
Human IGF2 Gene Epigenetic and Transcriptional Regulation: At the Core of Developmental Growth and Tumorigenic Behavior

Abstract
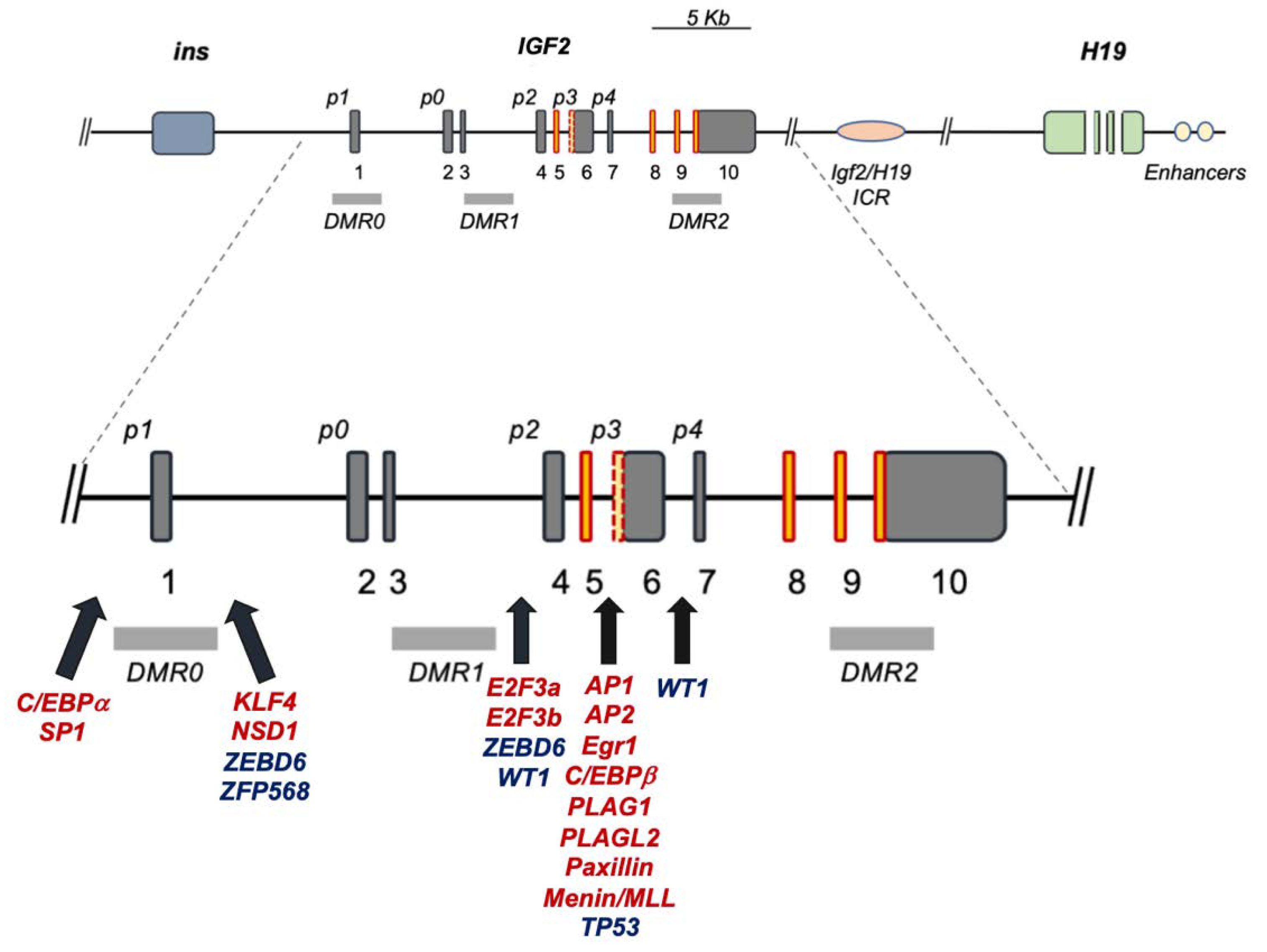
:1. IGF2 Gene Regulation at the Promoter and Transcript Level: An Unexploited View
2. The Human IGF2 Gene Structure: A Functional Overview
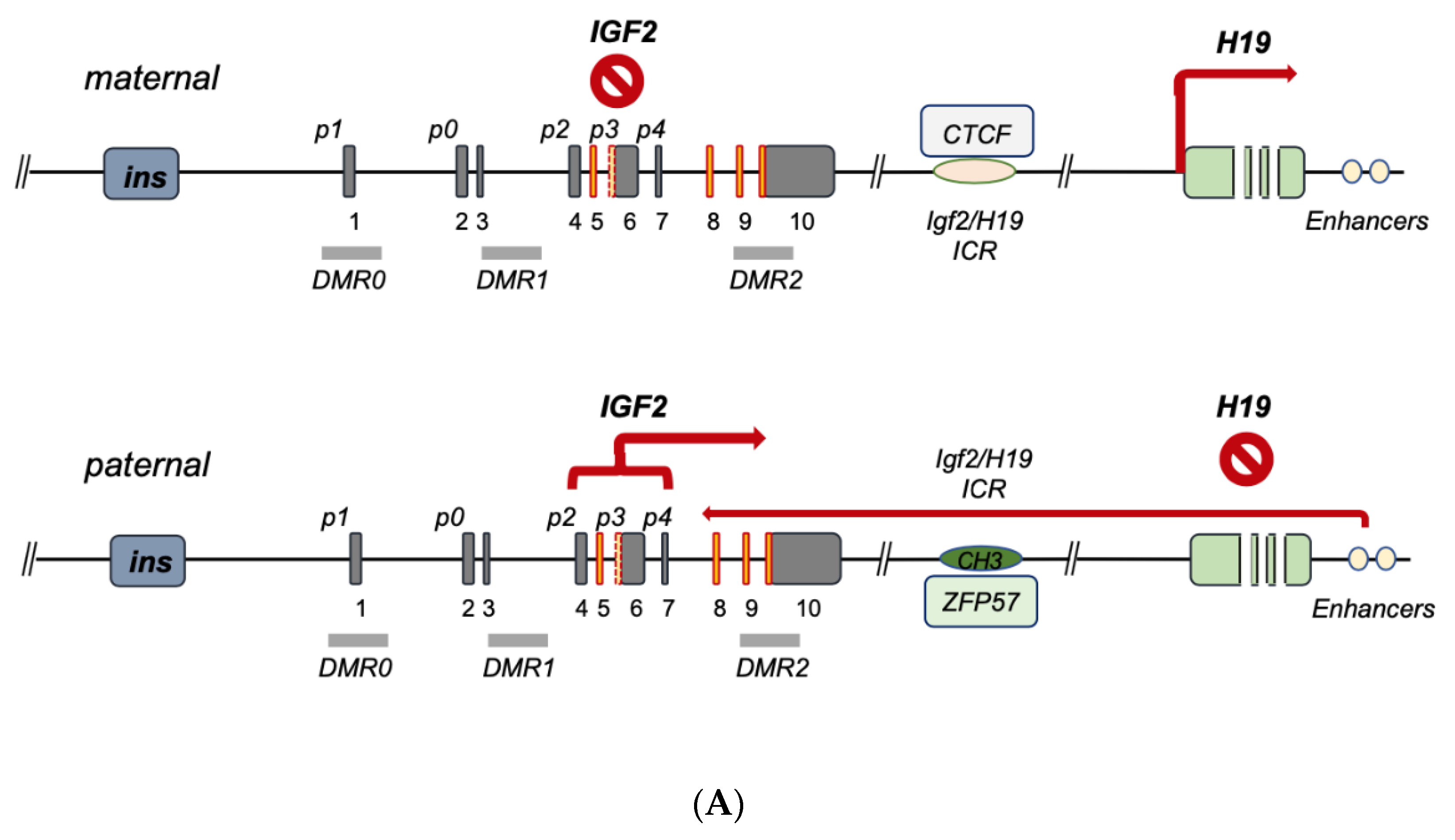
3. IGF2 Gene Regulation during Development and IGF2 Overexpression Syndromes

{kind=link}
{kind=link}
{kind=link}
| Imprinting Factor | Key Feature | Reference(s) |
|---|---|---|
| CTCF | binds maternal ICR and insulates IGF2-p activity | [35] |
| Cohesin | Cohesin is required for chromatin function at the H19/IGF2 locus | [42] |
| EZH2 | CH3-transferase component of PRC2 | [41] |
| SUZ12 | PRC2 component enabling ICR imprinting | [38] |
| Sox2/Oct3–4 | CTCF-like effect | [36] |
| Vigilin | ICR imprinting effect via CTCF binding | [39] |
| ZFP57 | Binds paternal ICR and maintains methylated status | [29] |
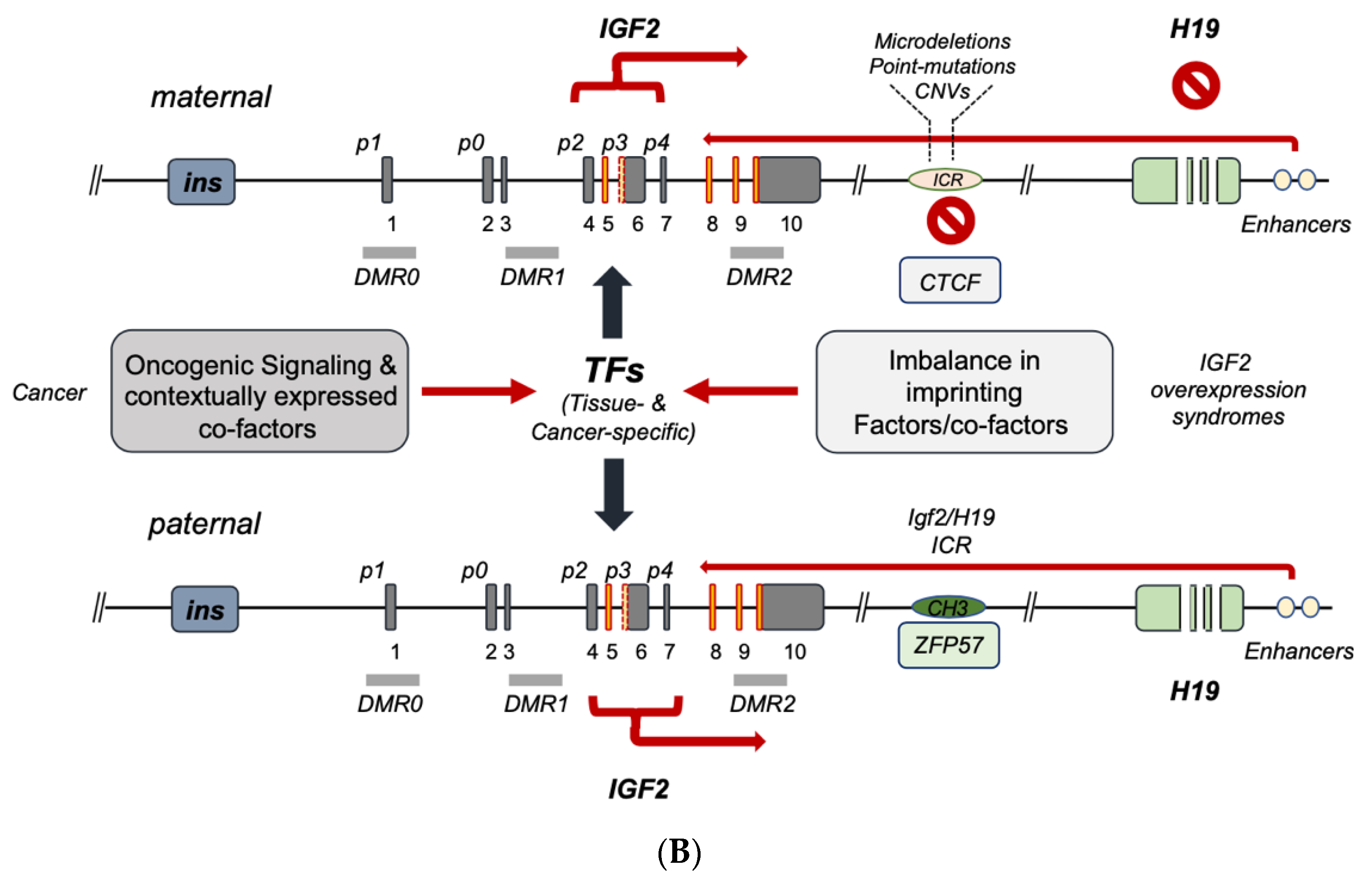
4. IGF2 Gene Transcriptional Control in Cancer
| Promoter Usage | Imprinting Control | Reference(s) |
|---|---|---|
| IGF2-p0 | Not imprinted Mostly active in fetal placenta | [15] |
| IGF2-p1 | Not imprinted—mostly active in postnatal Liver | [57,58] |
| IGF2-p2 | Imprinted- Mostly active during Fetal growth | [59] |
| IGF2-p3 & IGF2-p3/p4 (*) | Imprinted- Mostly active during Fetal growth, Widely reactivated in cancer | [51,57,58,60] |
4.1. Transcription Factors Regulating IGF2 through Its Fetal Promoters
4.2. Transcription Factors and Other Co-Factors Regulating IGF2 via Its Placental (p0) and Adult (p1) Promoters
4.3. Transcription Factors and Co-Factors Affecting IGF2 Transcription through Interactions Outside the IGF2 Direct Regulatory Cluster
4.4. Regulation of IGF2 Gene Expression via mRNA Stabilization and Beyond
4.5. Non-Coding RNA-Mediated Regulation of IGF2 Gene Expression
4.6. IGF2-p3 Functional Block: A Valuable Targeting Strategy for Cancer Gene Therapy?
