Evidence for the Involvement of the Master Transcription Factor NF-κB in Cancer Initiation and Progression

Abstract
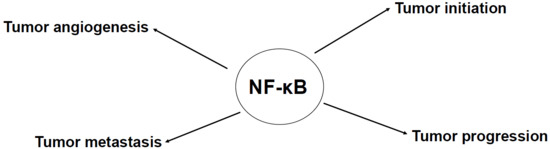
:
1. Introduction
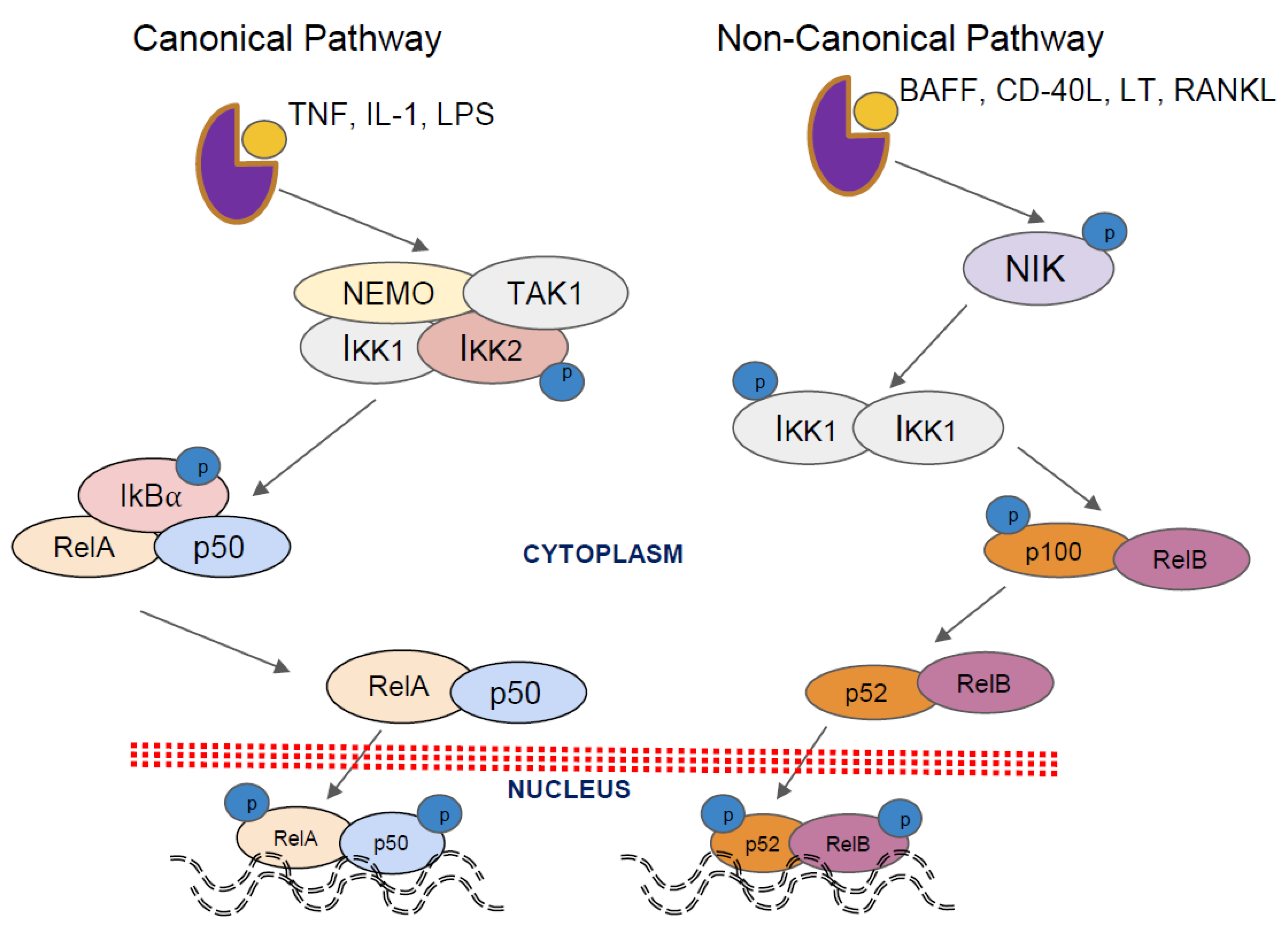
2. Activation Pathways
3. NF-κB Activation by Chemotherapeutic Agents
4. NF-κB Activation by Radiation
5. NF-κB Signaling Pathway in Inflammation
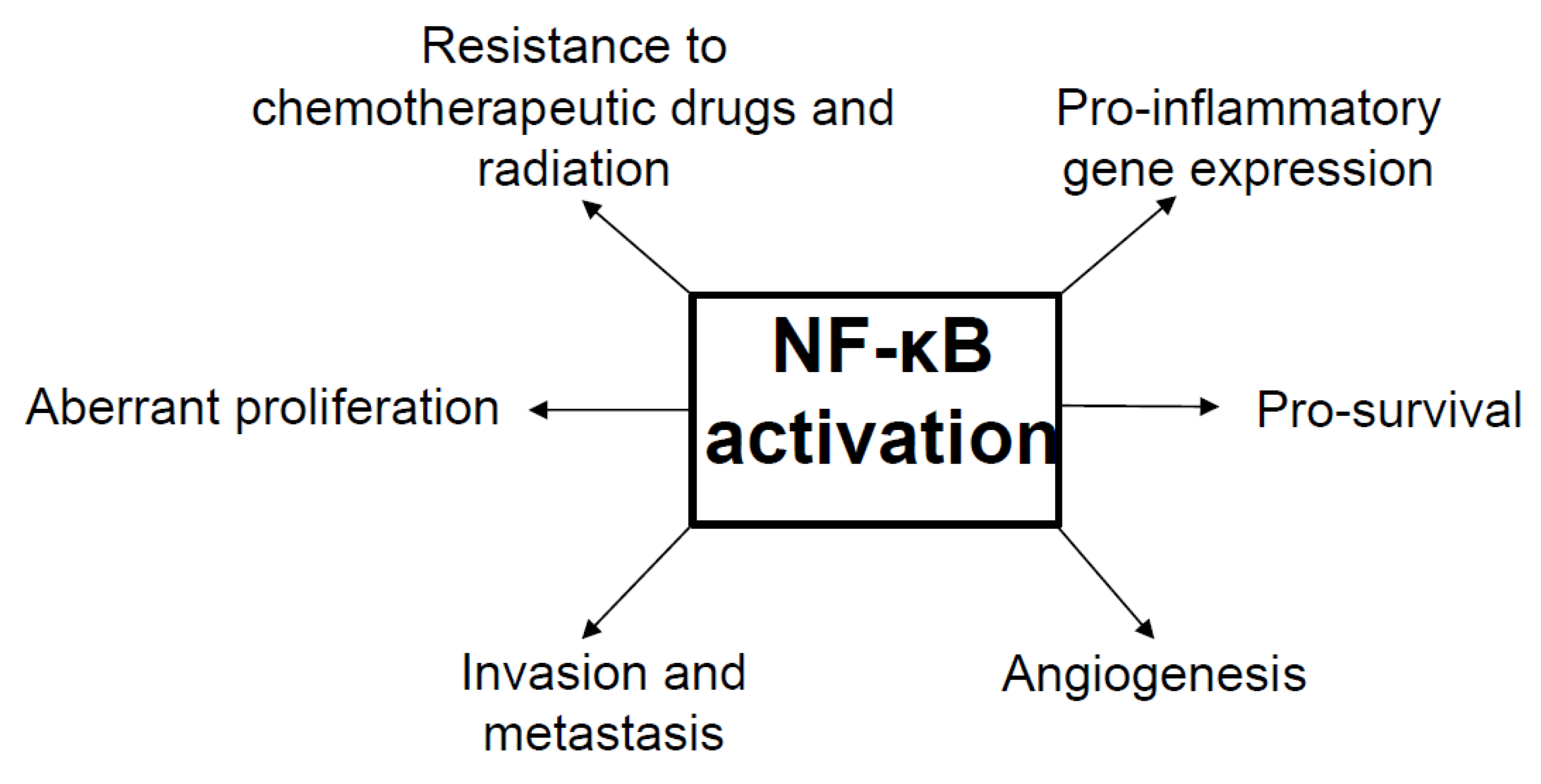
6. NF-κB as a Tumor Promoter and Suppressor
7. Inhibitors of NF-κB Function and Selected Pharmacological Strategies to Block NF-κB Function
7.1. Inhibitors That Act Upstream of the IKK Complex
7.2. IKK Inhibitors
7.3. Proteasomal Degradation of IκBα
7.4. NF-κB DNA Binding
7.5. Non-Steroidal Anti-Inflammatory Drugs and Antioxidants
7.6. Gene Therapy Approaches
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sethi, G.; Sung, B.; Aggarwal, B.B. Nuclear factor-kappab activation: From bench to bedside. Expe. Biol. Med. 2008, 233, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Tergaonkar, V. Potential pharmacological control of the NF-κB pathway. Trends Pharmacol. Sci. 2009, 30, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Antonucci, L.; Karin, M. Targeting inflammation in cancer prevention and therapy. Cancer Prev. Res. 2016, 9, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-κB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.M.; Hay, H.S.; Lee, M.H.; Goh, J.N.; Tan, T.Z.; Sen, Y.P.; Lim, S.W.; Yousef, E.M.; Ong, H.T.; Thike, A.A.; et al. Dead-box helicase dp103 defines metastatic potential of human breast cancers. J. Clin. Investig. 2014, 124, 3807–3824. [Google Scholar] [CrossRef] [PubMed]
- Tergaonkar, V. NF-κB pathway: A good signaling paradigm and therapeutic target. Int. J. Biochem. Cell Biol. 2006, 38, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Maubach, G.; Schmadicke, A.C.; Naumann, M. Nemo links nuclear factor-kappab to human diseases. Trends Mol. Med. 2017, 23, 1138–1155. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.B.; Silva, R.; Ferreira, G.M.; Abdelhay, E. NF-κB: Two sides of the same coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Colombo, F.; Zambrano, S.; Agresti, A. NF-κB, the importance of being dynamic: Role and insights in cancer. Biomedicines 2018, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and ikk function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Scheidereit, C. Ikappab kinase complexes: Gateways to NF-κB activation and transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Shanmugam, M.K.; Ramachandran, L.; Kumar, A.P.; Tergaonkar, V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012, 32, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chai, E.Z.; Siveen, K.S.; Shanmugam, M.K.; Arfuso, F.; Sethi, G. Analysis of the intricate relationship between chronic inflammation and cancer. Biochem. J. 2015, 468, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Tergaonkar, V. NF-κB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis 2009, 14, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Rogovskii, V.S. The linkage between inflammation and immune tolerance: Interfering with inflammation in cancer. Curr. Cancer Drug Targets 2017, 17, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Derudder, E.; Dejardin, E.; Pritchard, L.L.; Green, D.R.; Korner, M.; Baud, V. Relb/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: Critical roles for p100. J. Biol. Chem. 2003, 278, 23278–23284. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, O.; Naumann, M. NF-κB signaling in gastric cancer. Toxins 2017, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good cop, bad cop: The different faces of NF-κB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Durand, J.K.; Baldwin, A.S. Targeting ikk and NF-κB for therapy. Adv. Protein Chem. Struct. Biol. 2017, 107, 77–115. [Google Scholar] [CrossRef] [PubMed]
- Anto, R.J.; Mukhopadhyay, A.; Shishodia, S.; Gairola, C.G.; Aggarwal, B.B. Cigarette smoke condensate activates nuclear transcription factor-kappab through phosphorylation and degradation of IκBα: Correlation with induction of cyclooxygenase-2. Carcinogenesis 2002, 23, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, P.; Catalano, D.; Szabo, G. Alcohol-induced regulation of nuclear regulatory factor-kappa beta in human monocytes. Alcohol. Clin. Exp. Res. 1997, 21, 988–994. [Google Scholar] [PubMed]
- O’Dea, E.L.; Kearns, J.D.; Hoffmann, A. Uv as an amplifier rather than inducer of NF-κB activity. Mol. Cell 2008, 30, 632–641. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, C.; Roulston, A.; Koromilas, A.; Wainberg, M.A.; Hiscott, J. Chronic human immunodeficiency virus type 1 infection of myeloid cells disrupts the autoregulatory control of the NF-κB/rel pathway via enhanced IκBα degradation. J. Virol. 1996, 70, 5183–5193. [Google Scholar] [PubMed]
- Meyer, M.; Caselmann, W.H.; Schluter, V.; Schreck, R.; Hofschneider, P.H.; Baeuerle, P.A. Hepatitis b virus transactivator mhbst: Activation of NF-κB, selective inhibition by antioxidants and integral membrane localization. EMBO J. 1992, 11, 2991–3001. [Google Scholar] [PubMed]
- You, L.R.; Chen, C.M.; Lee, Y.H. Hepatitis c virus core protein enhances NF-κB signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J. Virol. 1999, 73, 1672–1681. [Google Scholar] [PubMed]
