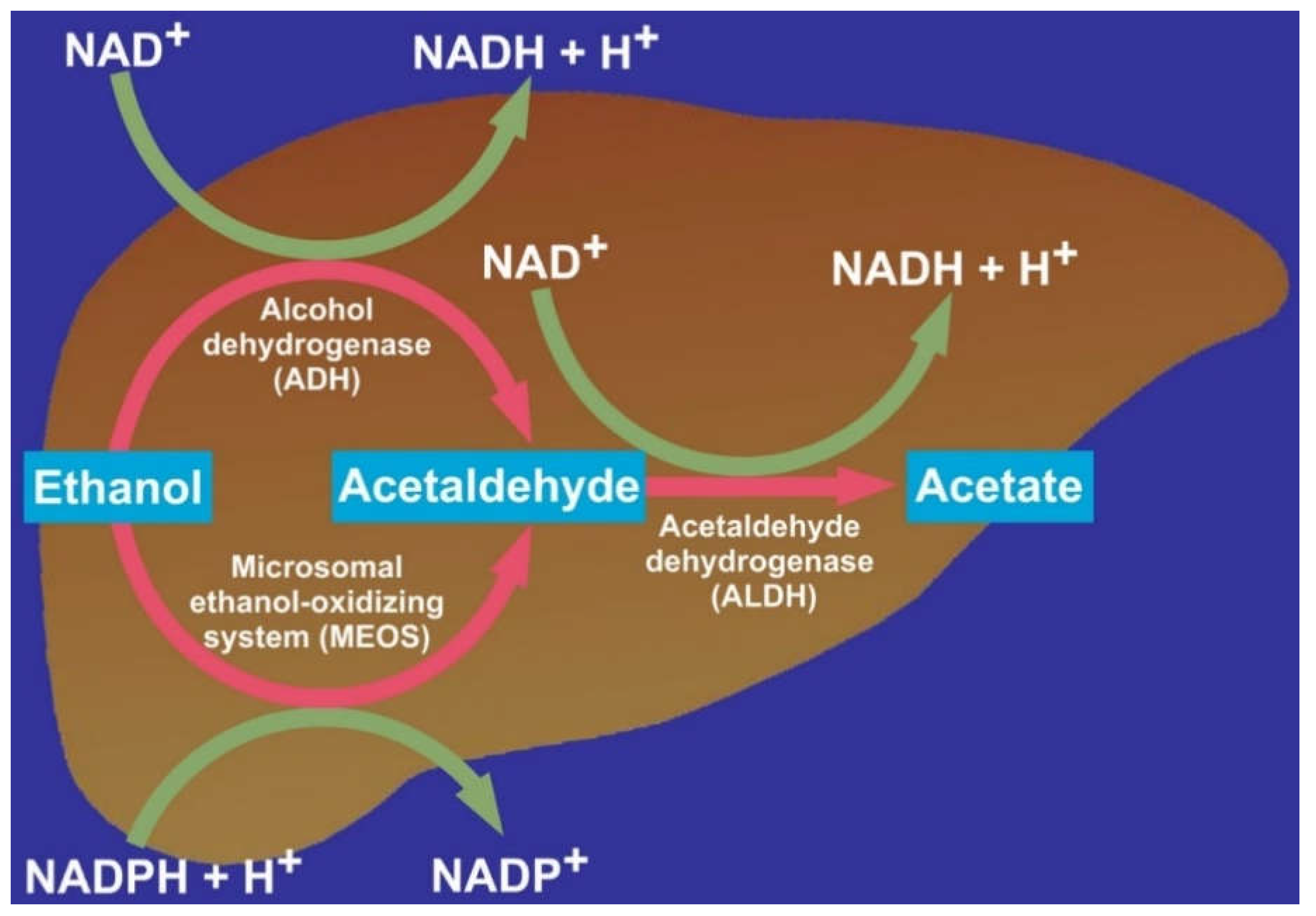{kind=link}
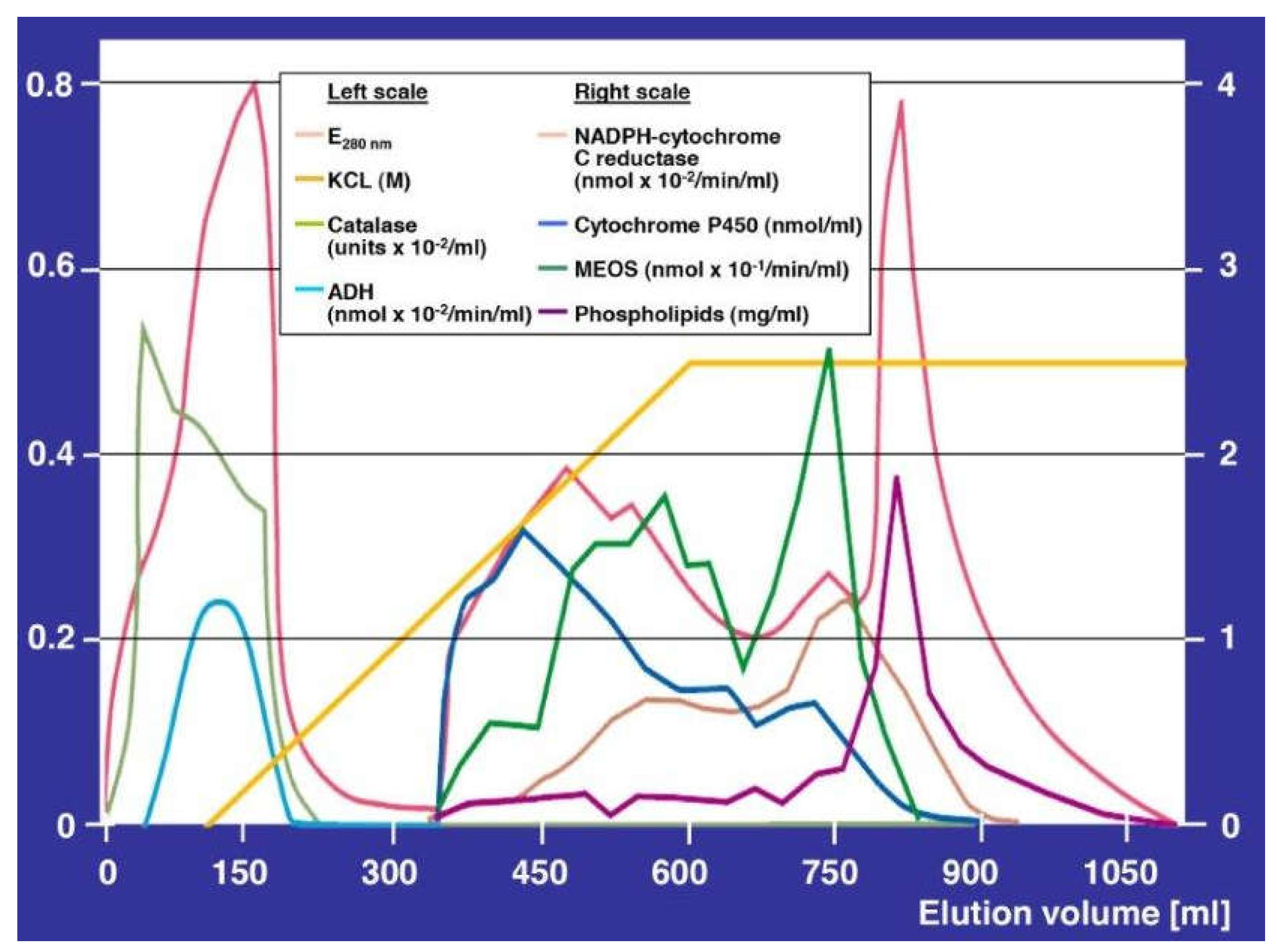{kind=link}
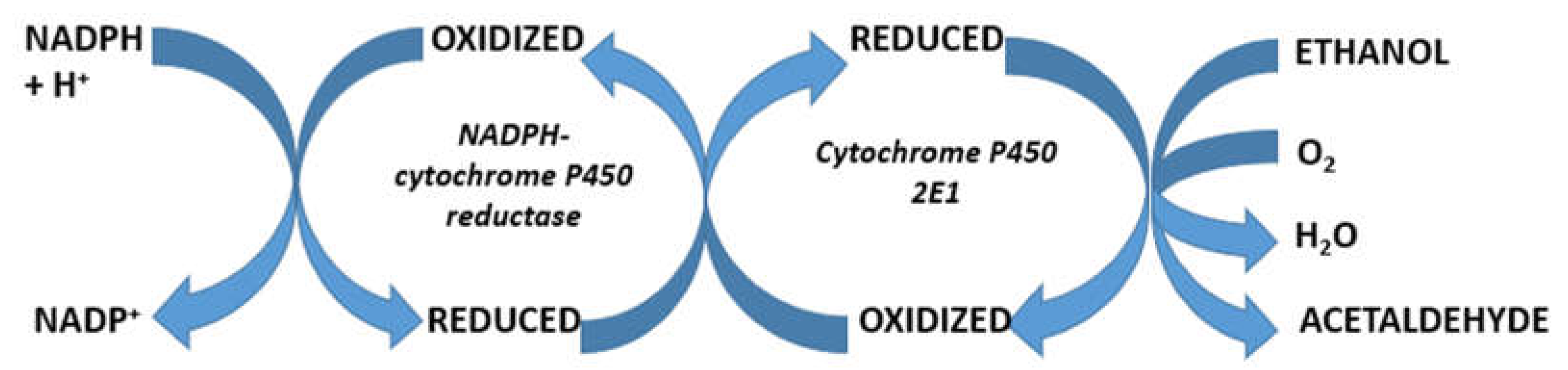{kind=link}
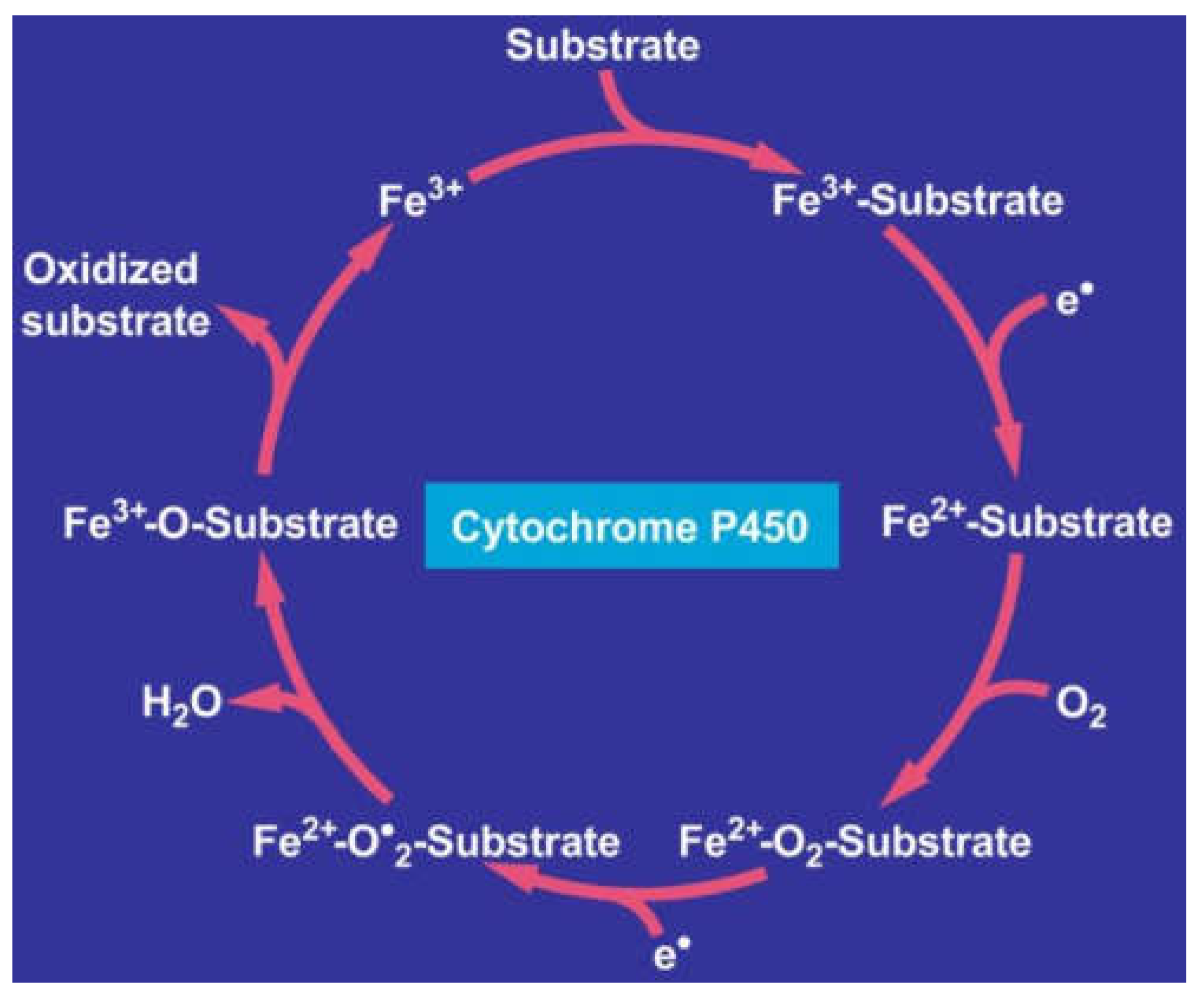{kind=link}
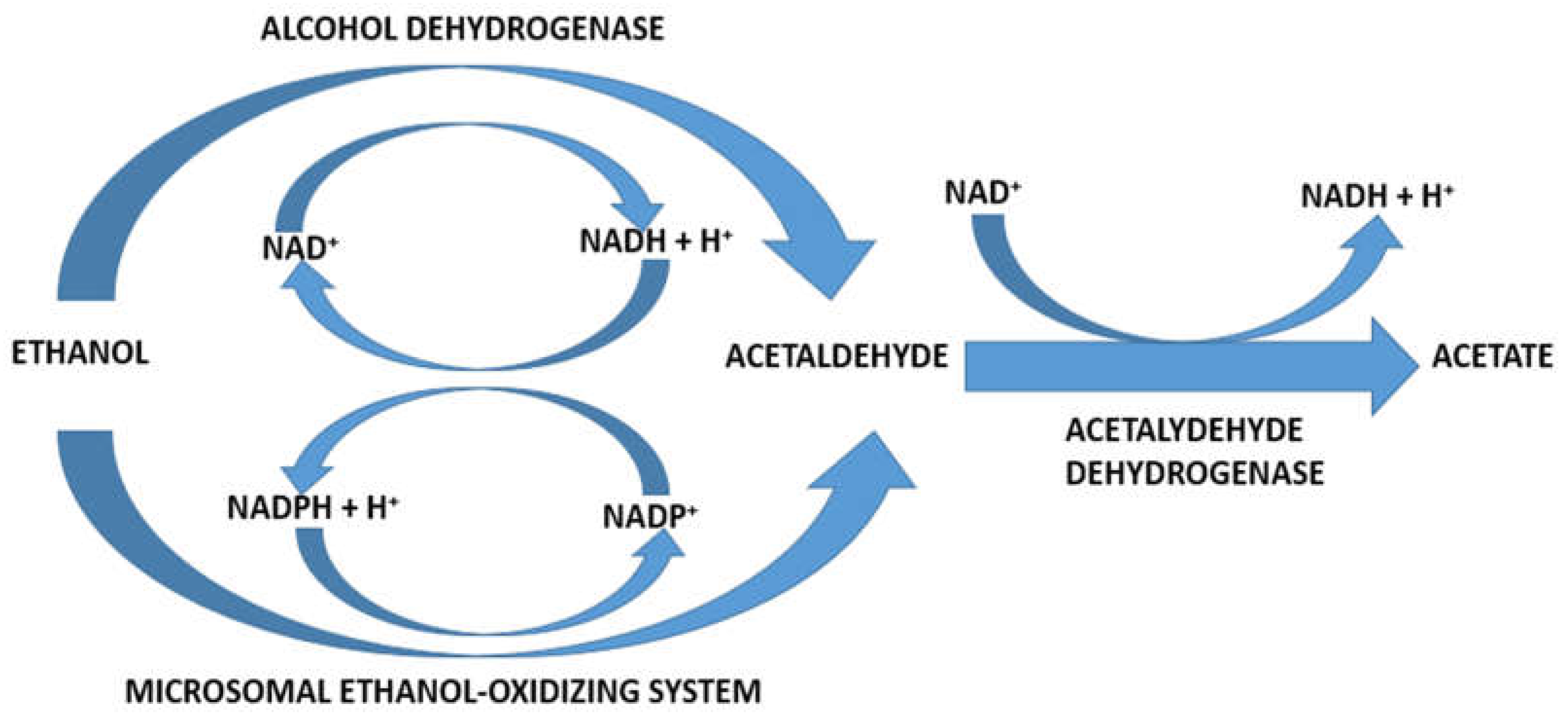{kind=link}
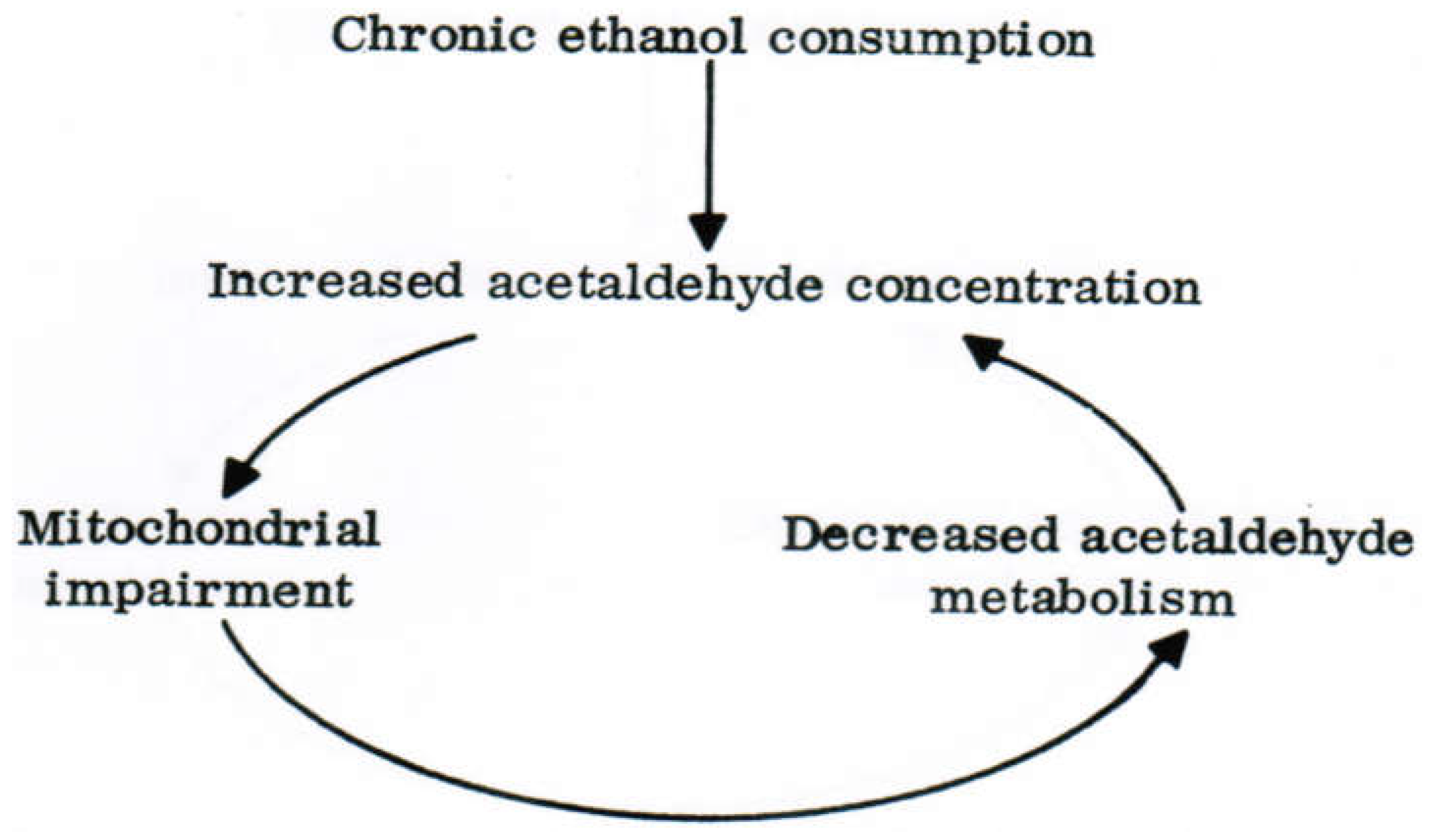{kind=link}
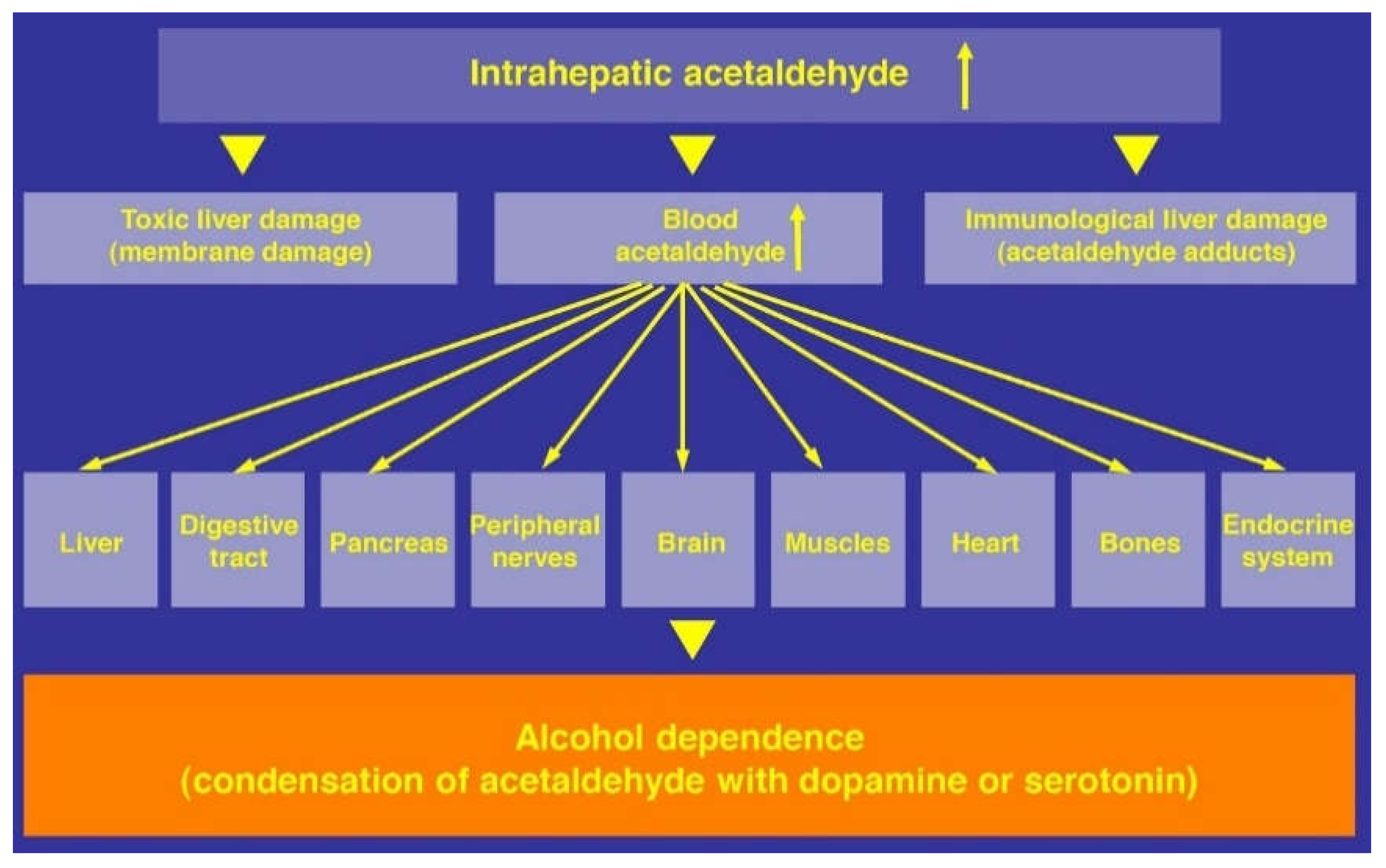{kind=link}
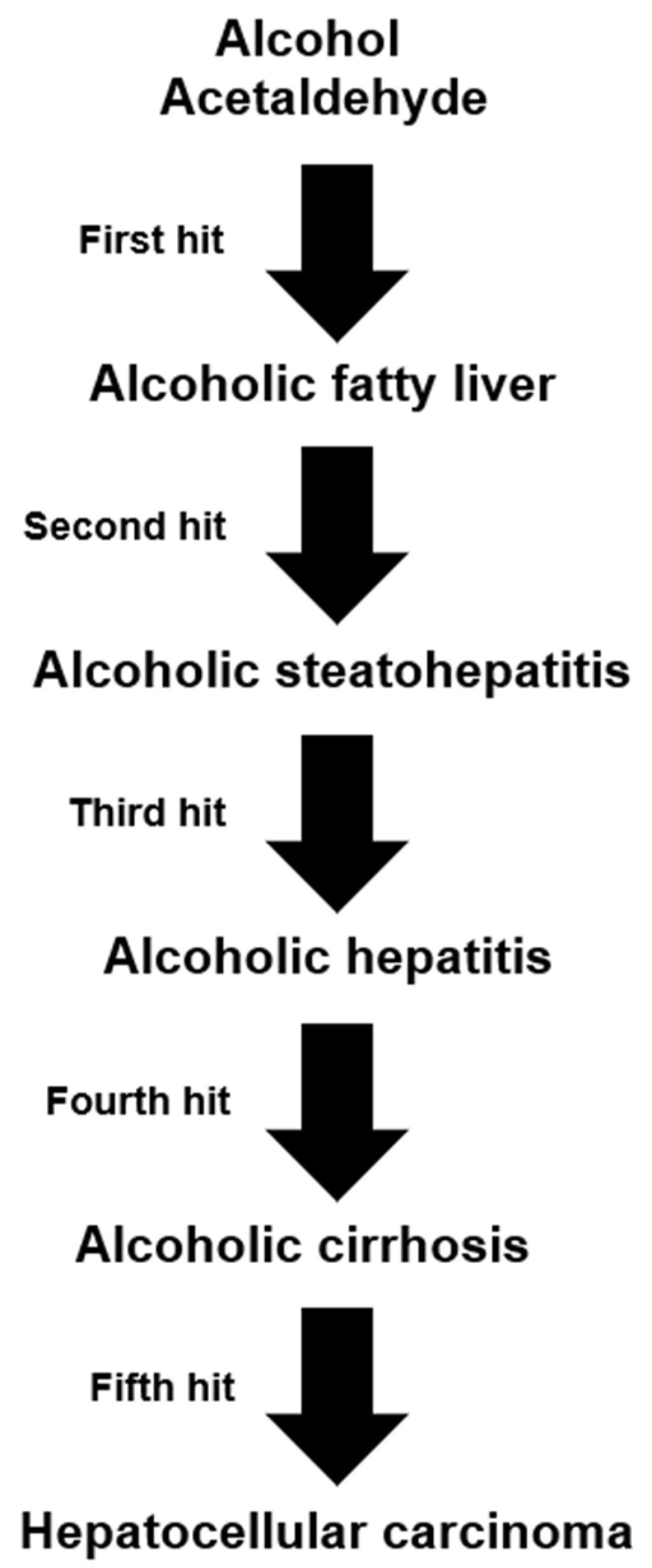{kind=link}
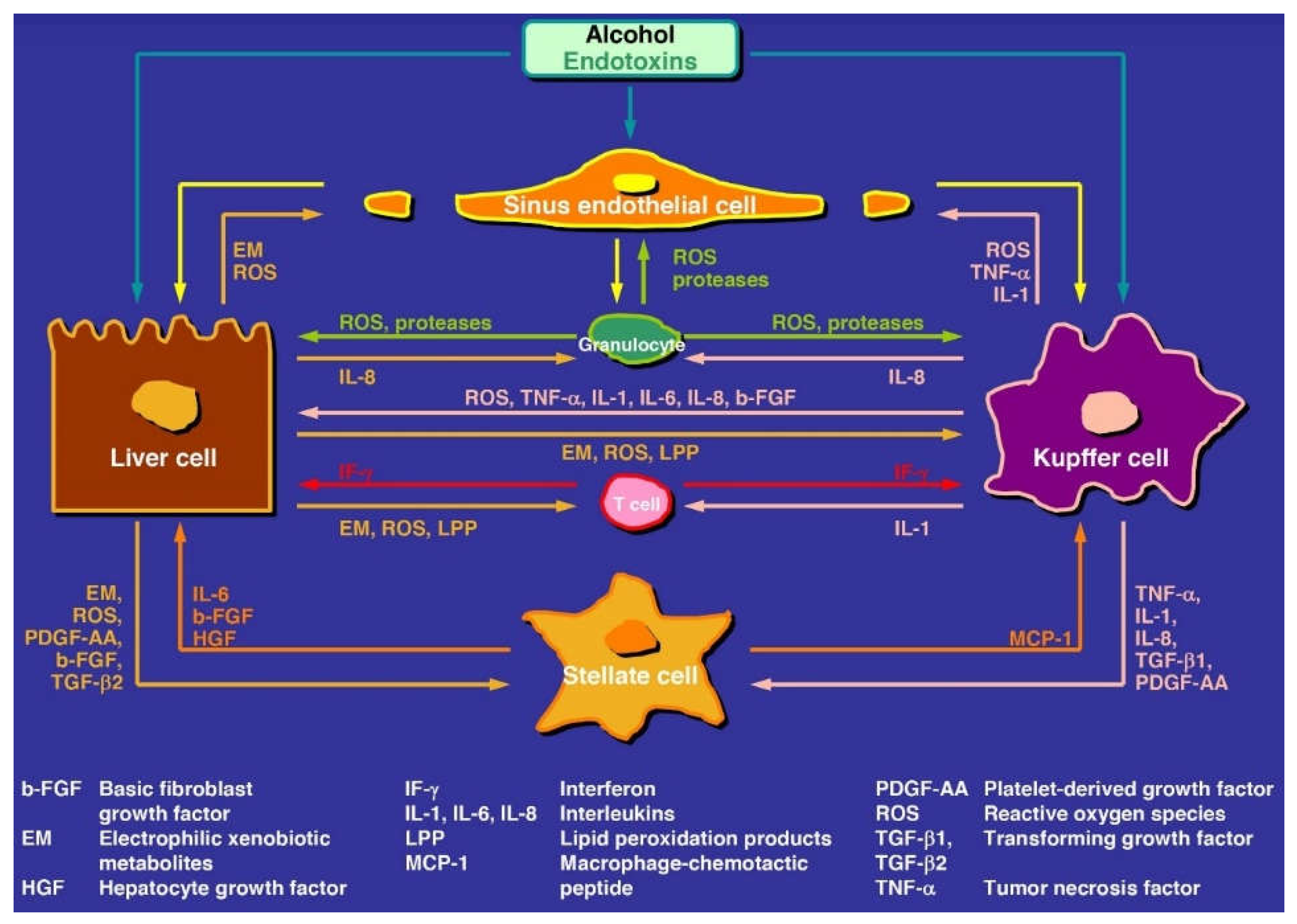{kind=link}
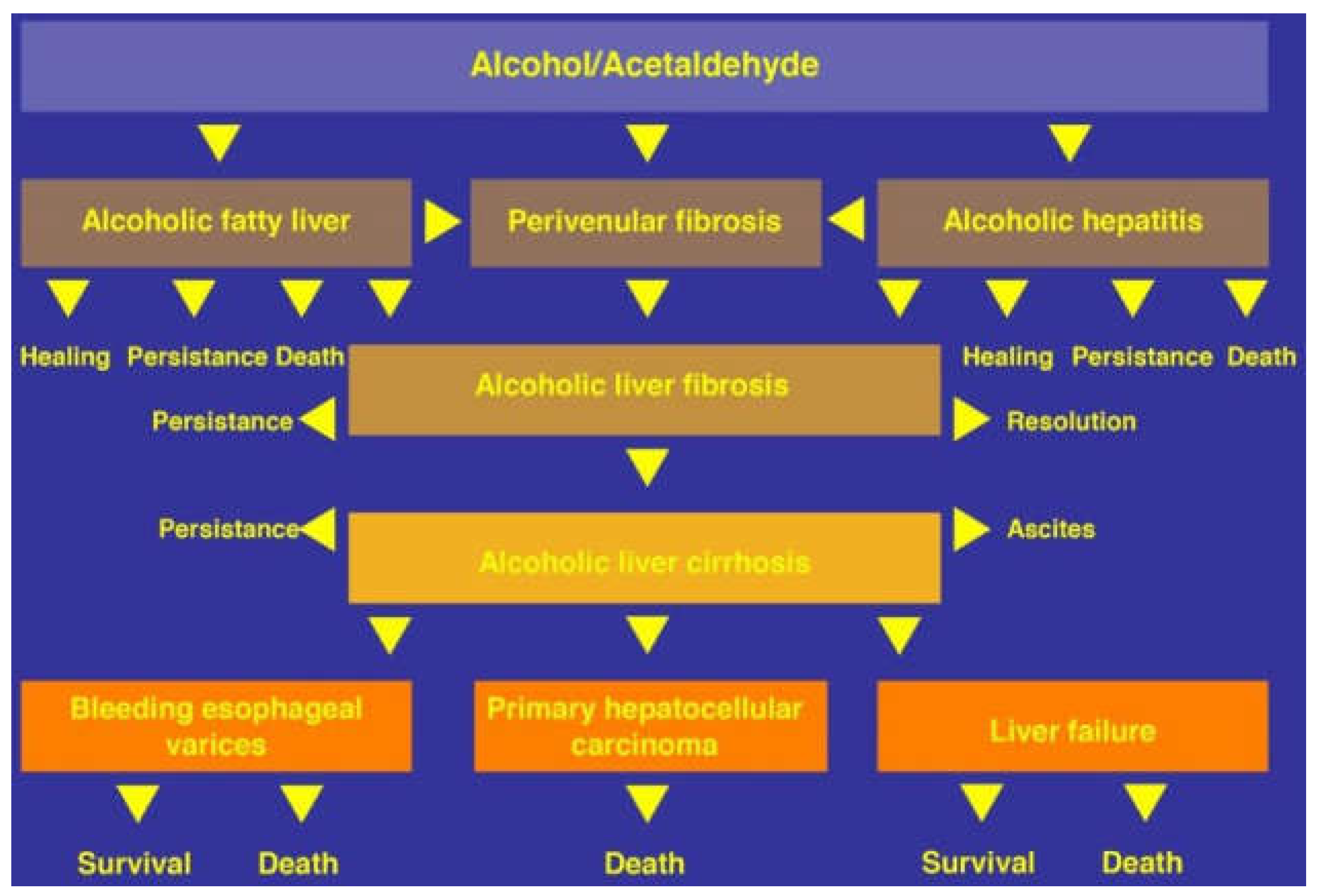{kind=link}
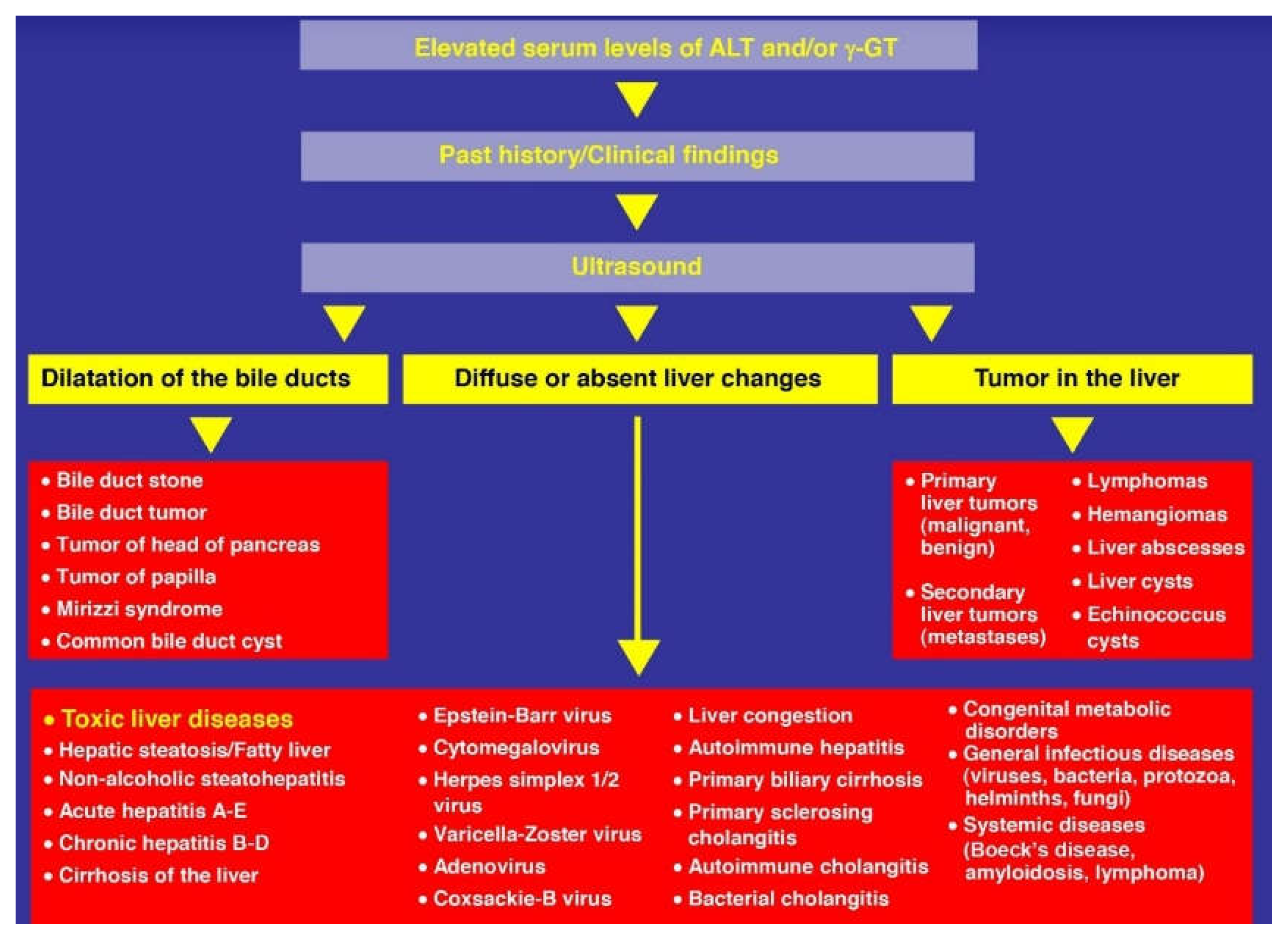{kind=link}
{kind=link}
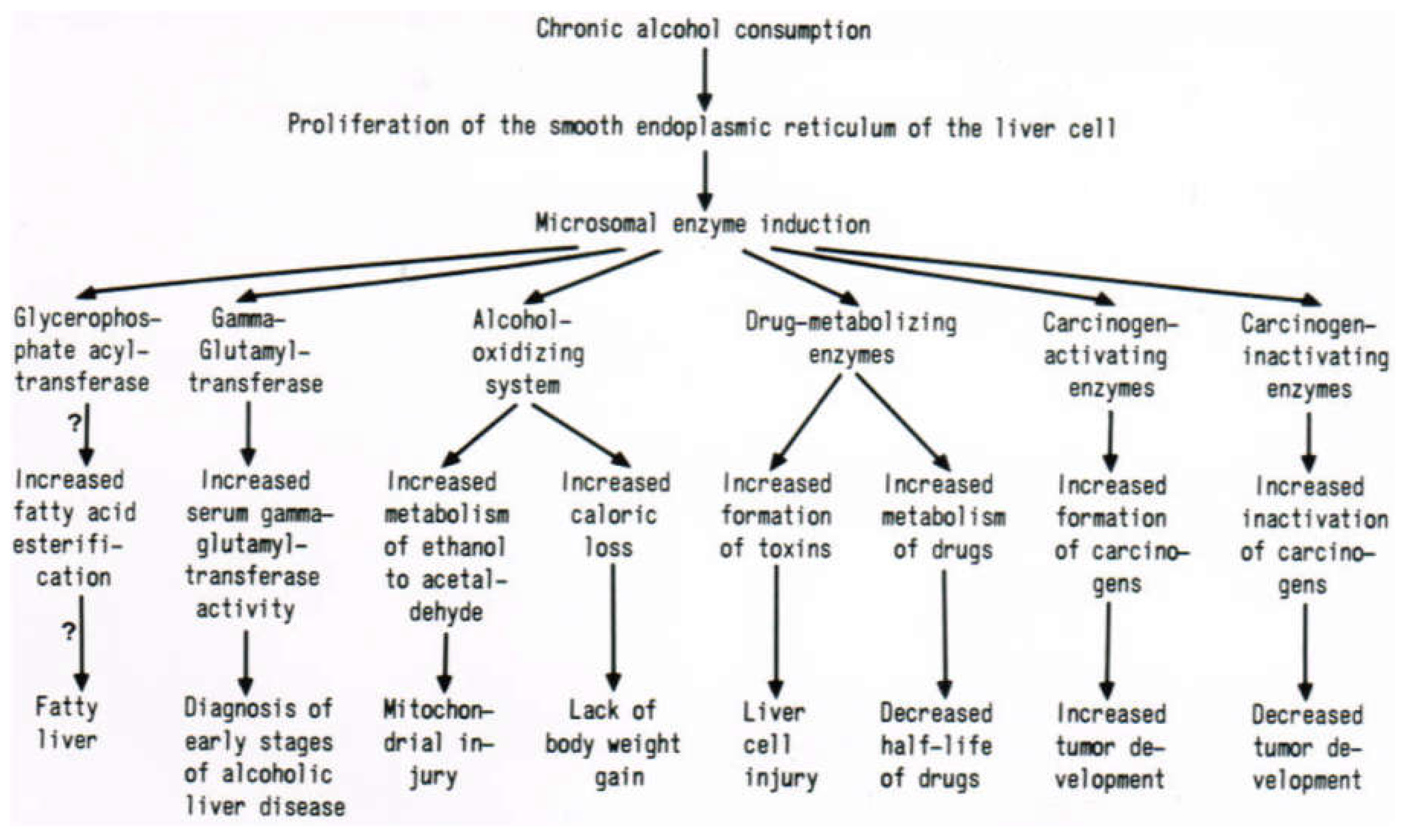{kind=link}
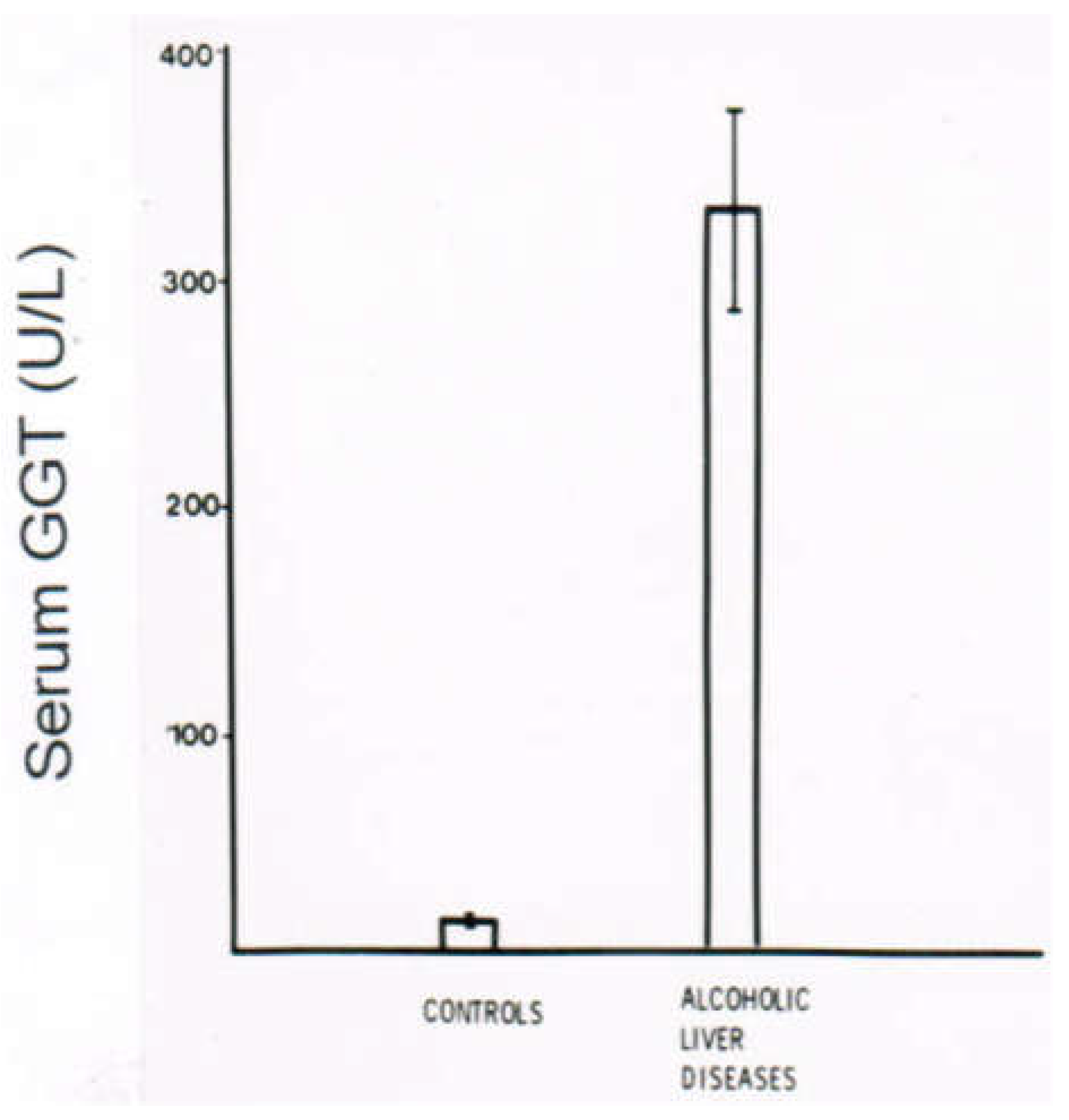{kind=link}
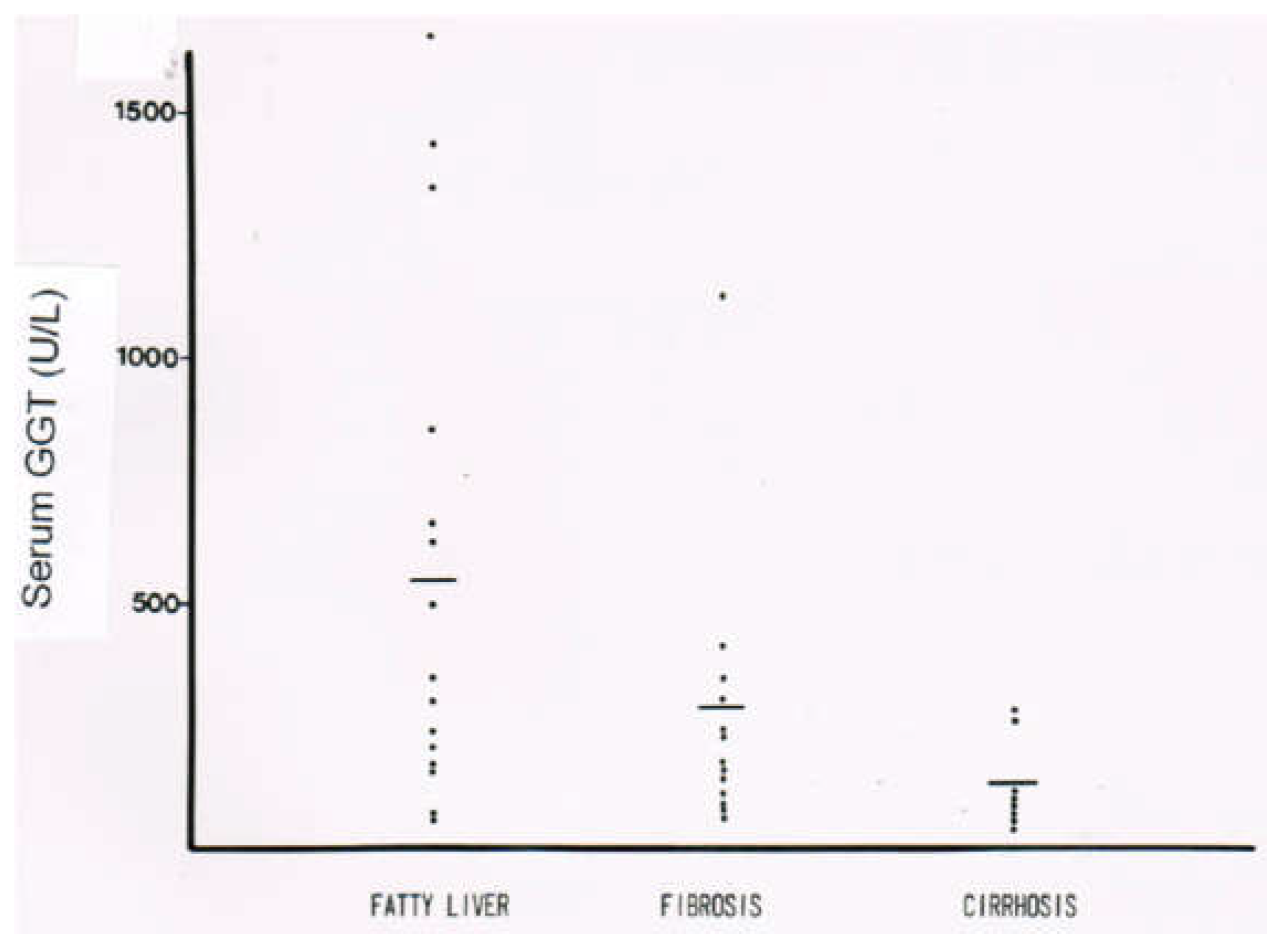{kind=link}
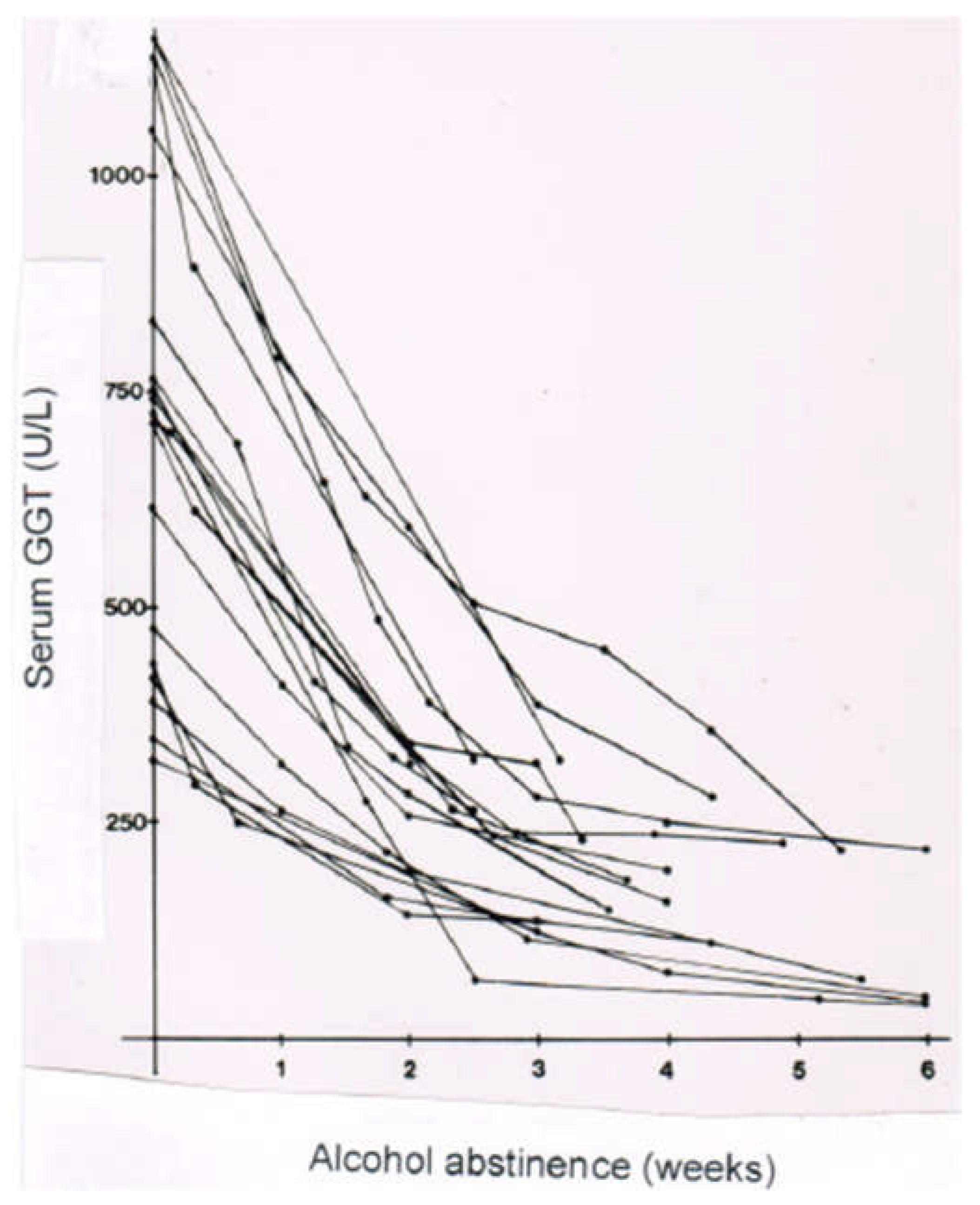{kind=link}
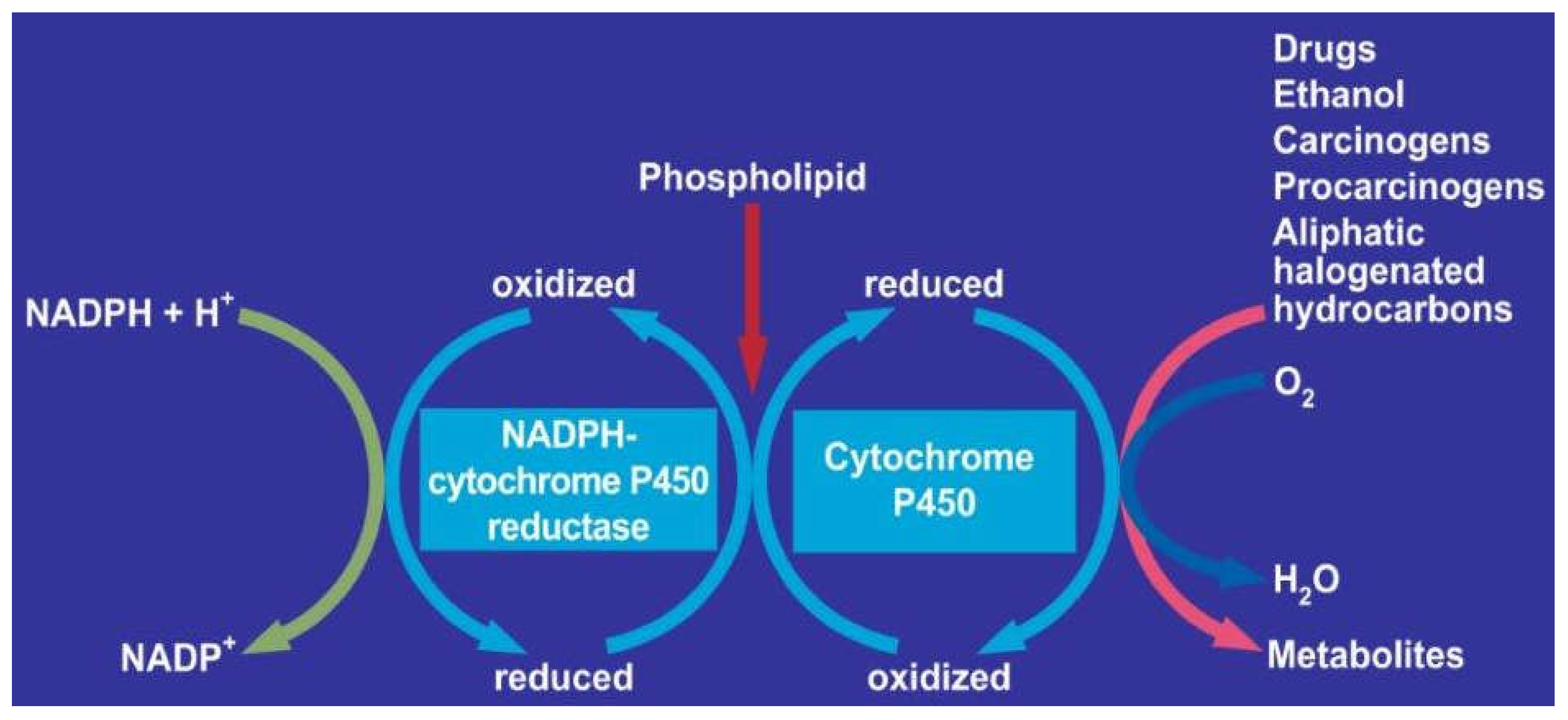{kind=link}
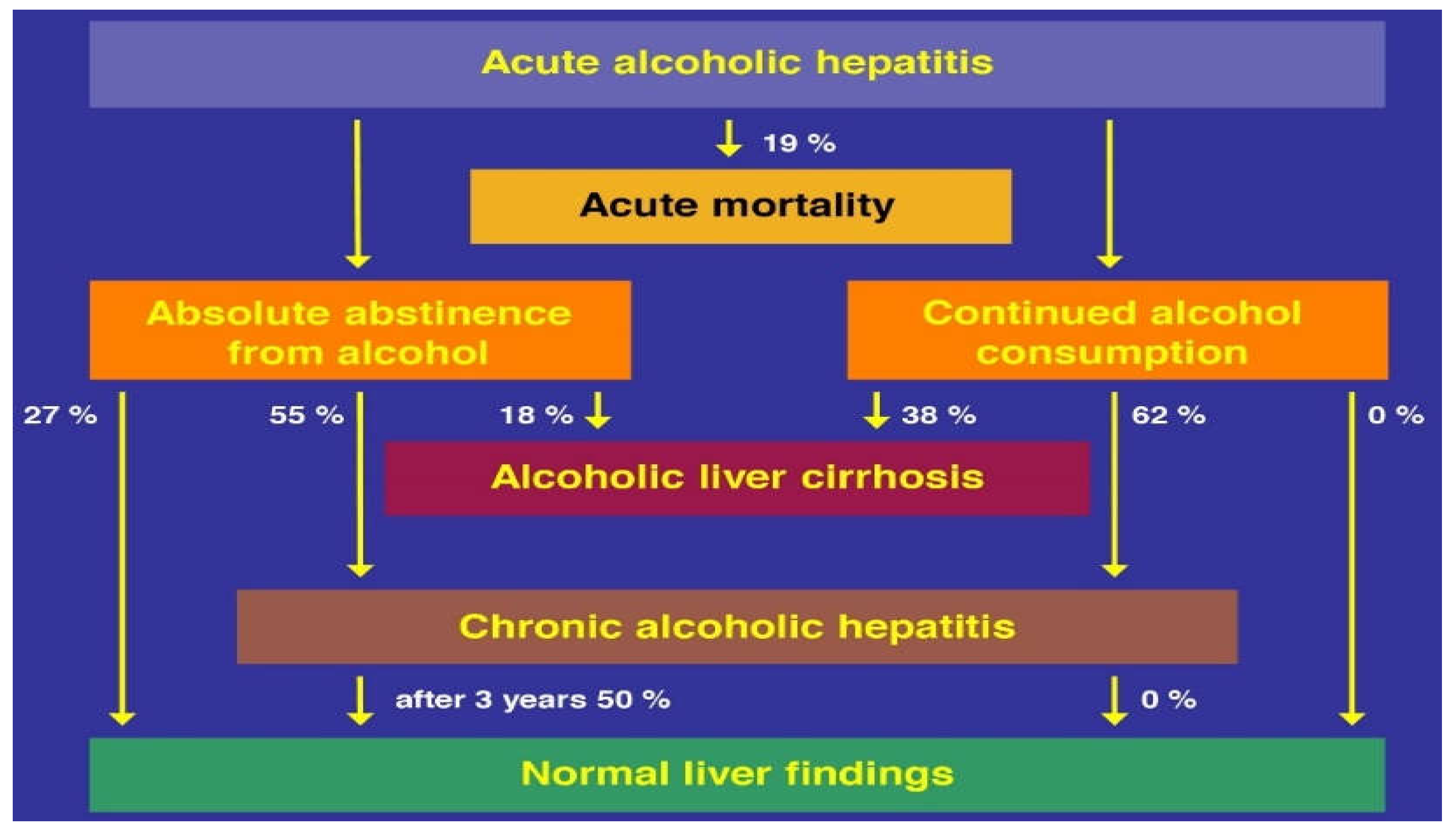{kind=link}
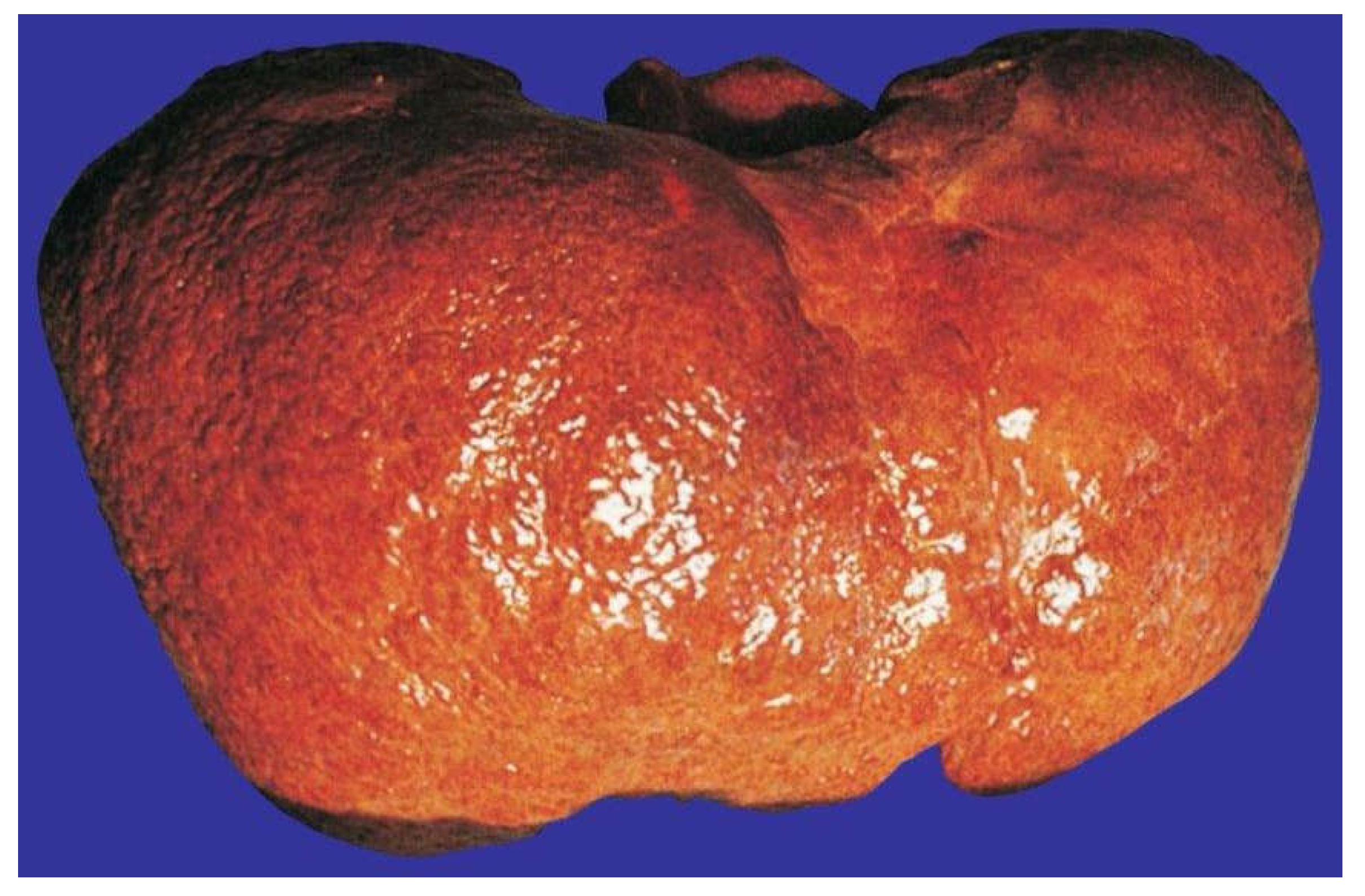{kind=link}
{kind=link}
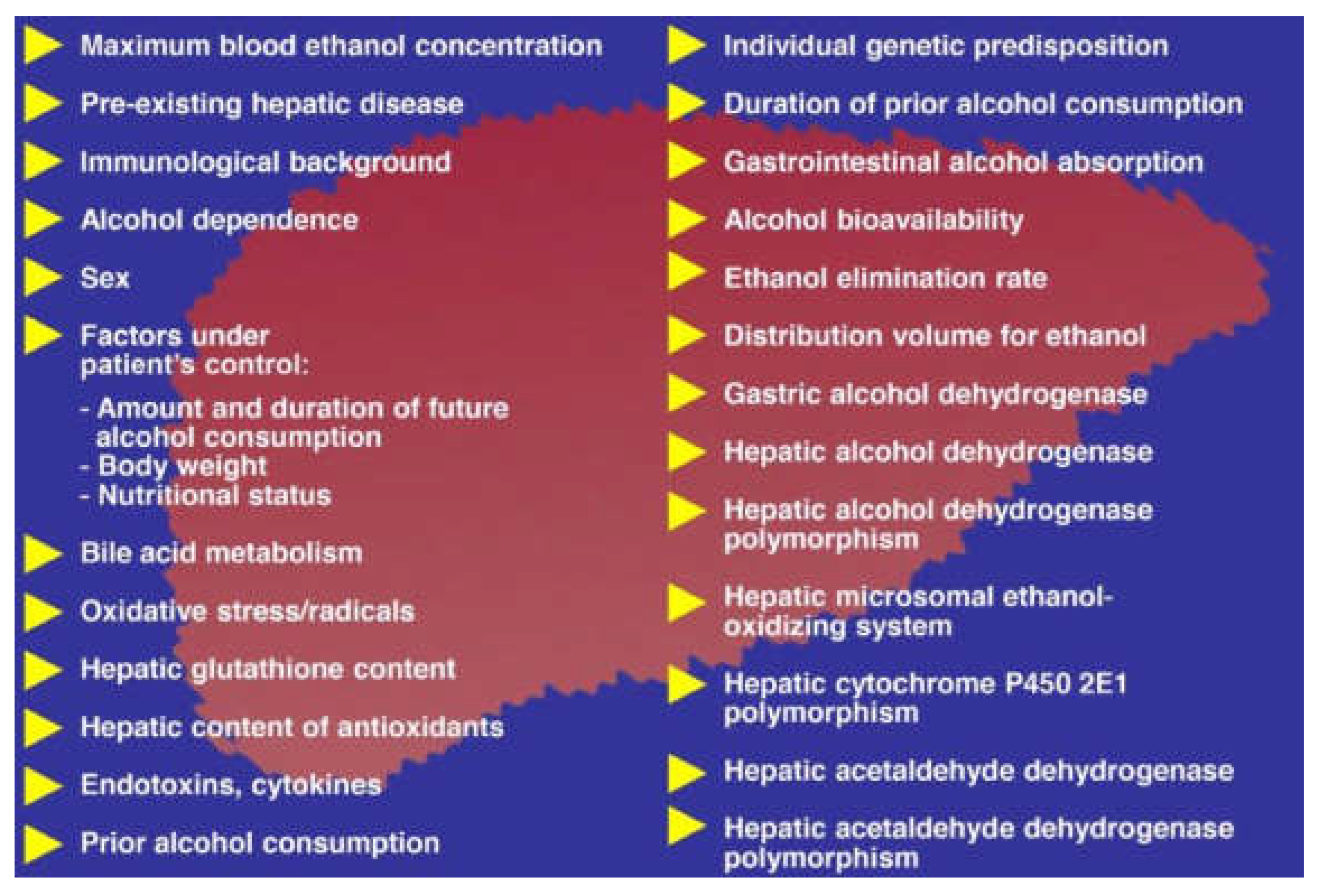{kind=link}
{kind=link}
| Characteristics | ADH | MEOS | Catalase |
|---|---|---|---|
| Intracellular localization | Cytosol | Endoplasmic reticulum | Peroxisomes |
| Co-factor | NAD+ | NADPH + H+ | N.D. |
| Co-substrate | None | Molecular oxygen | H2O2 |
| Reaction products | Acetaldehyde NADH + H+ | Acetaldehyde NADP+, H2O | Acetaldehyde H2O |
| Kinetics | |||
| Km (ethanol) | 0.5–2.0 mM | 7–11 mM | 0.6–10 mM |
| Km (O2) | N.D. | 8.3 μM | 50 μM |
| pH optimum | 11 | 6.9–7.5 | 5.5 |
| Inhibitory effect | |||
| Pyrazole (0.1 mM) | ++++ | 0 | (+) |
| Cyanide (0.1 mM) | N.D. | 0 | ++++ |
| Azide (0.1 mM) | 0 | 0 | ++++ |
| Substrate specificity | |||
| Methanol | ++ | ++ | ++++ |
| Ethanol | +++ | ++++ | ++++ |
| n-Propanol | ++++ | +++ | (+) |
| n-Butanol | ++++ | ++ | 0 |
| n-Pentanol | ++++ | + | 0 |
| i-Propanol | + | + | 0 |
| t-Butanol | 0 | + | 0 |
| Increase in activity following chronic ethanol consumption | 0 | ++++ | 0 |
| Enzyme isolation | + | + | + |
| Isoenzymes | + | + | + |
| Cytochrome P450 Isoenzyme | MEOS Activity/Cytochrome P450 |
|---|---|
| 1A2 | 10.90 |
| 2A6 | 3.75 |
| 2B6 | 2.89 |
| 2D6 | 0.70 |
| 2E1 | 11.51 |
| 3A4 | 3.38 |
| Year | In Short: Selected Details of MEOS, CYP2E1, and Related Aspects | Authors |
|---|---|---|
| 1968 | Discovery of ethanol oxidation by rat liver microsomal enzymes, now called the hepatic microsomal ethanol-oxidizing system (MEOS), which was different from ADH and catalase, using specific inhibitors, and induction by chronic alcohol consumption | Lieber and DeCarli [26] |
| 1970 | Detailed characterization of MEOS | Lieber and DeCarli [27] |
| 1970 | Dissociation of MEOS from NADPH oxidase | Lieber and DeCarli [28] |
| 1972 | Role of MEOS for ethanol metabolism in vivo | Lieber and DeCarli [29] |
| 1972 | Solubilization and purification of MEOS, and its separation from ADH and catalase by DEAE-cellulose ion exchange column chromatography, with the identification of CYP 450, reductase, and phospholipids as components of MEOS | Teschke et al. [30] |
| 1973 | Presence of induced MEOS in hepatic smooth and rough microsomes | Ishii et al. [31] |
| 1973 | Induced NADPH-cytochrome P450 reductase in hepatic smooth and rough microsomes | Joly et al. [32] |
| 1973 | Liver microsomal glycerophosphate acyltransferase activity following prolonged alcohol use | Joly et al. [33] |
| 1973 | Increased activity of glucose-6-phosphatase in liver microsomes due to prolonged alcohol consumption | Ishii et al. [34] |
| 1973 | A component of hepatic microsomes that is rich in CYP oxidizes ethanol | Mezey et al. [35] |
| 1974 | Broad substrate specificity of the microsomal alcohol-oxidizing system (MAOS) for methanol, ethanol, n-propyl alcohol, and n-butyl alcohol, inducible by chronic ethanol consumption | Teschke et al. [36] |
| 1974 | MEOS in acatalasemic mice | Lieber and DeCarli [37] |
| 1974 | Characterization of the solubilized, isolated, and purified MEOS | Teschke et al. [38] |
| 1974 | Enhanced liver injury by carbon tetrachloride after chronic ethanol use: its mechanism | Hasumura et al. [39] |
| 1975 | Role of dietary fat and caloric intake for the induction of MEOS by prolonged ethanol use | Joly and Hétu [40] |
| 1975 | Alteration of acetaldehyde metabolism after prolonged use of ethanol | Lieber et al. [41] |