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeChiara, T.M.; Robertson, E.J.; Efstratiadis, A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 1991, 64, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsen, J.L.; Duran, K.L.; Bartolomei, M.S. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998, 12, 3693–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Jones, P.A.; Chaillet, J.R.; Ferguson-Smith, A.C.; Barton, S.C.; Reik, W.; Surani, M.A. Parental imprinting: Potentially active chromatin of the repressed maternal allele of the mouse insulin-like growth factor II (Igf2) gene. Genes Dev. 1992, 6, 1843–1856. [Google Scholar] [CrossRef] [Green Version]
- Morison, I.M.; Becroft, D.M.; Taniguchi, T.; Woods, C.G.; Reeve, A.E. Somatic overgrowth associated with overexpression of insulin-like growth factor II. Nat. Med. 1996, 2, 311–316. [Google Scholar] [CrossRef]
- Ogawa, O.; Mishina, M.; Yoshida, O. Activation of imprinted genes in human carcinogenesis. Nihon rinsho. Jpn. J. Clin. Med. 1995, 53, 1009–1016. [Google Scholar]
- Taniguchi, T.; Sullivan, M.J.; Ogawa, O.; Reeve, A.E. Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proc. Natl. Acad. Sci. USA 1995, 92, 2159–2163. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.P.; Ng, E.K.O.; Ng, S.S.M.; Jin, H.; Yu, J.; Sung, J.J.Y.; Kwok, T.T. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2009, 31, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Constancia, M.; Dean, W.; Davies, K.; Bowden, L.; Murrell, A.; Feil, R.; Walter, J.; Kelsey, G. Igf2 imprinting in development and disease. Int. J. Dev. Biol. 2000, 44, 145–150. [Google Scholar]
- Sussenbach, J.S.; Rodenburg, R.J.T.; Scheper, W.; Holthuizen, P. Transcriptional and Post-Transcriptional Regulation of the Human IGF-II Gene Expression. In Current Directions in Insulin-Like Growth Factor Research; Le Roith, D., Raizada, M.K., Eds.; Springer US: Boston, MA, USA, 1993; pp. 63–71. [Google Scholar] [CrossRef]
- Mineo, R.; Fichera, E.; Liang, S.J.; Fujita-Yamaguchi, Y. Promoter usage for insulin-like growth factor-II in cancerous and benign human breast, prostate, and bladder tissues, and confirmation of a 10th exon. Biochem. Biophys. Res. Commun. 2000, 268, 886–892. [Google Scholar] [CrossRef]
- Baral, K.; Rotwein, P. The insulin-like growth factor 2 gene in mammals: Organizational complexity within a conserved locus. PLoS ONE 2019, 14, e0219155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holthuizen, P.; Van Dijk, M.A.; Rodenburg, R.J.; Koonen-Reemst, A.M.; Sussenbach, J.S. Transcriptional regulation of the major promoters of the human IGF-II gene. Mol. Reprod. Dev. 1993, 35, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Constancia, M.; Hemberger, M.; Hughes, J.; Dean, W.; Ferguson-Smith, A.; Fundele, R.; Stewart, F.; Kelsey, G.; Fowden, A.; Sibley, C.; et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002, 417, 945–948. [Google Scholar] [CrossRef]
