Non Melanoma Skin Cancer Pathogenesis Overview


{kind=link}
Abstract
:1. Introduction
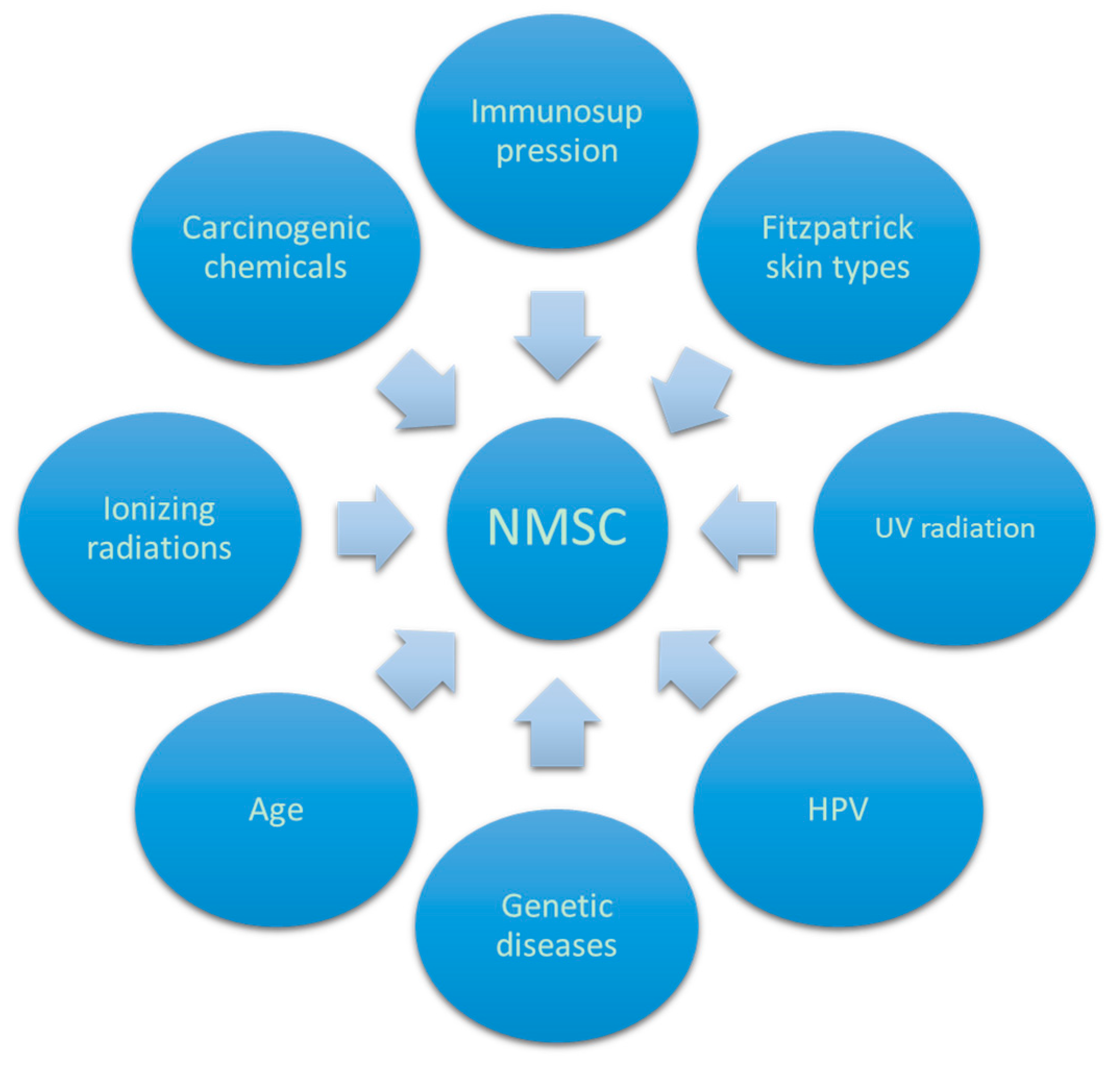
2. NMSC and AK: Factors Involved in the Pathogenesis
2.1. UV Role
2.2. X-rays Role
2.3. HPV Role
2.4. Carcinogenic Chemicals and Arsenic
2.5. Immunosuppression
3. AK and SCC Genetic Profile
3.1. SCC Proliferation
3.2. Abnormal Cell Surface Expression of HLA Protein in SCC
3.3. Alteration of APC Gene
4. BCC Risk Factors
4.1. Sporadic BCC
4.2. Recurrent BCC
5. Role of Keratinocytes-Specific Proteins
6. Role of ROS and NO
7. Role of Angiogenesis
8. Animal Models to Study in NMSC
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of skin cancer. Adv. Exp. Med. Biol. 2014, 810, 120–140. [Google Scholar] [PubMed]
- Apalla, Z.; Nashan, D.; Weller, R.B.; Castellsagué, X. Skin Cancer: Epidemiology, Disease Burden, Pathophysiology, Diagnosis, and Therapeutic Approaches. Dermatol. Ther. 2017, 7, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Calzavara-Pinton, P.; Ortel, B.; Venturini, M. Non-melanoma skin cancer, sun exposure and sun protection. G. Ital. Dermatol. Venereol. 2015, 150, 369–378. [Google Scholar] [PubMed]
- Gloster, H.M., Jr.; Brodland, D.G. The epidemiology of skin cancer. Dermatol. Surg. 1996, 22, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Paulitschke, V.; Gerner, C.; Hofstätter, E.; Mohr, T.; Mayer, R.L.; Pehamberger, H.; Kunstfeld, R. Proteome profiling of keratinocytes transforming to malignancy. Electrophoresis 2015, 36, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Konrad Muller, H.; Gorny, A.; Ananthaswamy, H.N. UVB-induced experimental carcinogenesis: Dysregulation of apoptosis and p53 signalling pathway. Redox Rep. 2000, 5, 128–129. [Google Scholar] [CrossRef] [PubMed]
- López-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Pérez-Plasencia, C.; Álvarez-Sánchez, E.; Marchat, L.A. Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis. Int. J. Mol. Sci. 2012, 13, 142–172. [Google Scholar] [CrossRef] [PubMed]
- Rittié, L.; Fisher, G.J. UV-light-induced signal cascades and skin aging. Ageing Res. Rev. 2002, 1, 705–720. [Google Scholar] [CrossRef]
- Iriyama, S.; Matsunaga, Y.; Takahashi, K.; Matsuzaki, K.; Kumagai, N.; Amano, S. Activation of heparanase by ultraviolet B irradiation leads to functional loss of basement membrane at the dermal-epidermal junction in human skin. Arch. Dermatol. Res. 2011, 303, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Lavker, R.M.; Zheng, P.S.; Dong, G. Morphology of aged skin. Clin. Geriatr. Med. 1989, 5, 53–67. [Google Scholar] [PubMed]
- Jeanloz, R.W. The nomenclature of mucopolysaccharides. Arthritis Rheum. 1960, 3, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Schönherr, E.; Hausser, H.J. Extracellular matrix and cytokines: A functional unit. Dev. Immunol. 2000, 7, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Edovitsky, E.; Lerner, I.; Zcharia, E.; Peretz, T.; Vlodavsky, I.; Elkin, M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood 2006, 107, 3609–3616. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Friedmann, Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J. Clin. Investig. 