| 1975 | Detailed description of the microsomal system oxidizing methanol, ethanol, n-propyl alcohol, and n-butyl alcohol as substrates | Teschke et al. [42] |
| 1975 | Isolation of the microsomal alcohol-oxidizing system with methanol, ethanol, n-propyl alcohol, n-butyl alcohol, and n-pentanol in normal and acatalasemic mice | Teschke et al. [43] |
| 1975 | Inhibition of the ethanol-induced cytochrome P450 by tetrahydrofurane | Ullrich et al. [44] |
| 1975 | Ethanol and acetaldehyde metabolism influenced by chronic alcohol use | Lieber et al. [45] |
| 1975 | Chronic alcohol consumption decreases acetaldehyde oxidation in liver mitochondria | Hasumura et al. [46] |
| 1976 | Characteristics of acetaldehyde oxidation in rat liver mitochondria | Hasumura et al. [47] |
| 1976 | Role of MEOS for ethanol metabolism in liver slices, using also n-butyl alcohol as inhibitor | Teschke et al. [48] |
| 1977 | Isolation and reconstitution of MEOS, with substrate specificity of the partially purified ethanol-induced CYP 2E1 for ethanol, n-propyl alcohol, and n-butyl alcohol, and characterization of the reconstituted MEOS | Ohnishi and Lieber [49] |
| 1977 | Involvement of hydroxyl radicals in MEOS | Cederbaum et al. [50] |
| 1977 | Spectral and catalytic properties of an ethanol-induced form of cytochrome P450 | Joly et al. [51] |
| 1977 | Details of MEOS isolation and reconstitution | Teschke et al. [52] |
| 1977 | Current status of MEOS characterization | Teschke et al. [53] |
| 1977 | Biochemical nature and role of MEOS | Teschke et al. [54] |
| 1977 | MEOS described in Methods in Enzymology | Lieber et al. [55] |
| 1978 | Role of superoxide and hydroxyl radicals in MEOS | Ohnishi and Lieber [56] |
| 1978 | Reconstitution of MEOS with highly purified microsomal cytochrome P450, reductase, and phospholipids, free of catalase and ADH | Miwa et al. [57] |
| 1978 | Photochemical action spectrum of MEOS | Fabry and Lieber [58] |
| 1979 | Induction of intestinal MEOS by chronic ethanol administration | Seitz et al. [59] |
| 1979 | Induction of MEOS by thyroid hormones | Moreno et al. [60] |
| 1979 | Prolonged ethanol use augments liver injury due to paracetamol (acetaminophen) | Teschke et al. [61] |
| 1980 | Enhanced chlorpromazine-induced cholestasis following chronic alcohol use | Teschke et al. [62] |
| 1980 | Existence and role of MEOS in deermice genetically lacking ADH | Burnett and Felder [63] |
| 1980 | Prolonged ethanol use ameliorates liver injury due to dimethylnitrosamine (DMN) | Gellert et al. [64] |
| 1980 | Oxidative demethylation of t-butyl alcohol in rat liver microsomes | Cederbaum and Cohen [65] |
| 1980 | Thyroid hormones induce MEOS activity and reduce ADH activity in rat liver | Moreno et al. [66] |
| 1981 | Microsomal system oxidizing isopropyl alcohol | Cederbaum et al. [67] |
| 1981 | Respective role of ethanol and carbohydrates for the induction of MEOS | Teschke et al. [68] |
| 1981 | Induction of pulmonary MEOS by chronic ethanol consumption | Seitz et al. [69] |
| 1981 | Induction of MEOS by propylthiouracil | Moreno et al. [70] |
| 1981 | Prolonged alcohol use potentiates experimental liver injury caused by paracetamol | Sato et al. [71] |
| 1982 | Liver enzymes metabolizing ethanol are altered in male rats treated by sex hormones | Teschke and Heymann [72] |
| 1982 | Purification and characterization of the ethanol-specific CYP 2E1 in rabbits metabolizing ethanol and aniline | Koop et al. [73] |
| 1982 | Substrate specificity of the purified ethanol-induced cytochrome P450 for methanol, ethanol, n-propyl alcohol, n-butyl alcohol, and aniline in rabbits | Morgan et al. [74] |
| 1982 | Induction of the ethanol-specific CYP 2E1 by benzene in rabbits | Ingelman-Sundberg and Hagbjörk [75] |
| 1982 | Increase of MEOS by a single dose of ethanol | Petersen et al. [76] |
| 1982 | Induction of MEOS by testosterone | Teschke and Wiese [77] |
| 1982 | Description of the isolated MEOS by electron microscopy and confirmation by method reproduction of the previous description of MEOS regarding its microsomal constituents and independency of ADH and catalase | Damgaard [78] |
| 1982 | Decreased hepatic vitamin A levels in patients with ALD | Leo and Lieber [79] |
| 1982 | Induction of colonic MEOS by chronic ethanol ingestion | Seitz et al. [80] |