- Monk, D.; Sanches, R.; Arnaud, P.; Apostolidou, S.; Hills, F.A.; Abu-Amero, S.; Murrell, A.; Friess, H.; Reik, W.; Stanier, P.; et al. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum. Mol. Genet. 2006, 15, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Christofori, G.; Naik, P.; Hanahan, D. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature 1994, 369, 414–418. [Google Scholar] [CrossRef]
- Scalia, P.; Giordano, A.; Williams, S.J. The IGF-II-Insulin Receptor Isoform-A Autocrine Signal in Cancer: Actionable Perspectives. Cancers 2020, 12, 366. [Google Scholar] [CrossRef] [Green Version]
- Mercader, J.M.; Liao, R.G.; Bell, A.D.; Dymek, Z.; Estrada, K.; Tukiainen, T.; Huerta-Chagoya, A.; Moreno-Macias, H.; Jablonski, K.A.; Hanson, R.L.; et al. A Loss-of-Function Splice Acceptor Variant in IGF2 Is Protective for Type 2 Diabetes. Diabetes 2017, 66, 2903–2914. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.; Bowden, L.; Smith, P.; Dean, W.; Hill, D.; Furuumi, H.; Sasaki, H.; Cattanach, B.; Reik, W. Disruption of mesodermal enhancers for Igf2 in the minute mutant. Development 2002, 129, 1657–1668. [Google Scholar] [CrossRef]
- Murrell, A.; Heeson, S.; Cooper, W.N.; Douglas, E.; Apostolidou, S.; Moore, G.E.; Maher, E.R.; Reik, W. An association between variants in the IGF2 gene and Beckwith-Wiedemann syndrome: Interaction between genotype and epigenotype. Hum. Mol. Genet. 2004, 13, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Charalambous, M.; Menheniott, T.R.; Bennett, W.R.; Kelly, S.M.; Dell, G.; Dandolo, L.; Ward, A. An enhancer element at the Igf2/H19 locus drives gene expression in both imprinted and non-imprinted tissues. Dev. Biol. 2004, 271, 488–497. [Google Scholar] [CrossRef]
- Kurukuti, S.; Tiwari, V.K.; Tavoosidana, G.; Pugacheva, E.; Murrell, A.; Zhao, Z.; Lobanenkov, V.; Reik, W.; Ohlsson, R. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc. Natl. Acad. Sci. USA 2006, 103, 10684–10689. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.K.; Erginer, E.; Huang, Z.; Visco, Z.; Hoyo, C. Genotype-Epigenotype Interaction at the IGF2 DMR. Genes 2015, 6, 777–789. [Google Scholar] [CrossRef] [Green Version]