2001, 108, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Karagas, M.R.; McDonald, J.A.; Greenberg, E.R.; Stukel, T.A.; Weiss, J.E.; Baron, J.A.; Stevens, M.M. Risk of basal cell and squamous cell skin cancers after ionizing radiation therapy. For The Skin Cancer Prevention Study Group. J. Natl. Cancer Inst. 1996, 88, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Lichter, M.D.; Karagas, M.R.; Mott, L.A.; Spencer, S.K.; Stukel, T.A.; Greenberg, E.R. Therapeutic ionizing radiation and the incidence of basal cell carcinoma and squamous cell carcinoma. The New Hampshire Skin Cancer Study Group. Arch. Dermatol. 2000, 136, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Ron, E.; Modan, B.; Preston, D.; Alfandary, E.; Stovall, M.; Boice, J.D., Jr. Radiation-induced skin carcinomas of the head and neck. Radiat. Res. 1991, 125, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R.P.; Bajdik, C.D.; Fincham, S.; Hill, G.B.; Keefe, A.R.; Coldman, A.; McLean, D.I. Chemical exposures, medical history, and risk of squamous and basal cell carcinoma of the skin. Cancer Epidemiol. Biomark. Prev. 1996, 5, 419–424. [Google Scholar]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Rusin, P.; Olszewski, J.; Morawiec-Bajda, A.; Przybylowska, K.; Kaczmarczyk, D.; Golinska, A.; Majsterek, I. Comparative study of DNA damage and repair in head and neck cancer after radiation treatment. Cell Biol. Int. 2009, 33, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J. Molecular mechanism of the recruitment of NBS1/hMRE11/hRAD50 complex to DNA double-strand breaks: NBS1 binds to γ-H2AX through FHA/BRCT domain. J. Radiat. Res. 2004, 45, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. The role of p53 in determining sensitivity to radiotherapy. Nat. Rev. Cancer 2003, 3, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Asgari, M.M.; Kiviat, N.B.; Critchlow, C.W.; Stern, J.E.; Argenyi, Z.B.; Raugi, G.J.; Berg, D.; Odland, P.B.; Hawes, S.E.; de Villiers, E. Detection of human papillomavirus DNA in cutaneous squamous cell carcinoma among immunocompetent individuals. J. Investig. Dermatol. 2008, 128, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Asgari, M.M.; Wang, W.; Ioannidis, N.M.; Itnyre, J.; Hoffmann, T.; Jorgenson, E.; Whittemore, A.S. Identification of susceptibility loci for cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2016, 136, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.A.; Surentheran, T.; McGregor, J.M.; Spink, P.J.; Leigh, I.M.; Breuer, J.; Proby, C.M. Human papillomavirus infection and non-melanoma skin cancer in immunosuppressed and immunocompetent individuals. J. Med. Virol. 2000, 61, 289–297. [Google Scholar] [CrossRef]
- Bouwes Bavinck, J.N.; Plasmeijer, E.I.; Feltkamp, M.C. β-papillomavirus infection and skin cancer. J. Investig. Dermatol. 2008, 128, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Aldabagh, B.; Angeles, J.G.; Cardones, A.R.; Arron, S.T. Cutaneous squamous cell carcinoma and human papillomavirus: Is there an association? Dermatol. Surg. 2014, 39, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Aldabagh, B.; Yu, J.; Arron, S.T. Role of human papillomavirus in cutaneous squamous cell carcinoma: A meta-analysis. J. Am. Acad. Dermatol. 