| 1983 | Induction of MEOS by hexachlorobenzene | Teschke et al. [81] |
| 1983 | Interaction of ethanol with vitamin A | Leo and Lieber [82] |
| 1983 | Tumor incidence caused by dimethylnitrosamine is influenced by prolonged alcohol use | Teschke et al. [83] |
| 1983 | Liver injury caused by carbon tetrachloride is modified by ethanol administered acutely | Teschke et al. [84] |
| 1983 | Liver injury due to chlorpromazine, paracetamol, and dimethylnitrosamine is modified by prolonged use of alcohol | Teschke [85] |
| 1983 | MEOS and ethanol metabolism in baboons | Nomura et al. [86] |
| 1983 | The alcohol dehydrogenase (ADH) independent pathway of ethanol metabolism in deermice lacking ADH | Shigeta et al. [87] |
| 1984 | Induction of the ethanol-specific CYP by imidazole in rabbits | Koop et al. [88] |
| 1984 | Induction of the ethanol-specific CYP 2E1 by isoniazid | Gadeholt [89] |
| 1984 | Circadian rhythm of MEOS | Sturtevant and Garber [90] |
| 1984 | Induction of the ethanol-specific CYP 2E1 by pyrazole in rabbits | Ingelman-Sundberg and Jörnvall [91] |
| 1984 | Formation of hydroxyl radical and oxidation of ethanol by CYP 2E1: studies of their mechanisms | Ingelman-Sundberg and Johansson [92] |
| 1984 | Reduced liver levels of vitamin A in humans and rats following drug treatment | Leo et al. [93] |
| 1985 | Induction of the ethanol-specific CYP 2E1 by trichloroethylene, acetone, pyrazole, and isoniazid in rabbit liver microsomes | Koop et al. [94] |
| 1985 | Involvement of the ethanol-specific CYP 2E1 in the microsomal metabolism of dimethylnitrosamine in rats, rabbits, mice, and guinea pigs | Yang et al. [95] |
| 1985 | Involvement of the ethanol-specific CYP in the microsomal metabolism of carbon tetrachloride in rabbits | Johansson and Ingelman-Sundberg [96] |
| 1985 | Ethanol-inducible CYP 2E1 identified as metabolizing acetone and acetol | Koop and Casazza [97] |
| 1985 | Details of liver microsomal CYP induced by isoniazid in the rat | Ryan et al. [98] |
| 1985 | Mixed function oxidation in deermice lacking alcohol dehydrogenase: Modification by acute alcohol administration and prolonged consumption of alcohol | Gellert et al. [99] |
| 1986 | Studies in deermice containing or missing ADH: Metabolic interactions of ethanol oxidation and mixed-function oxidation | Gellert et al. [100] |
| 1986 | Chronic administration of sex hormones and alcohol in female rats and the effect on liver enzymes metabolizing ethanol | Teschke et al. [101] |
| 1986 | Microsomal ethanol-oxidizing system of the liver: Biochemical nature and clinical aspects | Teschke [102] |
| 1986 | Drugs, retinol, and the relevance of their interactions | Leo et al. [103] |
| 1986 | Isoniazid and ethanol: induction of the same microsomal CYP isozyme 3a | Ryan et al. [104] |
| 1986 | Ethanol-inducible human liver demethylase for N-nitrosodimethylamine | Wrighton et al. [105] |
| 1986 | Ethanol-inducible CYP isozyme in rabbit nasal and kidney microsomes | Ding et al. [106] |
| 1986 | Hydroxylation of acetone catalyzed by ethanol- and acetone-inducible CYP in hepatic microsomes and reconstituted membranes | Johansson et al. [107] |
| 1986 | Complementary DNA and protein sequences of ethanol-inducible CYPs. A study in rats and humans | Song et al. [108] |
| 1987 | Induction of cytochrome P-450j: A study in the spontaneously diabetic BB rat, a strain in which about half of the animals develop insulin-dependent diabetes | Bellward et al. [109] |
| 1987 | Purification and characterization of human liver CYP 2E1 | Lasker et al. [110] |
| 1987 | Hepatic microsomal CYP 2E1 inducible by ethanol in rabbits: Details of cDNA and derived amino acid sequence | Khani et al. [111] |
| 1987 | Role of MEOS for interactions with other drugs, carcinogens, and vitamins | Lieber et al. [112] |
| 1987 | Pathways contributing to ethanol metabolism: ethanol-metabolizing pathways in deermice. A study on the estimation of flux calculated from isotope effects | Alderman et al. [113] |
| 1988 | Ethanol-inducible CYP 2E1 expressed in the centrilobular region of the rat liver | Ingelman-Sundberg et al. [114] |
| 1988 | Obesity is considered as a risk factor for drug-induced organ injury: Increased hepatic CYP levels and MEOS activity in the obese overfed rat | Salazar et al. [115] |
| 1988 | Acetaldehyde adducts formed with ethanol-inducible CYP 2E1 in vivo | Behrens et al. [116] |