- Papulino, C.; Chianese, U.; Nicoletti, M.M.; Benedetti, R.; Altucci, L. Preclinical and Clinical Epigenetic-Based Reconsideration of Beckwith-Wiedemann Syndrome. Front. Genet. 2020, 11, 563718. [Google Scholar] [CrossRef]
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000, 405, 486–489. [Google Scholar] [CrossRef]
- Pant, V.; Mariano, P.; Kanduri, C.; Mattsson, A.; Lobanenkov, V.; Heuchel, R.; Ohlsson, R. The nucleotides responsible for the direct physical contact between the chromatin insulator protein CTCF and the H19 imprinting control region manifest parent of origin-specific long-distance insulation and methylation-free domains. Genes Dev. 2003, 17, 586–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, A.; Murrell, A. Genomic imprinting: CTCF protects the boundaries. Curr. Biol. 2004, 14, R284–R286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Niu, B.; Hu, J.F.; Ge, S.; Wang, H.; Li, T.; Ling, J.; Steelman, B.N.; Qian, G.; Hoffman, A.R. Interruption of intrachromosomal looping by CCCTC binding factor decoy proteins abrogates genomic imprinting of human insulin-like growth factor II. J. Cell Biol. 2011, 193, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riso, V.; Cammisa, M.; Kukreja, H.; Anvar, Z.; Verde, G.; Sparago, A.; Acurzio, B.; Lad, S.; Lonardo, E.; Sankar, A.; et al. ZFP57 maintains the parent-of-origin-specific expression of the imprinted genes and differentially affects non-imprinted targets in mouse embryonic stem cells. Nucleic Acids Res. 2016, 44, 8165–8178. [Google Scholar] [CrossRef]
- Sparago, A.; Cerrato, F.; Vernucci, M.; Ferrero, G.B.; Silengo, M.C.; Riccio, A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat. Genet. 2004, 36, 958–960. [Google Scholar] [CrossRef]
- Poole, R.L.; Leith, D.J.; Docherty, L.E.; Shmela, M.E.; Gicquel, C.; Splitt, M.; Temple, I.K.; Mackay, D.J. Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur. J. Hum. Genet. 2012, 20, 240–243. [Google Scholar] [CrossRef]