2014, 70, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Yesantharao, P.; Wang, W.; Ioannidis, N.M.; Demehri, S.; Whittemore, A.S.; Asgari, M.M. Cutaneous squamous cell cancer (cSCC) risk and the human leukocyte antigen (HLA) system. Hum. Immunol. 2017, 78, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Prigge, E.S.; von Knebel Doeberitz, M.; Reuschenbach, M. Clinical relevance and implications of HPV-induced neoplasia in different anatomical locations. Mutat. Res. 2017, 772, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Hampras, S.S.; Reed, R.A.; Bezalel, S.; Cameron, M.; Cherpelis, B.; Fenske, N.; Sondak, V.K.; Messina, J.; Tommasino, M.; Gheit, T.; et al. Cutaneous Human Papillomavirus Infection and Development of Subsequent Squamous Cell Carcinoma of the Skin. J. Skin Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.M.; Srivastava, R.K.; Elmets, C.A.; Athar, M. The mechanistic basis of arsenicosis: Pathogenesis of skin cancer. Cancer Lett. 2014, 354, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.J.; Chen, H.; Chen, J.Q.; Lei, Q.H.; Zheng, M.; Shao, Z.R. Immunolocalization of vimentin, keratin 17, Ki-67, involucrin, β-catenin and E-cadherin in cutaneous squamous cell carcinoma. Pathol. Oncol. Res. 2014, 20, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Abel, E.L.; Riggs, P.K.; Repass, J.; Hensley, S.C.; Schroeder, L.J.; Temple, A.; Chau, A.; McClellan, S.A.; Rho, O.; et al. Proteomic and pathway analyses reveal a network of inflammatory genes associated with differences in skin tumor promotion susceptibility in DBA/2 and C57BL/6 mice. Carcinogenesis 2012, 33, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, X.H.; Xu, F.; Sharma, C.; Wang, H.X.; Knoblich, K.; Rabinovitz, I.; Granter, S.R.; Hemler, M.E. Tetraspanin CD151 plays a key role in skin squamous cell carcinoma. Oncogene 2013, 32, 1772–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moloney, F.J.; Comber, H.; O’Lorcain, P.; O’Kelly, P.; Conlon, P.J.; Murphy, G.M. A population-based study of skin cancer incidence and prevalence in renal transplant recipients. Br. J. Dermatol. 2006, 154, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Glover, M.T.; Brown, J.; Navarrete, C.; Kwan, J.T.C.; Bodmer, J.; Bodmer, W.; Kennedy, L.J.; Leigh, I.M. HLA antigen frequencies in renal transplant recipients and immunocompetent patients with non-melanoma skin cancer. Eur. J. Cancer 1993, 29, 520–524. [Google Scholar] [CrossRef]
- Bouwes Bavinck, J.N.; Claas, F.H.; Hardie, D.R.; Green, A.; Vermeer, B.J.; Hardie, I.R. Relation between HLA antigens and skin cancer in renal transplant recipients in Queensland, Australia. J. Investig. Dermatol. 1997, 108, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Bouwes Bavinck, J.N.; Vermeer, B.J.; van der Woude, F.J.; Vandenbroucke, J.P.; Schreuder, G.M.; Thorogood, J.; Claas, F.H. Relation between skin cancer and HLA antigens in renal-transplant recipients. N. Engl. J. Med. 1991, 325, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Campoli, M.; Ferrone, S. HLA class I defects in malignant lesions: What have we learned? Keio J. Med. 2003, 52, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Barysch, M.J.; Behnke, S.; Dziunycz, P.; Schmid, B.; Riter, E.; Gnjatic, S.; Kristiansen, G.; Moch, H.; Knuth, A.; et al. Cancer-Testis antigens and immunosurveillance in human cutaneous squamous cell and basal cell carcinomas. Clin. Cancer Res. 2010, 16, 3562–3570. [Google Scholar] [CrossRef] [PubMed]