| 1988 | CYP 2E1 in rabbit olfactory mucosa: its induction by ethanol and acetone | Ding et al. [117] |
| 1988 | Ethanol-inducible CYP 2E1: a study on molecular regulation in hamsters | Kubota et al. [118] |
| 1988 | Metabolism of benzene in microsomes obtained from rat and rabbit liver, and the role of CYP 2E1 induced by ethanol, acetone, and benzene | Johansson and Ingelman-Sundberg [119] |
| 1988 | Ligand-dependent maintenance of ethanol-inducible CYP: an experimental study using primary rat hepatocyte cell cultures | Eliasson et al. [120] |
| 1988 | Hepatic microsomal ethanol-inducible CYP 2E1 and its intralobular distribution of in liver | Tsutsumi et al. [121] |
| 1988 | Prolonged alcohol use enhances oxygen radical dependent inactivation of metabolic enzymes by liver microsomes | Dicker and Cederbaum [122] |
| 1989 | Induction and tissue-specific expression of rabbit CYP 2E1 genes | Porter et al. [123] |
| 1989 | The intralobular distribution of ethanol-inducible CYP 2E1; an experimental study in rat liver and a clinical analysis in human liver | Tsutsumi et al. [124] |
| 1990 | Ethanol-inducible CYP 2E1 and its regional distribution of in the central nervous system: an experimental study in rats | Hansson et al. [125] |
| 1990 | Modification of hepatic CYP 2E1 by pituitary hormones in rats and mice | Hong et al. [126] |
| 1990 | Ethanol-inducible CYP 2E1: multiple mechanisms are involved in its regulation | Koop and Tierney [127] |
| 1990 | Lymphocytes from patients with poorly controlled insulin-dependent diabetes exhibit increased CYP 2E1 | Song et al. [128] |
| 1990 | Induction of rat hepatic CYP 2E1 by pyridine | Kim et al. [129] |
| 1990 | Solvents enhance the translational efficiency in the course of CYP 2E1 induction | Kim et al. [130] |
| 1990 | CYP 2E1 influences the interactions of ethanol with enflurane metabolism and their toxicity | Tsutsumi et al. [131] |
| 1990 | CYP 2E1 is involved in nitrosamine metabolism and regulation | Yang et al. [132] |
| 1990 | Chlorzoxazone hydroxylation is a specific probe for CYP 2E1 in human liver | Peter et al. [133] |
| 1990 | Localization of ethanol-inducible CYP 2E1 assessed by immunohistochemistry in the alimentary tract of rats | Shimizu et al. [134] |
| 1991 | Post-translational reduction of CYP 2E1 by CCl4 | Sohn et al. [135] |
| 1991 | Role of hormones in the phosphorylation and degradation of CYP 2B1 and 2E1: a study in in isolated rat hepatocytes | Johansson et al. [136] |
| 1991 | Dietary lipids and carbohydrates modify the levels of CYP 2E1 in microsomes obtained from rat liver | Yoo et al. [137] |
| 1991 | CYP 2E1 is induced in the in the experimental obese rat model | Raucy et al. [138] |
| 1991 | Acetaldehyde as another substrate for ethanol-inducible CYP 2E1 | Terelius et al. [139] |
| 1991 | Identification and induction of CYP 2E1 in Kupffer cells of an experimental model of rats | Koop et al. [140] |
| 1991 | Genetic polymorphism in the 5′-flanking region change transcriptional regulation of the CYP 2E1 gene: a study in humans | Hayashi et al. [141] |
| 1992 | Interaction of ethanol with β-carotene: Delayed blood clearance and evidence of increased liver injury | Leo et al. [142] |
| 1992 | Intracellular degradation of CYP 2E1 is controlled by hormones and substrates | Eliasson et al. [143] |
| 1992 | Distribution the ethanol-inducible CYP 2E1 in the pancreas of rats fed ethanol combined with high fat or low fat diet | Sohda et al. [144] |
| 1992 | Oxidative and reductive metabolic pathways by CYP 2E1 | Koop [145] |
| 1992 | CYP 2E1 and 2A6 enzymes are the preferred catalysts for metabolic activation of N-nitrosodialkylamines and nitrosamines in the microsomes of human liver | Yamazaki et al. [146] |
| 1993 | Inhibition of chlorzoxazone metabolism by a single dose of disulfiram, and its potential role as a clinical probe for CYP 2E1 | Kharasch et al. [147] |
| 1993 | Enflurane defluorination catalyzed by CYP 2E1 in microsomes of human liver | Thummel et al. [148] |
| 1993 | Human CYP 2E1 stability in HepG2 cells | Day et al. [149] |
| 1993 | DraI and RsaI restriction fragment length polymorphisms analyzed in a study from Finland | Hirvonen et al. [150] |
| 1993 | Pathogenesis of alcoholic liver disease and the role of CYP 2E1 | Morimoto et al. [151] |