- Munn, L.L. Cancer and inflammation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1370. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Ahn, K.S.; Chaturvedi, M.M.; Aggarwal, B.B. Epidermal growth factor (EGF) activates nuclear factor-kappab through IκBα kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IκBα. Oncogene 2007, 26, 7324–7332. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; O’Shea, J.M.; Perkins, N.D. Differential regulation of NF-κB activation and function by topoisomerase II inhibitors. BMC Cancer 2006, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sethi, G. Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 2010, 1805, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Mustafa, N.; Li, F.; Kannaiyan, R.; Ahn, K.S.; Kumar, A.P.; Chng, W.J.; Sethi, G. Thymoquinone overcomes chemoresistance and enhances the anticancer effects of bortezomib through abrogation of NF-κB regulated gene products in multiple myeloma xenograft mouse model. Oncotarget 2014, 5, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Shanmugam, M.K.; Siveen, K.S.; Wang, F.; Ong, T.H.; Loo, S.Y.; Swamy, M.M.; Mandal, S.; Kumar, A.P.; Goh, B.C.; et al. Garcinol sensitizes human head and neck carcinoma to cisplatin in a xenograft mouse model despite downregulation of proliferative biomarkers. Oncotarget 2015, 6, 5147–5163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piret, B.; Piette, J. Topoisomerase poisons activate the transcription factor NF-κB in ACH-2 and CEM cells. Nucleic Acids Res. 1996, 24, 4242–4248. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Busuttil, V.; Loubat, A.; Magne, N.; Fischel, J.L.; Milano, G.; Peyron, J.F. Activation of nuclear factor kappab through the IKK complex by the topoisomerase poisons SN38 and doxorubicin: A brake to apoptosis in Hela human carcinoma cells. Cancer Res. 2001, 61, 7785–7791. [Google Scholar] [PubMed]
- Andriollo, M.; Favier, A.; Guiraud, P. Adriamycin activates NF-κB in human lung carcinoma cells by IκBα degradation. Arch. Biochem. Biophys. 2003, 413, 75–82. [Google Scholar] [CrossRef]
- Baumann, P.; Mandl-Weber, S.; Oduncu, F.; Schmidmaier, R. Alkylating agents induce activation of NF-κB in multiple myeloma cells. Leuk. Res. 2008, 32, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Tergaonkar, V.; Pando, M.; Vafa, O.; Wahl, G.; Verma, I. P53 stabilization is decreased upon NF-κB activation: A role for NF-κB in acquisition of resistance to chemotherapy. Cancer Cell 2002, 1, 493–503. [Google Scholar] [CrossRef]
- Tergaonkar, V.; Bottero, V.; Ikawa, M.; Li, Q.; Verma, I.M. Ikappab kinase-independent IκBα degradation pathway: Functional NF-κB activity and implications for cancer therapy. Mol. Cell. Biol. 2003, 23, 8070–8083. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Fan, Y.; Cheng, J.; Cheng, D.; Zhao, Y.; Cao, B.; Ma, L.; An, L.; Jia, W.; Su, X.; et al. Tak1 ubiquitination regulates doxorubicin-induced NF-κB activation. Cell. Signal. 2013, 25, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.C.; Dickson, K.M.; Barker, P.A. Nuclear factor-kappab induced by doxorubicin is deficient in phosphorylation and acetylation and represses nuclear factor-kappab-dependent transcription in cancer cells. Cancer Res. 2005, 65, 4273–4281. [Google Scholar] [CrossRef] [PubMed]
- Das, K.C.; White, C.W. Activation of NF-κB by antineoplastic agents. Role of protein kinase C. J. Biol. Chem. 1997, 272, 14914–14920. [Google Scholar] [CrossRef] [PubMed]
- Brach, M.A.; Hass, R.; Sherman, M.L.; Gunji, H.; Weichselbaum, R.; Kufe, D. Ionizing radiation induces expression and binding activity of the nuclear factor kappa b. J. Clin. Investig. 1991, 88, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Rosenzweig, K.R.; Youmell, M.; Price, B.D. The DNA-dependent protein kinase participates in the activation of NF-κB following DNA damage. Biochem. Biophys. Res. Commun. 1998, 247, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Dimtchev, A.; Lavin, M.F.; Dritschilo, A.; Jung, M. A novel ionizing radiation-induced signaling pathway that activates the transcription factor NF-κB. Oncogene 1998, 17, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Brown, S.A.; Yu, T.; Chen, G.; Barve, S.; Kang, B.C.; Thompson, J.S. A high dose of ionizing radiation induces tissue-specific activation of nuclear factor-kappab in vivo. Radiat. Res. 1999, 151, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Karin, M. Ionizing radiation and short wavelength UV activate NF-κB through two distinct mechanisms. Proc. Natl. Acad. Sci. USA 1998, 95, 13012–13017. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, B.; Xia, L.; Khaletzkiy, A.; Chu, D.; Wong, J.Y.; Li, J.J. Activation of nuclear factor kappab in radioresistance of tp53-inactive human keratinocytes. Cancer Res. 2002, 62, 1213–1221. [Google Scholar] [PubMed]