- Amiot, L. Biology of HLA-G in cancer: A candidate molecule for therapeutic intervention? Cell. Mol. Life Sci. 2011, 68, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Halliday, G.M.; Damian, D.L. Non-Melanoma Skin Cancer: Carcinogenesis and Chemoprevention. Pathology 2013, 45, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.E. Mechanisms underlying UV-induced immune suppression. Mutat. Res. 2005, 571, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Noordegraaf, M.; Maeda, A.; Torii, K.; Clausen, B.E.; Schwarz, T. Langerhans cells are required for UVR-induced immunosuppression. J. Investig. Dermatol. 2010, 130, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Glogau, R.G. The risk of progression to invasive disease. J. Am. Acad. Dermatol. 2000, 42, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 2012, 122, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2001, 344, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Kushida, Y.; Miki, H.; Ohmori, M. Loss of heterozygosity in actinic keratosis, squamous cell carcinoma and sun-exposed normal-appearing skin in Japanese: Difference between Japanese and Caucasians. Cancer Lett. 1999, 140, 169–175. [Google Scholar] [CrossRef]
- Krishnan, K.J.; Birch-Machin, M.A. The incidence of both tandem uplications and the common deletion in mtDNA from three distinct categories of sun exposed human skin and in prolonged culture of fibroblasts. J. Investig. Dermatol. 2006, 126, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; Murphy, G.; Ralph, N.; O’Gorman, S.M.; Murphy, J.E. Mitochondrial DNA deletion percentage in sun exposed and non sun exposed skin. J. Photochem. Photobiol. B 2016, 165, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Figueras, M.T.; Carrato, C.; Sáenz, X.; Puig, L.; Musulen, E.; Ferrándiz, C.; Ariza, A. Actinic keratosis with atypical basal cells (AK I) is the most common lesion associated with invasive squamous cell carcinoma of the skin. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.M.; Nissinen, L.M.; Ala-Aho, R.; Kallajoki, M.; Grénman, R.; Meri, S.; Peltonen, S.; Peltonen, J.; Kähäri, V.M. Complement factor H: A biomarker for progression of cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2014, 134, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Orlandi, A.; Costanza, G.; Di Stefani, A.; Piccioni, A.; Di Cesare, A.; Chiricozzi, A.; Ferlosio, A.; Peris, K.; Fargnoli, M.C. Expression of IL-23/Th17-related cytokines in basal cell carcinoma and in the response to medical treatments. PLoS ONE 2017, 12, e0183415. [Google Scholar] [CrossRef] [PubMed]
- Farshchian, M.; Kivisaari, A.; Ala-Aho, R.; Riihilä, P.; Kallajoki, M.; Grénman, R.; Peltonen, J.; Pihlajaniemi, T.; Heljasvaara, R.; Kähäri, V.M. Serpin peptidase inhibitor clade a member 1 (SerpinA1) is a novel biomarker for progression of cutaneous squamous cell carcinoma. Am. J. Pathol. 2011, 179, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Urosevic, M.; Dummer, R. Immunotherapy for nonmelanoma skin cancer: Does it have a future? Cancer 2002, 94, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Al-Dujaili, Z.; Henry, M.; Dorizas, A.S.; Sadick, N.S. Skin cancer concerns particular to women. Int. J. Womens Dermatol. 