- Abi Habib, W.; Brioude, F.; Azzi, S.; Salem, J.; Das Neves, C.; Personnier, C.; Chantot-Bastaraud, S.; Keren, B.; Le Bouc, Y.; Harbison, M.D.; et al. 11p15 ICR1 Partial Deletions Associated with IGF2/H19 DMR Hypomethylation and Silver-Russell Syndrome. Hum. Mutat. 2017, 38, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Arai, Y.; Sugawara, W.; Watanabe, N.; Honda, S.; Ohshima, J.; Soejima, H.; Nakadate, H.; Okita, H.; Hata, J.; et al. Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes Chromosomes Cancer 2008, 47, 712–727. [Google Scholar] [CrossRef]
- Hubertus, J.; Lacher, M.; Rottenkolber, M.; Müller-Höcker, J.; Berger, M.; Stehr, M.; von Schweinitz, D.; Kappler, R. Altered expression of imprinted genes in Wilms tumors. Oncol. Rep. 2011, 25, 817–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Hu, J.F.; Qiu, X.; Ling, J.; Chen, H.; Wang, S.; Hou, A.; Vu, T.H.; Hoffman, A.R. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol. Cell. Biol. 2008, 28, 6473–6482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, N.; Yamane, M.; Kouno, K.; Sato, K. Induction of DNA demethylation depending on two sets of Sox2 and adjacent Oct3/4 binding sites (Sox-Oct motifs) within the mouse H19/insulin-like growth factor 2 (Igf2) imprinted control region. J. Biol. Chem. 2012, 287, 44006–44016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashimoto, K.; Jozaki, K.; Kosho, T.; Matsubara, K.; Fuke, T.; Yamada, D.; Yatsuki, H.; Maeda, T.; Ohtsuka, Y.; Nishioka, K.; et al. A novel de novo point mutation of the OCT-binding site in the IGF2/H19-imprinting control region in a Beckwith-Wiedemann syndrome patient. Clin. Genet. 2014, 86, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ge, S.; Qian, G.; Li, W.; Cui, J.; Wang, G.; Hoffman, A.R.; Hu, J.F. Restoration of IGF2 imprinting by polycomb repressive complex 2 docking factor SUZ12 in colon cancer cells. Exp. Cell Res. 2015, 338, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yang, B.; Xie, X.; Wei, L.; Liu, W.; Yang, W.; Ge, Y.; Zhu, Q.; Zhang, J.; Jiang, L.; et al. Vigilin interacts with CCCTC-binding factor (CTCF) and is involved in CTCF-dependent regulation of the imprinted genes Igf2 and H19. FEBS J. 2014, 281, 2713–2725. [Google Scholar] [CrossRef]
- Hu, J.F.; Oruganti, H.; Vu, T.H.; Hoffman, A.R. The role of histone acetylation in the allelic expression of the imprinted human insulin-like growth factor II gene. Biochem. Biophys. Res. Commun. 1998, 251, 403–408. [Google Scholar] [CrossRef]