- Criswell, T.; Leskov, K.; Miyamoto, S.; Luo, G.; Boothman, D.A. Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene 2003, 22, 5813–5827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodel, F.; Hantschel, M.; Hildebrandt, G.; Schultze-Mosgau, S.; Rodel, C.; Herrmann, M.; Sauer, R.; Voll, R.E. Dose-dependent biphasic induction and transcriptional activity of nuclear factor kappa b (NF-κB) in EA.hy.926 endothelial cells after low-dose X-irradiation. Int. J. Radiat. Biol. 2004, 80, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Rithidech, K.N.; Tungjai, M.; Arbab, E.; Simon, S.R. Activation of NF-κB in bone marrow cells of balb/cj mice following exposure in vivo to low doses of 137Cs gamma-rays. Radiat. Environ. Biophys. 2005, 44, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Dong, S.; Fan, M.; Li, J.J. Nuclear factor-kappab p65 inhibits mitogen-activated protein kinase signaling pathway in radioresistant breast cancer cells. Mol. Cancer Res. 2006, 4, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Spandau, D.F. Uvb activation of NF-κB in normal human keratinocytes occurs via a unique mechanism. Arch. Dermatol. Res. 2007, 299, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, C.F.; Wu, S. Mechanism of uv-induced IκBα-independent activation of NF-κB. Photochem. Photobiol. 2008, 84, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Li, S.; Wang, Z.; Ahmed, K.M.; Degnan, M.E.; Fan, M.; Dynlacht, J.R.; Li, J.J. NF-κB-mediated HER2 overexpression in radiation-adaptive resistance. Radiat. Res. 2009, 171, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Veuger, S.J.; Hunter, J.E.; Durkacz, B.W. Ionizing radiation-induced NF-κB activation requires PARP-1 function to confer radioresistance. Oncogene 2009, 28, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Pordanjani, S.M.; Hosseinimehr, S.J. The role of nf-kb inhibitors in cell response to radiation. Curr. Med. Chem. 2016, 23, 3951–3963. [Google Scholar] [CrossRef] [PubMed]
- Sahijdak, W.M.; Yang, C.R.; Zuckerman, J.S.; Meyers, M.; Boothman, D.A. Alterations in transcription factor binding in radioresistant human melanoma cells after ionizing radiation. Radiat. Res. 1994, 138, S47–S51. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, B.P.; Shackelford, R.E.; Baldwin, A.S., Jr.; Paules, R.S. Lack of involvement of ataxia telangiectasia mutated (ATM) in regulation of nuclear factor-kappab (NF-κB) in human diploid fibroblasts. Cancer Res. 1999, 59, 5456–5460. [Google Scholar] [PubMed]
- Li, N.; Banin, S.; Ouyang, H.; Li, G.C.; Courtois, G.; Shiloh, Y.; Karin, M.; Rotman, G. ATM is required for IκB kinase (IKK) activation in response to DNA double strand breaks. J. Biol. Chem. 2001, 276, 8898–8903. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Wuerzberger-Davis, S.M.; Seufzer, B.J.; Shumway, S.D.; Kurama, T.; Boothman, D.A.; Miyamoto, S. NF-κB activation by camptothecin. A linkage between nuclear DNA damage and cytoplasmic signaling events. J. Biol. Chem. 2000, 275, 9501–9509. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Kondratyev, A.; Lee, S.A.; Dimtchev, A.; Dritschilo, A. ATM gene product phosphorylates I kappa B-alpha. Cancer Res. 1997, 57, 24–27. [Google Scholar] [PubMed]
- Raju, U.; Gumin, G.J.; Noel, F.; Tofilon, P.J. IκBα degradation is not a requirement for the x-ray-induced activation of nuclear factor kappab in normal rat astrocytes and human brain tumour cells. Int. J. Radiat. Biol. 1998, 74, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Curry, H.A.; Clemens, R.A.; Shah, S.; Bradbury, C.M.; Botero, A.; Goswami, P.; Gius, D. Heat shock inhibits radiation-induced activation of NF-κB via inhibition of I-κB kinase. J. Biol. Chem. 1999, 274, 23061–23067. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, J.; Yagi, K. Inhibition of I κB-α phosphorylation at serine and tyrosine acts independently on sensitization to DNA damaging agents in human glioma cells. Br. J. Cancer 2000, 82, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, V.A.; Barani, I.J.; Rabender, C.S.; Black, S.M.; Leach, J.K.; Graves, P.R.; Kellogg, G.E.; Mikkelsen, R.B. Tyrosine nitration of IκBα: A novel mechanism for NF-κB activation. Biochemistry 2007, 46, 11671–11683. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Chapman, N.R.; Perkins, N.D. UV stimulation induces nuclear factor κB (NF-κB) DNA-binding activity but not transcriptional activation. Biochem. Soc. Trans. 2001, 29, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Gilroy, D.W.; Colville-Nash, P.R.; Willoughby, D.A. Possible new role for NF-κB in the resolution of inflammation. Nat. Med. 2001, 7, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappab in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Wong, E.; Kua, N.; Teo, H.L.; Tergaonkar, V.; Lane, D. Hexamethylene bisacetamide (HMBA) simultaneously targets akt and mapk pathway and represses NF-κB activity: Implications for cancer therapy. Cell Cycle 2008, 7, 3759–3767. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; de Matos Simoes, R.; Dhimolea, E.; Sheffer, M.; Gandolfi, S.; Dashevsky, O.; Sorrell, J.D.; Mitsiades, C.S. NF-κB dysregulation in multiple myeloma. Semin. Cancer Biol. 2016, 39, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, L.; Medeiros, L.J.; Young, K.H. NF-κB signaling pathway and its potential as a target for therapy in lymphoid neoplasms. Blood Rev. 2017, 31, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.J.; Ratnam, N.M.; Byrd, J.C.; Guttridge, D.C. NF-κB functions in tumor initiation by suppressing the surveillance of both innate and adaptive immune cells. Cell Rep. 2014, 9, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Pettaway, C.A.; Uehara, H.; Bucana, C.D.; Fidler, I.J. Blockade of NF-κB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001, 20, 4188–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Inda, M.E.; Bagam, P.; Sahoo, M.K.; Osorio, D.; Batra, S. Transcription factor NF-κB: An update on intervention strategies. Arch. Immunol. Ther. Exp. 2016, 64, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Q.L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.Y.; et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 2015, 17, 1327–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tergaonkar, V. NF-κB drives tert promoter reactivation in cancer. Cell Cycle 2016, 15, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, H.S.; Chng, W.J.; Tergaonkar, V. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proc. Natl. Acad. Sci. USA 2016, 113, 14402–14407. [Google Scholar] [CrossRef] [PubMed]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akincilar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting trna expression. J. Clin. Investig. 2016, 126, 4045–4060. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Zeng, S.; Xie, R.; Hu, C.J.; Wang, S.M.; Wu, Y.Y.; Xiao, Y.F.; Yang, S.M. Htert promotes gastric intestinal metaplasia by upregulating CDX2 via NF-κB signaling pathway. Oncotarget 2017, 8, 26969–26978. [Google Scholar] [CrossRef] [PubMed]
- Fusella, F.; Secli, L.; Busso, E.; Krepelova, A.; Moiso, E.; Rocca, S.; Conti, L.; Annaratone, L.; Rubinetto, C.; Mello-Grand, M.; et al. The IKK/NF-κB signaling pathway requires morgana to drive breast cancer metastasis. Nat. Commun. 2017, 8, 1636. [Google Scholar] [CrossRef] [PubMed]
- Manu, K.A.; Shanmugam, M.K.; Ong, T.H.; Subramaniam, A.; Siveen, K.S.; Perumal, E.; Samy, R.P.; Bist, P.; Lim, L.H.; Kumar, A.P.; et al. Emodin suppresses migration and invasion through the modulation of CXCR4 expression in an orthotopic model of human hepatocellular carcinoma. PLoS ONE 2013, 8, e57015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, M.K.; Ahn, K.S.; Lee, J.H.; Kannaiyan, R.; Mustafa, N.; Manu, K.A.; Siveen, K.S.; Sethi, G.; Chng, W.J.; Kumar, A.P. Celastrol attenuates the invasion and migration and augments the anticancer effects of bortezomib in a xenograft mouse model of multiple myeloma. Front. Pharmacol. 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Helbig, G.; Christopherson, K.W., 2nd; Bhat-Nakshatri, P.; Kumar, S.; Kishimoto, H.; Miller, K.D.; Broxmeyer, H.E.; Nakshatri, H. NF-κB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J. Biol. Chem. 2003, 278, 21631–21638. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Manu, K.A.; Ong, T.H.; Ramachandran, L.; Surana, R.; Bist, P.; Lim, L.H.; Kumar, A.P.; Hui, K.M.; Sethi, G. Inhibition of CXCR4/CXCL12 signaling axis by ursolic acid leads to suppression of metastasis in transgenic adenocarcinoma of mouse prostate model. Int. J. Cancer 2011, 129, 1552–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Ahn, K.S.; Wang, L.Z.; Kim, C.; Deivasigamni, A.; Arfuso, F.; Um, J.Y.; Kumar, A.P.; Chang, Y.C.; Kumar, D.; et al. Ascochlorin enhances the sensitivity of doxorubicin leading to the reversal of epithelial-to-mesenchymal transition in hepatocellular carcinoma. Mol. Cancer Ther. 2016, 15, 2966–2976. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-mediated metastasis: From epithelial-mesenchymal transition to escape from immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Tao, D.; Fang, Y.; Deng, C.; Xu, Q.; Zhou, J. TNF-alpha promotes invasion and metastasis via NF-κB pathway in oral squamous cell carcinoma. Med. Sci. Monit. Basic Res. 2017, 23, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.P.; Pober, J.S.; Bradley, J.R. Tumour necrosis factor and cancer. J. Pathol. 2013, 230, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.R.; Jo, K.; Lee, Y.; Sung, B.J.; Park, Y.W.; Lee, J.H. Upregulation of CD9 in ovarian cancer is related to the induction of TNF-alpha gene expression and constitutive NF-κB activation. Carcinogenesis 2012, 33, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. TNF-alpha/NF-κB/snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of nemo/IKKγgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-κB addiction and its role in cancer: ‘One size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Maeda, G.; Chiba, T.; Kawashiri, S.; Satoh, T.; Imai, K. Epigenetic inactivation of IkappaB Kinase-alpha in oral carcinomas and tumor progression. Clin. Cancer Res. 2007, 13, 5041–5047. [Google Scholar] [CrossRef] [PubMed]
- Van Waes, C.; Yu, M.; Nottingham, L.; Karin, M. Inhibitor-kappaB kinase in tumor promotion and suppression during progression of squamous cell carcinoma. Clin. Cancer Res. 2007, 13, 4956–4959. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.T.; Bertrand, M.J.M. More to life than NF-κB in tnfr1 signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Manu, K.A.; Chen, L.; Li, F.; Rajendran, P.; Subramaniam, A.; Lam, P.; Kumar, A.P.; Sethi, G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-jun n-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis 2011, 16, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wang, L.; Deivasigamni, A.; Looi, C.Y.; Karthikeyan, C.; Trivedi, P.; Chinnathambi, A.; Alharbi, S.A.; Arfuso, F.; Dharmarajan, A.; et al. A novel benzimidazole derivative, MBIC inhibits tumor growth and promotes apoptosis via activation of ROS-dependent JNK signaling pathway in hepatocellular carcinoma. Oncotarget 2017, 8, 12831–12842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Castranova, V. Nuclear factor-kappab, an unappreciated tumor suppressor. Cancer Res. 2007, 67, 11093–11098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, F. Reactive oxygen species (ROS), troublemakers between nuclear factor-kappaB (NF-κB) and c-Jun NH2-terminal kinase (JNK). Cancer Res. 2004, 64, 1902–1905. [Google Scholar] [CrossRef] [PubMed]