| 1993 | NADPH- and NADH-dependent production of superoxide and hydroxyl radical is enhanced in hepatic microsomes obtained following prolonged alcohol use | Rashba-Step et al. [152] |
| 1993 | Increased enzyme synthesis is responsible for the in vivo induction of hepatic CYP 2E1 | Tsutsumi et al. [153] |
| 1993 | Formation of 19(S)-, 19(R)-, and 18(R)-hydroxyeicosatetraenoic acids by alcohol-inducible CYP 2E1 | Laethem et al. [154] |
| 1993 | Induction of CYP 2E1 during prolonged alcohol use is due to the transcription of the CYP 2E1 gene when blood alcohol concentrations are high | Badger et al. [155] |
| 1993 | Contribution of cytochrome P-450s to MEOS: assessed by a specific and sensitive assay of MEOS activity using HPLC with fluorescence labeling | Kunitoh et al. [156] |
| 1993 | Levels of CYP 1A2 and CYP 2E1, and their related monooxygenase activities in human liver obtained as surgical samples | Lucas et al. [157] |
| 1993 | CYP 2E1 induction during chronic ethanol exposure occurs by a two-step mechanism, depending on blood alcohol levels: a study in rats | Ronis et al. [158] |
| 1993 | CYP 2E1 is the preferred enzyme catalyzing the defluorination of sevoflurane, isoflurane, and methoxyflurane in human liver microsomes | Kharasch et al. [159] |
| 1993 | Inhibition of CYP 2E1 by ethanol is caused in the human liver by corresponding increase in encoding messenger RNA | Takahashi et al. [160] |
| 1993 | Use of 4-nitrophenol as an in vitro substrate probe was validated for human liver CYP 2E1 | Tassaneeyakul et al. [161] |
| 1994 | Alcohol-derived radicals and their spin trapping in liver microsomes and reconstituted systems | Albano et al. [162] |
| 1994 | Ethanol augments the content and activity of human CYP 2E1 in a transduced HEPG2 cell line | Carrocio A [163] |
| 1994 | Significance of tissue-specific expression and methylation of the human CYP 2E1 gene | Botto et al. [164] |
| 1994 | Role of genetic CYP 2E1 polymorphism for the development of alcoholic liver disease | Tsutsumi et al. [165] |
| 1994 | Ethnic variation in the CYP 2E1 gene: Polymorphism analysis of 695 African-Americans, European-Americans and Taiwanese | Stephens et al. [166] |
| 1994 | Relationship between CYP 2E1 and acetone catabolism in rats as studied with the inhibitor diallyl sulfide | Chen et al. [167] |
| 1994 | Association between restriction fragment-length polymorphism of the human CYP 2E1 gene and susceptibility to alcoholic liver cirrhosis | Maezawa et al. [168] |
| 1994 | Involvement of CYP 2E1 in the (omega-1)-hydroxylation of lauric acid in rat liver microsomes | Amet et al. [169] |
| 1994 | CYP 2E1 induction by ethanol in a rat hepatoma FGC-4 cell model | McGehee et al. [170] |
| 1994 | Piperine modifies the expression of P4502E1, P4502B, and P4501A in rats | Kang et al. [171] |
| 1994 | Restriction fragment-length polymorphism of the human CYP 2E1 gene and susceptibility to lung cancer: Possible relevance to low smoking exposure | Uematsu et al. [172] |
| 1994 | Differences of regulation and expression of the human CYP 2E1 gene due to the RsaI polymorphism in the 5’ flanking region | Watanabe et al. [173] |
| 1995 | An RsaI polymorphism in the CYP 2E1 gene does not affect lung cancer risk in a Japanese population | Watanabe et al. [174] |
| 1995 | Ethanol induces CYP 2E1 by a mechanism involving protein stabilization | Roberts et al. [175] |
| 1995 | Renal tumorigenicity of 1,1-dichloroethene in mice: the role of male-specific expression of CYP 2E1 in the renal bioactivation of 1,1-dichloroethene | Speerschneider and Dekant [176] |
| 1995 | Intestinal toxicity of acrylonitrile: in vitro metabolism by intestinal CYP 2E1 | Subramanian and Ahmed [177] |
| 1995 | Stable expression of human CYP 2E1 in V79 Chinese hamster cells | Schmalix et al. [178] |
| 1995 | Lacking association of polymorphism at the CYP 2E1 locus with alcoholic liver disease in Caucasian men | Carr et al. [179] |
| 1995 | Modulation of experimental alcoholic liver injury by inhibitors of CYP 2E1 | Morimoto et al. [180] |
| 1995 | Genetic polymorphism of CYP 1A1, 2D6 and 2E1: Regulation and toxicological significance | Rannug et al. [181] |
| 1995 | Genetic polymorphism of CYP 2E1 and risk of alcoholic liver disease in Caucasians | Pirmohamed et al. [182] |