2017, 3, S49–S51. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.S.; Strange, R.C.; Lear, J.T. Basal cell carcinoma. Br. Med. J. 2003, 327, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Lupu, M.; Caruntu, C.; Ghita, M.A.; Voiculescu, V.; Voiculescu, S.; Rosca, A.E.; Caruntu, A.; Moraru, L.; Popa, I.M.; Calenic, B.; et al. Gene Expression and Proteome Analysis as Sources of Biomarkers in Basal Cell Carcinoma. Dis. Markers 2016. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Oro, A.E.; Higgins, K. Hair cycle regulation of Hedgehog signal reception. Dev. Biol. 2003, 255, 238–248. [Google Scholar] [CrossRef]
- Gorlin, R.J. Nevoid basal cell carcinoma syndrome. Dermatol. Clin. 1995, 13, 113–125. [Google Scholar] [PubMed]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.R. The hedgehog signalling pathway and its role in basal cell carcinoma. Cancer Metastasis Rev. 1999, 18, 261–284. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Undèn, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgård, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef] [PubMed]
- Asplund, A.; Gry Björklund, M.; Sundquist, C.; Strömberg, S.; Edlund, K.; Ostman, A.; Nilsson, P.; Pontén, F.; Lundeberg, J. Expression profiling of microdissected cell populations selected from basal cells in normal epidermis and basal cell carcinoma. Br. J. Dermatol. 2008, 158, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.A.; Bishop, G.A.; Lowes, M.A.; Cooke, B.; Barnetson, R.S.; Halliday, G.M. Cytokine profiles in spontaneously regressing basal cell carcinomas. Br. J. Dermatol. 2000, 143, 91–98. [Google Scholar] [CrossRef] [PubMed]
- El-Khalawany, M.A.; Abou-Bakr, A.A. Role of cyclooxygenase-2, ezrin and matrix metalloproteinase-9 as predictive markers for recurrence of basal cell carcinoma. J. Cancer Res. Ther. 2013, 9, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G.; Aznavoorian, S.; Liotta, L.A. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 1993, 9, 541–573. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, M.; McLintock, S.; Weerakoon, R.; Lomatschinsky, N.; Jones, S.; Rossi, A.G.; Weller, R.B. Enzyme-independent NO stores in human skin: Quantification and influence of UV radiation. J. Investig. Dermatol. 2009, 129, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Villiotou, V.; Deliconstantinos, G. Nitric oxide, peroxynitrite and nitrosocompounds formation by ultraviolet A (UVA) irradiated human squamous cell carcinoma: Potential role of nitric oxide in cancer prognosis. Anticancer Res. 1995, 15, 931–942. [Google Scholar] [PubMed]
- Maeda, A.; Nakata, M.; Yasuda, K.; Yukawa, T.; Saisho, S.; Okita, R.; Hirami, Y.; Shimizu, K. Influence of vascular endothelial growth factor single nucleotide polymorphisms on non-small cell lung cancer tumor angiogenesis. Oncol. Rep. 2013, 29, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Wang, Q.; Li, C.; Wang, Y.; Chen, X. VEGF stimulated the angiogenesis by promoting the mitochondrial functions. Oncotarget 2017, 8, 77020–77027. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.T.; Biselli, P.M.; Maniglia, J.V.; Pavarino-Bertelli, E.C.; Goloni-Bertollo, E.M. Genetic variability of vascular endothelial growth factor and prognosis of head and neck cancer in a Brazilian population. Braz. J. Med. Biol. Res. 2010, 43, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Jain, L.; Vargo, C.A.; Danesi, R.; Sissung, T.M.; Price, D.K.; Venzon, D.; Venitz, J.; Figg, W.D. The role of vascular endothelial growth factor SNPs as predictive and prognostic markers for major solid tumors. Mol. Cancer Ther. 2009, 8, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, Y.; Yu, X.; Ye, L.; Jiang, Y.; Cheng, Y. Expression of TP53, BCL-2, and VEGFA Genes in Esophagus Carcinoma and its Biological Significance. Med. Sci. Monit. 2015, 21, 3016–3022. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.J.; Liu, W.M.; Zhang, L. Association of VEGF Gene Polymorphisms with the Risk and Prognosis of Cutaneous Squamous Cell Carcinoma. Med. Sci. Monit. 2016, 22, 3658–3665. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.D.; Silva, C.; Rocha, A.; Nogueira, C.; Castro-Poças, F.; Araujo, A.; Matos, E.; Pereira, C.; Medeiros, R.; Lopes, C. Predictive clinical model of tumor response after chemoradiation in rectal cancer. Oncotarget 2017, 8, 58133–58151. [Google Scholar] [CrossRef] [PubMed]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, G.; Fletcher, A.; Slater, D.N. Basal cell carcinoma: A dermatopathological and molecular biological update. Br. J. Dermatol. 2003, 148, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Aszterbaum, M.; Epstein, J.; Oro, A.; Douglas, V.; LeBoit, P.E.; Scott, M.P.; Epstein, E.H. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat. Med. 1999, 5, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.G.; Kastan, M.B. DNA strand breaks: The DNA template alterations that trigger p53-dependent DNA damage response. Mol. Cell. Biol. 1994, 14, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Dinsdale, D.; Rufini, A.; Salomoni, P.; Knight, R.A.; Mueller, M.; Krammer, P.H.; Melino, G. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle 2007, 6, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, F.; Buechner, S.A.; Wernli, M.; Strebel, S.; Erb, P. Ultraviolet light downregulates CD95 ligand and TRAIL receptor expression facilitating actinic keratosis and squamous cell carcinoma formation. J. Investig. Dermatol. 2001, 117, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Ananthaswamy, H.N.; Price, J.E.; Goldberg, L.H.; Bale, E.S. Simultaneous transfer of tumorigenic and metastatic phenotypes by transfection with genomic DNA from a human cutaneous squamous cell carcinoma. J. Cell. Biochem. 1988, 36, 137–146. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Didona, D.; Paolino, G.; Bottoni, U.; Cantisani, C. Non Melanoma Skin Cancer Pathogenesis Overview. Biomedicines 2018, 6, 6. https://doi.org/10.3390/biomedicines6010006
Didona D, Paolino G, Bottoni U, Cantisani C. Non Melanoma Skin Cancer Pathogenesis Overview. Biomedicines. 2018; 6(1):6. https://doi.org/10.3390/biomedicines6010006
Chicago/Turabian StyleDidona, Dario, Giovanni Paolino, Ugo Bottoni, and Carmen Cantisani. 2018. "Non Melanoma Skin Cancer Pathogenesis Overview" Biomedicines 6, no. 1: 6. https://doi.org/10.3390/biomedicines6010006