- Neelgundmath, M.; Dinesh, K.R.; Mohan, C.D.; Li, F.; Dai, X.; Siveen, K.S.; Paricharak, S.; Mason, D.J.; Fuchs, J.E.; Sethi, G.; et al. Novel synthetic coumarins that targets NF-κB in hepatocellular carcinoma. Bioorg. Med. Chem. Lett. 2015, 25, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Anilkumar, N.C.; Rangappa, S.; Shanmugam, M.K.; Mishra, S.; Chinnathambi, A.; Alharbi, S.A.; Bhattacharjee, A.; Sethi, G.; Kumar, A.P.; et al. Novel 1,3,4-oxadiazole induces anticancer activity by targeting NF-κB in hepatocellular carcinoma cells. Front. Oncol. 2018, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Jain, H.; Dhingra, N.; Narsinghani, T.; Sharma, R. Insights into the mechanism of natural terpenoids as NF-κB inhibitors: An overview on their anticancer potential. Exp. Oncol. 2016, 38, 158–168. [Google Scholar] [PubMed]
- De Castro Barbosa, M.L.; da Conceicao, R.A.; Fraga, A.G.M.; Camarinha, B.D.; de Carvalho Silva, G.C.; Lima, A.G.F.; Cardoso, E.A.; de Oliveira Freitas Lione, V. NF-κB signaling pathway inhibitors as anticancer drug candidates. Anticancer Agents Med. Chem. 2017, 17, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Kunnumakkara, A.B.; Aggarwal, B.B. Targeting TNF for treatment of cancer and autoimmunity. Adv. Exp. Med. Biol. 2009, 647, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Keerthy, H.K.; Mohan, C.D.; Sivaraman Siveen, K.; Fuchs, J.E.; Rangappa, S.; Sundaram, M.S.; Li, F.; Girish, K.S.; Sethi, G.; Basappa; et al. Novel synthetic biscoumarins target tumor necrosis factor-alpha in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 31879–31890. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp. Biol. Med. 2015, 240, 760–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirvanappa, A.C.; Mohan, C.D.; Rangappa, S.; Ananda, H.; Sukhorukov, A.Y.; Shanmugam, M.K.; Sundaram, M.S.; Nayaka, S.C.; Girish, K.S.; Chinnathambi, A.; et al. Novel synthetic oxazines target NF-κB in colon cancer in vitro and inflammatory bowel disease in vivo. PLoS ONE 2016, 11, e0163209. [Google Scholar] [CrossRef] [PubMed]
- Ningegowda, R.; Shivananju, N.S.; Rajendran, P.; Basappa; Rangappa, K.S.; Chinnathambi, A.; Li, F.; Achar, R.R.; Shanmugam, M.K.; Bist, P.; et al. A novel 4,6-disubstituted-1,2,4-triazolo-1,3,4-thiadiazole derivative inhibits tumor cell invasion and potentiates the apoptotic effect of tnfalpha by abrogating NF-κB activation cascade. Apoptosis 2017, 22, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Song, X.Y.; Torphy, T.J.; Griswold, D.E.; Shealy, D. Coming of age: Anti-cytokine therapies. Mol. Interv. 2002, 2, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, S.A.; Shealy, D.J.; Nakada, M.T.; Le, J.; Woulfe, D.S.; Probert, L.; Kollias, G.; Ghrayeb, J.; Vilcek, J.; Daddona, P.E. The mouse/human chimeric monoclonal antibody cA2 neutralizes TNF in vitro and protects transgenic mice from cachexia and TNF lethality in vivo. Cytokine 1995, 7, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Scallon, B.J.; Moore, M.A.; Trinh, H.; Knight, D.M.; Ghrayeb, J. Chimeric anti-TNF-alpha monoclonal antibody cA2 binds recombinant transmembrane TNF-alpha and activates immune effector functions. Cytokine 1995, 7, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; van der Heijde, D.; de Jager, J.P.; Gough, A.; Kalden, J.; Malaise, M.; Martin Mola, E.; Pavelka, K.; Sany, J.; Settas, L.; et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: Double-blind randomised controlled trial. Lancet 2004, 363, 675–681. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Klareskog, L.; Rodriguez-Valverde, V.; Codreanu, C.; Bolosiu, H.; Melo-Gomes, J.; Tornero-Molina, J.; Wajdula, J.; Pedersen, R.; Fatenejad, S. Comparison of etanercept and methotrexate, alone and combined, in the treatment of rheumatoid arthritis: Two-year clinical and radiographic results from the tempo study, a double-blind, randomized trial. Arthritis Rheum. 2006, 54, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Lethaby, A.; Lopez-Olivo, M.A.; Maxwell, L.; Burls, A.; Tugwell, P.; Wells, G.A. Etanercept for the treatment of rheumatoid arthritis. Cochrane Database Syst. Rev. 2013, 5, CD004525. [Google Scholar] [CrossRef] [PubMed]
- Van Schouwenburg, P.A.; van de Stadt, L.A.; de Jong, R.N.; van Buren, E.E.; Kruithof, S.; de Groot, E.; Hart, M.; van Ham, S.M.; Rispens, T.; Aarden, L.; et al. Adalimumab elicits a restricted anti-idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann. Rheum. Dis. 2013, 72, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Nanda, K.S.; Cheifetz, A.S.; Moss, A.C. Impact of antibodies to infliximab on clinical outcomes and serum infliximab levels in patients with inflammatory bowel disease (IBD): A meta-analysis. Am. J. Gastroenterol. 2013, 108, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Steenholdt, C.; Svenson, M.; Bendtzen, K.; Thomsen, O.O.; Brynskov, J.; Ainsworth, M.A. Acute and delayed hypersensitivity reactions to infliximab and adalimumab in a patient with crohn’s disease. J. Crohns Colitis 2012, 6, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Garces, S.; Demengeot, J.; Benito-Garcia, E. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: A systematic review of the literature with a meta-analysis. Ann. Rheum. Dis. 2013, 72, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Petro, J.B.; Rahman, S.M.; Ballard, D.W.; Khan, W.N. Bruton’s tyrosine kinase is required for activation of IκB kinase and nuclear factor kappaB in response to B cell receptor engagement. J. Exp. Med. 2000, 191, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. The NF-κB and IκB proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-κB/IκB family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Novero, A.; Ravella, P.M.; Chen, Y.; Dous, G.; Liu, D. Ibrutinib for B cell malignancies. Exp. Hematol. Oncol. 2014, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.Q.; Smith, S.M.; Zhang, S.Y.; Lynn Wang, Y. Mechanisms of ibrutinib resistance in chronic lymphocytic leukaemia and non-hodgkin lymphoma. Br. J. Haematol. 2015, 170, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Jeelall, Y.S.; Ferguson, L.L.; Horikawa, K. Toll-like receptors and cancer: MYD88 mutation and inflammation. Front. Immunol. 2014, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Brenner, L.; Arbeit, R.D.; Sullivan, T. IMO-8400, an antagonist of toll-like receptors 7, 8, and 9, in development for genetically defined B-cell lymphomas: Safety and activity in phase 1 and phase 2 clinical trials. Blood 2014, 124, 3101. [Google Scholar]
- Balak, D.M.; van Doorn, M.B.; Arbeit, R.D.; Rijneveld, R.; Klaassen, E.; Sullivan, T.; Brevard, J.; Thio, H.B.; Prens, E.P.; Burggraaf, J.; et al. IMO-8400, a toll-like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo-controlled trial in patients with moderate-to-severe plaque psoriasis. Clin. Immunol. 2017, 174, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Basseres, D.S.; Baldwin, A.S. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef] [PubMed]
- Awasthee, N.; Rai, V.; Chava, S.; Nallasamy, P.; Kunnumakkara, A.B.; Bishayee, A.; Chauhan, S.C.; Challagundla, K.B.; Gupta, S.C. Targeting IκB kinases for cancer therapy. Semin. Cancer Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-κB conundrum: Embracing complexity to achieve specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory kappa b kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-κB system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Pande, V.; Ramos, M.J. NF-κB in human disease: Current inhibitors and prospects for de novo structure based design of inhibitors. Curr. Med. Chem. 2005, 12, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Bhattacharjee, A.; Ali, A.; Mandal, N.C.; Mandal, S.C.; Pal, M. Chronic inflammation and cancer: Potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism. J. Inflamm. 2014, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- De Falco, F.; Di Giovanni, C.; Cerchia, C.; De Stefano, D.; Capuozzo, A.; Irace, C.; Iuvone, T.; Santamaria, R.; Carnuccio, R.; Lavecchia, A. Novel non-peptide small molecules preventing IKKβ/NEMO association inhibit NF-κB activation in LPS-stimulated J774 macrophages. Biochem. Pharmacol. 2016, 104, 83–94. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-activated IκB kinases essential for NF-κB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Cao, Z.; Goeddel, D.V. NF-κB-inducing kinase activates IKK-alpha by phosphorylation of ser-176. Proc. Natl. Acad. Sci. USA 1998, 95, 3792–3797. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.A.; Zuvanich, E.G.; Khan, D.A.; Mushtaq, S.; Silswal, N.; Qureshi, N. Proteasome inhibitors modulate anticancer and anti-proliferative properties via NF-κB signaling, and ubiquitin-proteasome pathways in cancer cell lines of different organs. Lipids Health Dis. 2018, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- Freitas, R.; Fraga, C.A.M. NF-κB-IKKβ pathway as a target for drug development: Realities, challenges and perspectives. Curr. Drug Targets 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Monack, D.M.; Kayagaki, N.; Wertz, I.; Yin, J.; Wolf, B.; Dixit, V.M. Yersinia virulence factor Yopj acts as a deubiquitinase to inhibit NF-κB activation. J. Exp. Med. 2005, 202, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Chatterjee, S.; Kauer, J.C.; Das, M.; Messina, P.; Freed, B.; Biazzo, W.; Siman, R. Potent inhibitors of proteasome. J. Med. Chem. 1995, 38, 2276–2277. [Google Scholar] [CrossRef] [PubMed]
- Grisham, M.B.; Palombella, V.J.; Elliott, P.J.; Conner, E.M.; Brand, S.; Wong, H.L.; Pien, C.; Mazzola, L.M.; Destree, A.; Parent, L.; et al. Inhibition of NF-κB activation in vitro and in vivo: Role of 26S proteasome. Methods Enzymol. 1999, 300, 345–363. [Google Scholar] [PubMed]
- Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004, 5, 417–421. [Google Scholar] [CrossRef]
- Staudt, L.M. Gene expression profiling of lymphoid malignancies. Annu. Rev. Med. 2002, 53, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, J.B.; Chen, Z.; Dong, G.; Yeh, N.; Crowl Bancroft, C.; Sausville, E.; Adams, J.; Elliott, P.; Van Waes, C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa b, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 2001, 7, 1419–1428. [Google Scholar] [PubMed]
- Lun, M.; Zhang, P.L.; Pellitteri, P.K.; Law, A.; Kennedy, T.L.; Brown, R.E. Nuclear factor-kappab pathway as a therapeutic target in head and neck squamous cell carcinoma: Pharmaceutical and molecular validation in human cell lines using velcade and siRNA/NF-κB. Ann. Clin. Lab. Sci. 2005, 35, 251–258. [Google Scholar] [PubMed]