| 1995 | CYP 2E1 genotype and chlorzoxazone metabolism in healthy and alcoholic Caucasians | Lucas et al. [183] |
| 1995 | Decreased CYP 2E1 as assessed by the rate of chlorzoxazone hydroxylation in alcoholics during the withdrawal phase | Lucas et al. [184] |
| 1995 | Respective roles of CYP 2E1 and CYP A2 in chlorzoxazone, and ethanol metabolism in mammalian liver microsomes | Mishin et al. [185] |
| 1995 | CYP 2E1 is not the sole catalyst of chlorzoxazone hydroxylation in rat liver microsomes | Jayyosi et al. [186] |
| 1995 | Selectivity of CYP 2E1 in catalyzing chlorzoxazone 6-hydroxylation | Yamazaki et al. [187] |
| 1995 | Insulin down-regulates CYP 2B and 2E expression at the posttranscriptional level in the rat hepatoma cell line | De Waziers et al. [188] |
| 1995 | Evidence for a tissue-specific induction of cutaneous CYP 2E1 by dexamethasone | Sampo et al. [189] |
| 1995 | Ethanol oxidizing enzymes: Roles in alcohol metabolism and alcoholic liver disease | Crabb [190] |
| 1995 | CYP 2E1 changes in rat liver, kidney and lung microsomes after prolonged alcohol application, either orally or by inhalation | Zerilli et al. [191] |
| 1996 | Microsomal ethanol oxidizing system activity by human hepatic cytochrome P-450s and involvement of CYP 1A2, 2A6, 2B6, 2D6, 2E1, and 3A4 | Asai et al. [192] |
| 1996 | High inducibility of mouse renal CYP 2E1 gene by tobacco smoke and its possible effect on DNA single strand breaks | Seree et al. [193] |
| 1996 | Effects of diet and ethanol on the expression and localization of CYP 2E1 and 2C7 in the colon of male rats | Hakkak et al. [194] |
| 1996 | Induction of CYP 2E1 by ethanol in rat Kupffer cells | Koivisto et al. [195] |
| 1996 | Human CYP 2E1: From genotype to phenotype | Carriere et al. [196] |
| 1996 | Expression, catalytic activity, and inducibility of CYP 2E1 in the rat central nervous system | Tindberg and Ingelman-Sundberg [197] |
| 1997 | Enzymatic degradation of chlorzoxazone by hepatic microsomes from humans and 10 other mammalian species | Court et al. [198] |
| 1997 | Regulation of the hepatic CYP 2E1 gene during prolonged ethanol exposure: Lack of an ethanol response element in the proximal 5′-flanking sequence | McGehee et al. [199] |
| 1997 | Immunohistochemical determination of hepatic CYP 2E1 in formalin-fixed, paraffin-embedded sections | Cohen et al. [200] |
| 1997 | Effect of fatty acids and ketone bodies on CYP 2B, 4A, and 2E1 expression in primary cultured rat hepatocytes | Zangar and Novak [201] |
| 1997 | Ethanol metabolism in the brain | Zimatkin and Deitrich [202] |
| 1997 | Inhibition of CYP 2E1 expression by 2-(allylthio) pyrazine, a potential chemoprotective agent, and considerations on hepatoprotective effects | Kim et al. [203] |
| 1997 | Insulin effects on CYP 2E1, 2B, 3A, and 4A expression in primary cultured rat hepatocytes | Woodcroft and Novak [204] |
| 1997 | Lipid peroxidation, CYP 2E1 and arachidonoid acid metabolism in alcoholic liver disease in rats | French et al. [205] |
| 1997 | Chlormethiazole inhibition of CYP 2E1 as assessed by chlorzoxazone hydroxylation in humans | Gebhardt et al. [206] |
| 1998 | CYP 2E1 activity as assessed by chlorzoxazone hydroxylation: studies in patients with diabetes and obesity | Lucas et al. [207] |
| 1998 | Expression of CYP 2E1 in human liver: Assessment by mRNA, genotype, and phenotype | Powell et al. [208] |
| 1998 | Increased hepatic CYP 2E1 in patients with nonalcoholic steatohepatitis | Weltman et al. [209] |
| 1998 | Respective roles of human CYP 2E1 and 3A4 in the hepatic microsomal ethanol oxidizing system | Salmela et al. [210] |
| 1998 | Microsomal acetaldehyde oxidation is negligible in the presence of ethanol | Wu et al. [211] |
| 1998 | Selective inhibition of CYP 2E1 in vivo and in vitro with trans-1, 2-dichloroethylene | Matthews et al. [212] |
| 1998 | CYP 2E1 and its catalytic activity in rat testis | Jiang et al. [213] |
| 1998 | CYP 2E1 and 1A1 in the rat pancreas | Kessova et al. [214] |
| 1998 | CYP 2E1 is present in the rat pancreas and induced by prolonged alcohol consumption | Norton et al. [215] |
| 1998 | Polyenylphosphatidylcholine opposes the increase of CYP 2E1 by ethanol, and corrects the iron-induced decrease | Aleynik et al. [216] |
| 1998 | Involvement of CYP 2E1 in the (omega-1)-hydroxylation of oleic acid in human and rat liver microsomes | Adas et al. [217] |