- Li, T.; Chen, H.; Li, W.; Cui, J.; Wang, G.; Hu, X.; Hoffman, A.R.; Hu, J. Promoter histone H3K27 methylation in the control of IGF2 imprinting in human tumor cell lines. Hum. Mol. Genet. 2014, 23, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Nativio, R.; Wendt, K.S.; Ito, Y.; Huddleston, J.E.; Uribe-Lewis, S.; Woodfine, K.; Krueger, C.; Reik, W.; Peters, J.M.; Murrell, A. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009, 5, e1000739. [Google Scholar] [CrossRef]
- Issa, J.P.; Vertino, P.M.; Boehm, C.D.; Newsham, I.F.; Baylin, S.B. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 11757–11762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, H.T.; Brown, L.J.; Fallin, M.D.; Rongione, M.A.; Bibikova, M.; Wickham, E.; Fan, J.B.; Feinberg, A.P. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J. Natl. Cancer Inst. 2007, 99, 1270–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Cai, M.; Peng, M.; Wang, F.; Zhai, X. miR-491-5p functions as a tumor suppressor by targeting IGF2 in colorectal cancer. Cancer Manag. Res. 2019, 11, 1805–1816. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Adam, G.; Cui, H.; Sandstedt, B.; Ohlsson, R.; Ekstrom, T.J. Expression, promoter usage and parental imprinting status of insulin-like growth factor II (IGF2) in human hepatoblastoma: Uncoupling of IGF2 and H19 imprinting. Oncogene 1995, 11, 221–229. [Google Scholar]
- Douc-Rasy, S.; Barrois, M.; Fogel, S.; Ahomadegbe, J.C.; Stéhelin, D.; Coll, J.; Riou, G. High incidence of loss of heterozygosity and abnormal imprinting of H19 and IGF2 genes in invasive cervical carcinomas. Uncoupling of H19 and IGF2 expression and biallelic hypomethylation of H19. Oncogene 1996, 12, 423–430. [Google Scholar]
- Vu, T.H.; Hoffman, A. Alterations in the promoter-specific imprinting of the insulin-like growth factor-II gene in Wilms’ tumor. J. Biol. Chem. 1996, 271, 9014–9023. [Google Scholar] [CrossRef] [Green Version]