- Allen, C.; Saigal, K.; Nottingham, L.; Arun, P.; Chen, Z.; Van Waes, C. Bortezomib-induced apoptosis with limited clinical response is accompanied by inhibition of canonical but not alternative nuclear factor-{kappa}B subunits in head and neck cancer. Clin. Cancer Res. 2008, 14, 4175–4185. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.V.; Hertlein, E.; Lu, Y.; Sass, E.J.; Lapalombella, R.; Chen, T.L.; Davis, M.E.; Woyach, J.A.; Lehman, A.; Jarjoura, D.; et al. The proteasome inhibitor carfilzomib functions independently of p53 to induce cytotoxicity and an atypical NF-κB response in chronic lymphocytic leukemia cells. Clin. Cancer Res. 2013, 19, 2406–2419. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Ikeda, H.; Chauhan, D.; Okawa, Y.; Raje, N.; Podar, K.; Mitsiades, C.; Munshi, N.C.; Richardson, P.G.; Carrasco, R.D.; et al. Bortezomib induces canonical nuclear factor-kappab activation in multiple myeloma cells. Blood 2009, 114, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.H.; Wong, H.R.; Odoms, K.; Deitch, E.A.; Szabo, C.; Vizi, E.S.; Hasko, G. Proteasome inhibitors induce inhibitory kappa b (IκB) kinase activation, IκBα degradation, and nuclear factor kappa B activation in HT-29 cells. Mol. Pharmacol. 2004, 65, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Z.; Yao, S.Y.; Veach, R.A.; Torgerson, T.R.; Hawiger, J. Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 1995, 270, 14255–14258. [Google Scholar] [CrossRef] [PubMed]
- Torgerson, T.R.; Colosia, A.D.; Donahue, J.P.; Lin, Y.Z.; Hawiger, J. Regulation of NF-κB, AP-1, NFAT, and STAT1 nuclear import in t lymphocytes by noninvasive delivery of peptide carrying the nuclear localization sequence of NF-κB p50. J. Immunol. 1998, 161, 6084–6092. [Google Scholar] [PubMed]
- Horie, R. Molecularly-targeted strategy and NF-κB in lymphoid malignancies. J. Clin. Exp. Hematop. 2013, 53, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.D.; Cleaveland, J.; Nadler, S.G. An intracellular targeted NLS peptide inhibitor of karyopherin alpha: NF-κB interactions. Biochem. Biophys. Res. Commun. 2003, 300, 403–407. [Google Scholar] [CrossRef]
- Mallavia, B.; Recio, C.; Oguiza, A.; Ortiz-Munoz, G.; Lazaro, I.; Lopez-Parra, V.; Lopez-Franco, O.; Schindler, S.; Depping, R.; Egido, J.; et al. Peptide inhibitor of NF-κB translocation ameliorates experimental atherosclerosis. Am. J. Pathol. 2013, 182, 1910–1921. [Google Scholar] [CrossRef] [PubMed]
- Lyss, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-κB by directly targeting p65. J. Biol. Chem. 1998, 273, 33508–33516. [Google Scholar] [CrossRef] [PubMed]
- Morishita, R.; Sugimoto, T.; Aoki, M.; Kida, I.; Tomita, N.; Moriguchi, A.; Maeda, K.; Sawa, Y.; Kaneda, Y.; Higaki, J.; et al. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat. Med. 1997, 3, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Khaled, A.R.; Butfiloski, E.J.; Sobel, E.S.; Schiffenbauer, J. Use of phosphorothioate-modified oligodeoxynucleotides to inhibit NF-κB expression and lymphocyte function. Clin. Immunol. Immunopathol. 1998, 86, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Kupatt, C.; Wichels, R.; Deiss, M.; Molnar, A.; Lebherz, C.; Raake, P.; von Degenfeld, G.; Hahnel, D.; Boekstegers, P. Retroinfusion of NF-κB decoy oligonucleotide extends cardioprotection achieved by cd18 inhibition in a preclinical study of myocardial ischemia and retroinfusion in pigs. Gene Ther. 2002, 9, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Tomita, N.; Ogihara, T.; Morishita, R. Transcription factors as molecular targets: Molecular mechanisms of decoy ODN and their design. Curr. Drug Targets 2003, 4, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Crinelli, R.; Bianchi, M.; Gentilini, L.; Palma, L.; Magnani, M. Locked nucleic acids (LNA): Versatile tools for designing oligonucleotide decoys with high stability and affinity. Curr. Drug Targets 2004, 5, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Isomura, I.; Morita, A. Regulation of NF-κB signaling by decoy oligodeoxynucleotides. Microbiol. Immunol. 2006, 50, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Bhardwaj, A.; Potdar, P.; Aggarwal, B.B. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-κB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 2004, 23, 9247–9258. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.R.; Loveridge, C.J.; Dunlop, M.G.; Stark, L.A. C-src dependency of nsaid-induced effects on NF-κB-mediated apoptosis in colorectal cancer cells. Carcinogenesis 2011, 32, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Callejas, N.A.; Casado, M.; Bosca, L.; Martin-Sanz, P. Absence of nuclear factor kappab inhibition by nsaids in hepatocytes. Hepatology 2002, 35, 341–348. [Google Scholar] [CrossRef] [PubMed]