| 1998 | Chlorzoxazone pharmacogenetics, a potential marker of hepatic CYP 2E1 in humans | Mishin et al. [218] |
| 1998 | Inhibition of CYP 2E1 by chlormethiazole as measured by chlorzoxazone pharmacokinetics in patients with alcoholism and in healthy volunteers | Eap et al. [219] |
| 1998 | Regulation of rabbit CYP 2E1 expression in HepG2 cells by insulin and thyroid hormones | Peng and Coon [220] |
| 1998 | CYP 2E1 inducibility and hydroxyethyl radical formation among alcoholics | Dupont et al. [221] |
| 1999 | Alcohol, vitamin A, and beta-carotene: adverse interactions, including hepatotoxicity and carcinogenicity | Leo and Lieber [222] |
| 1999 | Expression of CYP 2E1 by human monocyte-derived macrophages | Hutson and Wickramasinghe [223] |
| 1999 | Carbon monoxide, cigarette smoking, and CYP 2E1 activity | Benowitz et al. [224] |
| 1999 | Chlorzoxazone, a selective probe for phenotyping CYP 2E1 in humans | Lucas et al. [225] |
| 2001 | Effects of alcohol and diallylsulphide on CYP 2E1 activity in humans: a phenotyping study using chlorzoxazone | Loizou and Cocker [226] |
| 2001 | Inhibition of CYP 2E1 with natural agents may be a feasible strategy for minimizing liver injury by ethanol | McCarty [227] |
| 2001 | Ethanol and oxidative stress | Sun et al. [228] |
| 2002 | Effect of chronic disulfiram administration on CYP 1A2, CYP 2C19, CYP 2D6, CYP 2E1, and N-acetyltransferase in healthy humans | Frye and Branch [229] |
| 2003 | Rapid determination of enzyme activities of recombinant human CYPs, human liver microsomes, and hepatocytes | Ghosal et al. [230] |
| 2004 | CYP 2E1: biochemistry, toxicology, regulation, and function in alcoholic liver injury | Kessova and Cederbaum [231] |
| 2004 | Robustness of chlorzoxazone as an in vivo measure of CYP 2E1 activity | Ernstgard et al. [232] |
| 2006 | Effect of high-dosed aspirin on CYP 2E1 in healthy humans measured using chlorzoxazone as a probe | Park et al. [233] |
| 2008 | CYP 2E1 and oxidative liver injury caused by alcohol | Choi et al. [234] |
| 2010 | CYP-mediated differential oxidative modification of proteins: Albumin, apolipoprotein E, and CYP 2E1 as targets | Wellman and Siest [235] |
| 2014 | Association studies of CYP, family 2, subfamily E, and polypeptide 1 (CYP 2E1) gene polymorphisms with acute rejection in kidney transplantation recipients | Kim et al. [236] |
| 2014 | Pathogenesis of alcoholic liver disease: Significance of oxidative metabolism | Ceni et al. [237] |
| 2016 | Resveratrol pretreatment affects CYP 2E1 activity in healthy volunteers | Bedada and Neerati [238] |
| 2017 | Effect of piperine on CYP 2E1 enzyme activity in healthy volunteers | Bedada and Boga [239] |
| 2017 | The role of human CYP 2E1 in liver inflammation and fibrosis | Xu et al. [240] |
| 2017 | CYP 2E1 is involved in aging-related kidney damage in mice through increased nitroxidative stress | Abdelmegeed et al. [241] |
| 2018 | Vinyl chloride, CYP 2E1, and liver injury | Fujiwara [242] |
| 2018 | Vinyl chloride, diet, and liver injury | Lang et al. [243] |
| First Hit. | The first hit is dependent on ADH and occurs at low alcohol levels through the generation not only of NADH + H+ leading to an increased NADH + H+/NAD+ ratio, which stimulates hepatic fatty acid synthesis [22] and increases α-glycerophosphate-trapping fatty acids [22,33], but also of acetaldehyde, which impairs hepatic mitochondrial functions including hepatic mitochondrial fatty acid oxidation [22]. This first hit fully explains at least in part the development of alcoholic fatty liver. |
| Second Hit. | The second hit is classified as a transition from alcoholic fatty liver to alcoholic steatohepatitis, most likely triggered by the increased production of acetaldehyde via MEOS [22,23] and of reactive oxygen species (ROS) with its capacity for irreversible covalently binding to cellular macromolecules, including membrane proteins and phospholipids [45,50,56,92,152,205,231,234,237,244,245,246,247,248,249,250,251,252]. These injurious alterations at the molecular and cellular level cause some necrosis, apoptosis, and inflammatory cells in the fatty liver, justifying the term alcoholic steatohepatitis, as it includes toxic hepatitis in steatosis [25]. Further stages are characterized by perisinusoidal and pericentral fibrosis due to participation of non-hepatocytes such as Kupffer cells, stellate cells, and sinus endothelial cells. Mediators such growth factors, interferons, interleukins, tumor necrosis factor and endotoxins, as well as hepatic iron, are considered as possible active promoters of liver injury, but considering the multiplicity of proposed mediators, it is difficult to predict how they interact with each other and modify the course of liver injury. |