- Hodzic, D.; Frey, B.; Marechal, D.; Scarcez, T.; Grooteclaes, M.; Winkler, R. Cloning of breakpoints in and downstream the IGF2 gene that are associated with overexpression of IGF2 transcripts in colorectal tumours. Oncogene 1999, 18, 4710–4717. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, T.; Frisk, T.; Gray, S.G.; von Schweinitz, D.; Pietsch, T.; Larsson, C.; Sandstedt, B.; Ekström, T.J. Methylation Changes in the Human IGF2 P3 Promoter Parallel IGF2 Expression in the Primary Tumor, Established Cell Line, and Xenograft of a Human Hepatoblastoma. Exp. Cell Res. 2001, 270, 88–95. [Google Scholar] [CrossRef]
- Kim, S.J.; Park, S.E.; Lee, C.; Lee, S.Y.; Jo, J.H.; Kim, J.M.; Oh, Y.K. Alterations in promoter usage and expression levels of insulin-like growth factor-II and H19 genes in cervical carcinoma exhibiting biallelic expression of IGF-II. Biochim. Biophys. Acta 2002, 1586, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wylie-Sears, J.; Boscolo, E.; Mulliken, J.B.; Bischoff, J. Genomic imprinting of IGF2 is maintained in infantile hemangioma despite its high level of expression. Mol. Med. 2004, 10, 117–123. [Google Scholar] [CrossRef]
- Murphy, S.K.; Huang, Z.; Wen, Y.; Spillman, M.A.; Whitaker, R.S.; Simel, L.R.; Nichols, T.D.; Marks, J.R.; Berchuck, A. Frequent IGF2/H19 domain epigenetic alterations and elevated IGF2 expression in epithelial ovarian cancer. Mol. Cancer Res. 2006, 4, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.; Katsaros, D.; Lu, L.; Canuto, E.M.; Benedetto, C.; Beeghly-Fadiel, A.; Yu, H. IGF-II promoter specific methylation and expression in epithelial ovarian cancer and their associations with disease characteristics. Oncol. Rep. 2011, 25, 203–213. [Google Scholar] [PubMed]
- Küffer, S.; Gutting, T.; Belharazem, D.; Sauer, C.; Michel, M.S.; Marx, A.; Trojan, L.; Ströbel, P. Insulin-like growth factor 2 expression in prostate cancer is regulated by promoter-specific methylation. Mol. Oncol. 2018, 12, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Romano, M.; Calabrò, A.; Pedone, P.V.; de Sio, I.; Persico, M.; Budillon, G.; Bruni, C.B.; Riccio, A.; Zarrilli, R. Activation of fetal promoters of insulin-like growth factors II gene in hepatitis C virus-related chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Hepatology 1996, 23, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nong, Z.; Ekström, C.; Larsson, E.; Nordlinder, H.; Hofmann, W.J.; Trautwein, C.; Odenthal, M.; Dienes, H.P.; Ekström, T.J.; et al. Disrupted IGF2 promoter control by silencing of promoter P1 in human hepatocellular carcinoma. Cancer Res. 1997, 57, 2048–2054. [Google Scholar] [PubMed]