- De Cicco, P.; Panza, E.; Ercolano, G.; Armogida, C.; Sessa, G.; Pirozzi, G.; Cirino, G.; Wallace, J.L.; Ianaro, A. Atb-346, a novel hydrogen sulfide-releasing anti-inflammatory drug, induces apoptosis of human melanoma cells and inhibits melanoma development in vivo. Pharmacol. Res. 2016, 114, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Ghanghas, P.; Jain, S.; Rana, C.; Sanyal, S.N. Chemopreventive action of non-steroidal anti-inflammatory drugs on the inflammatory pathways in colon cancer. Biomed. Pharmacother. 2016, 78, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Erstad, D.J.; Cusack, J.C., Jr. Targeting the NF-κB pathway in cancer therapy. Surg. Oncol. Clin. N. Am. 2013, 22, 705–746. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Adriaansen, J.; Vervoordeldonk, M.J.; Tak, P.P. Gene therapy as a therapeutic approach for the treatment of rheumatoid arthritis: Innovative vectors and therapeutic genes. Rheumatology 2006, 45, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Van de Loo, F.A.; Smeets, R.L.; van den Berg, W.B. Gene therapy in animal models of rheumatoid arthritis: Are we ready for the patients? Arthritis Res. Ther. 2004, 6, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Vervoordeldonk, M.J.; Aalbers, C.J.; Tak, P.P. Advances in local targeted gene therapy for arthritis: Towards clinical reality. Future 2008, 3, 307–309. [Google Scholar] [CrossRef]
- Tak, P.P.; Gerlag, D.M.; Aupperle, K.R.; van de Geest, D.A.; Overbeek, M.; Bennett, B.L.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. Inhibitor of nuclear factor kappab kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 2001, 44, 1897–1907. [Google Scholar] [CrossRef]
- Sanlioglu, S.; Luleci, G.; Thomas, K.W. Simultaneous inhibition of Rac1 and IKK pathways sensitizes lung cancer cells to TNFα-mediated apoptosis. Cancer Gene Ther. 2001, 8, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanlioglu, A.D.; Koksal, I.T.; Karacay, B.; Baykara, M.; Luleci, G.; Sanlioglu, S. Adenovirus-mediated IKKβKA expression sensitizes prostate carcinoma cells to trail-induced apoptosis. Cancer Gene Ther. 2006, 13, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Karacay, B.; Sanlioglu, S.; Griffith, T.S.; Sandler, A.; Bonthius, D.J. Inhibition of the NF-κB pathway enhances trail-mediated apoptosis in neuroblastoma cells. Cancer Gene Ther. 2004, 11, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Horwitz, M.S. Inhibition of tumor necrosis factor alpha-induced NF-κB activation by the adenovirus e3-10.4/14.5k complex. J. Virol. 2002, 76, 5515–5521. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Q.; Shuai, X.R.; Yan, M.L.; Zhang, M.M.; Yan, L.N. Nuclear factor-kappab decoy oligodeoxynucleotides attenuates ischemia/reperfusion injury in rat liver graft. World J. Gastroenterol. 2005, 11, 6960–6967. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Kole, R.; Juliano, R.L. Stability of antisense DNA oligodeoxynucleotide analogs in cellular extracts and sera. Life Sci. 1991, 49, 1793–1801. [Google Scholar] [CrossRef]
- De Stefano, D. Oligonucleotides decoy to NF-κB: Becoming a reality? Discov. Med. 2011, 12, 97–105. [Google Scholar] [PubMed]
- Surabhi, R.M.; Gaynor, R.B. RNA interference directed against viral and cellular targets inhibits human immunodeficiency virus type 1 replication. J. Virol. 2002, 76, 12963–12973. [Google Scholar] [CrossRef] [PubMed]
- Takaesu, G.; Surabhi, R.M.; Park, K.J.; Ninomiya-Tsuji, J.; Matsumoto, K.; Gaynor, R.B. TAK1 is critical for I κB kinase-mediated activation of the NF-κB pathway. J. Mol. Biol. 2003, 326, 105–115. [Google Scholar] [CrossRef]
- Duan, J.; Friedman, J.; Nottingham, L.; Chen, Z.; Ara, G.; Van Waes, C. Nuclear factor-kappaB p65 small interfering RNA or proteasome inhibitor bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2007, 6, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cancers | Inflammatory Diseases |
|---|---|
| Acute lymphoblastic leukemia Anaplastic large-cell lymphoma Breast Burkitt lymphoma Cervical Colorectal Diffuse large B-cell lymphoma Fibrosarcoma Head and neck Hodgkin’s lymphoma Mammary carcinoma Mantle cell lymphoma Melanoma Multiple myeloma Lung Ovarian Pancreatic Prostate Squamous-cell carcinoma Thyroid Vulva | Alzheimer’s disease Rheumatoid arthritis Atherosclerosis Multiple sclerosis Chronic inflammatory demyelinating Polyradiculoneuritis Asthma Inflammatory bowel disease Helicobacter pylori-associated gastritis Systemic inflammatory response syndrome Parkinson’s disease |
| Class | Inducing Stimuli |
|---|---|
| Viruses | Human immunodeficiency virus Hepatitis B virus Human herpes virus 6 Adenovirus |
| Viral products | Double-stranded RNA Latent membrane protein Hepatitis B viral protein HBx Middle hepatitis Bvirussurfaceprotein MHBs |
| Inflammatory cytokines | Tumor necrosis factor-α Lymphotoxin Interleukin-1 Interleukin-2 Leukotriene B4 |
| Bacterial products | Lipopolysaccharide Exotoxin B Toxic shock syndrome toxin 1 Muramyl peptides |
| Physical stress | UV light |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puar, Y.R.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Sethi, G.; Tergaonkar, V. Evidence for the Involvement of the Master Transcription Factor NF-κB in Cancer Initiation and Progression. Biomedicines 2018, 6, 82. https://doi.org/10.3390/biomedicines6030082
Puar YR, Shanmugam MK, Fan L, Arfuso F, Sethi G, Tergaonkar V. Evidence for the Involvement of the Master Transcription Factor NF-κB in Cancer Initiation and Progression. Biomedicines. 2018; 6(3):82. https://doi.org/10.3390/biomedicines6030082
Chicago/Turabian StylePuar, Yu Rou, Muthu K Shanmugam, Lu Fan, Frank Arfuso, Gautam Sethi, and Vinay Tergaonkar. 2018. "Evidence for the Involvement of the Master Transcription Factor NF-κB in Cancer Initiation and Progression" Biomedicines 6, no. 3: 82. https://doi.org/10.3390/biomedicines6030082