| Third Hit. | The third hit initiates a more severe liver injury stage, whereby alcoholic steatohepatitis is the precursor in most, but certainly not all patients with alcoholic hepatitis. Steatosis is no more a characteristic feature, but is now replaced by necrosis, apoptosis, and inflammation. At this stage, injury becomes more severe and presents with more fibrosis and as a self-perpetuating process, immunity aspects gain additional relevance, because alcohol modifies the innate and adapted immune system, which may explain the individual differences of susceptibility for ALD. With the third hit, the disease may approach a point of no return. |
| Fourth Hit. | The fourth hit is dominated by increased fibrosis, due to increased collagen formation. This allows for a clinically unrecognizable transition from alcoholic hepatitis with fibrosis to irreversible cirrhosis. However, AC can also develop without ASH or AH. |
| Fifth Hit. | In rare cases, a fifth hit initiates the development of a hepatocellular carcinoma (HCC), mostly occurring in patients with cirrhosis. This final hit scenario of carcinogenesis is triggered by acetaldehyde and ROS through the generation of DNA adducts, which promote mutagenesis, and interference with methylation, synthesis, and repair of DNA. Suggested is a possible role of SIRT1. These overall events will enhance AHCC susceptibility, keeping in mind that ethanol itself is not a carcinogenetic chemical. |
| Selected Potentially Toxic Metabolites and Reactive O2-Species due to Hepatic Ethanol Degradation |
|---|
| Acetaldehyde C2H4O |
| Ethoxy radical CH3CH2O |
| Hydroxyethyl radical CH3C(·)HOH |
| Acetyl radical CH3CHO |
| Singlet radical 1O2 |
| Superoxide radical HO2 |
| Hydrogen peroxide H2O2 |
| Hydroxyl radical HO |
| Alkoxyl radical RO |
| Peroxyl radical ROO |
| Lipid peroxides |
| Study Cohort | Alcoholic Fatty Liver | Controls | Significance |
|---|---|---|---|
| Patients | |||
| Serum GGT (U/L) | 195.0 ± 93.7 | 13.7 ± 2.0 | p < 0.025 |
| Liver GGT | |||
| (U/g wet weight) | 4.78 ± 0.4 | 1.91 ± 0.2 | p < 0.025 |
| (U/g protein) | 35.9 ± 16.1 | 16.4 ± 6.6 | p < 0.025 |
| Animals | |||
| Serum GGT (U/L) | 4.41 ± 1.64 | 2.19 ± 0.31 | p < 0.025 |
| Liver GGT | |||
| (U/g wet weight) | 0.14 ± 0.06 | 0.07 ± 0.03 | p < 0.001 |
| (U/g protein) | 1.19 ± 0.23 | 0.79 ± 0.19 | p < 0.0.25 |
| (U/100 g body weight) | 0.80 ± 0.28 | 0.34 ± 0.09 | p < 0.001 |
| Patient with AFL | Degree of Steatosis | AST (U/L) | ALT (U/L) | Ratio AST/ALT | GDH (U/L) |
|---|---|---|---|---|---|
| 1 | 50% | 12.4 | 17.7 | 0.70 | 53.9 |
| 2 | 60% | 100.7 | 22.5 | 4.47 | 35.8 |
| 3 | 60% | 50.3 | 19.9 | 2.52 | 16.8 |
| 4 | 80% | 54.7 | 9.2 | 5.94 | 7.7 |
| 5 | 10–15% | 20.6 | 31.7 | 0.65 | 2.0 |
| 6 | 50% | 25.6 | 33.9 | 0.76 | 7.9 |
| 7 | 60–70% | 61.4 | 62.4 | 0.98 | 9.1 |
| 8 | 60–70% | 11.6 | 7.9 | 1.47 | 1.8 |
| 9 | 30–40% | 33.2 | 61.1 | 0.54 | 4.1 |
| 10 | 80% | 53.6 | 19.0 | 2.82 | 1.9 |
| 11 | 80–90% | 34.6 | 46.8 | 0.74 | 9.2 |
| 12 | 30% | 16.0 | 30.6 | 0.52 | 3.3 |
| 13 | 30–40% | 32.9 | 19.9 | 1.65 | 4.8 |
| 14 | 20–30% | 11.3 | 33.8 | 0.33 | 7.3 |
| 15 | 50–60% | 25.7 | 62.2 | 0.41 | 7.2 |
| 16 | 10% | 70.1 | 19.3 | 3.63 | 7.2 |
| 17 | 10–15% | 9.2 | 6.7 | 1.37 | 0.8 |
| 18 | 50% | 26.1 | 68.1 | 0.38 | 7.5 |
| 19 | 50% | 10.9 | 9.2 | 1.19 | 2.1 |
| Means ± SEM | 34.8 ± 5.7 | 30.2 ± 4.6 | 1.53 ± 1.51 | 9.9 ± 3.0 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teschke, R. Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects. Biomedicines 2018, 6, 106. https://doi.org/10.3390/biomedicines6040106
Teschke R. Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects. Biomedicines. 2018; 6(4):106. https://doi.org/10.3390/biomedicines6040106
Chicago/Turabian StyleTeschke, Rolf. 2018. "Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects" Biomedicines 6, no. 4: 106. https://doi.org/10.3390/biomedicines6040106