- Lui, J.C.; Baron, J. Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc. Natl. Acad. Sci. USA 2013, 110, 6181–6186. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Meng, G.; Huang, L.; Guo, Q.N. Hypomethylation of the P3 promoter is associated with up-regulation of IGF2 expression in human osteosarcoma. Hum. Pathol. 2009, 40, 1441–1447. [Google Scholar] [CrossRef]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Caricasole, A.; Ward, A. Transactivation of mouse insulin-like growth factor II (IGF-II) gene promoters by the AP-1 complex. Nucleic Acids Res. 1993, 21, 1873–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, D.; Buhl, S.; Weber, S.; Jäger, R.; Schorle, H. The AP-2 family of transcription factors. Genome Biol. 2005, 6, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rietveld, L.E.; Koonen-Reemst, A.M.; Sussenbach, J.S.; Holthuizen, P.E. Dual role for transcription factor AP-2 in the regulation of the major fetal promoter P3 of the gene for human insulin-like growth factor II. Biochem. J. 1999, 338 Pt 3, 799–806. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, M.A.; Rodenburg, R.J.; Holthuizen, P.; Sussenbach, J.S. The liver-specific promoter of the human insulin-like growth factor II gene is activated by CCAAT/enhancer binding protein (C/EBP). Nucleic Acids Res. 1992, 20, 3099–3104. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, X.; Li, T.; Liu, C.; Zhao, Y. Increased hepatic Igf2 gene expression involves C/EBPβ in TCDD-induced teratogenesis in rats. Reprod. Toxicol. 2011, 32, 313–321. [Google Scholar] [CrossRef]
- Lekstrom-Himes, J.; Xanthopoulos, K.G. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J. Biol. Chem. 1998, 273, 28545–28548. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Yano, M.; Liu, S.Y.; Matsunaga, E.; Johnson, P.F.; Gonzalez, F.J. A novel cis-acting element controlling the rat CYP2D5 gene and requiring cooperativity between C/EBP beta and an Sp1 factor. Mol. Cell. Biol. 1994, 14, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.K.; Bae, M.H.; Ahn, M.Y.; Son, M.J.; Lee, Y.M.; Bae, M.K.; Lee, O.H.; Park, B.C.; Kim, K.W. Egr-1 mediates transcriptional activation of IGF-II gene in response to hypoxia. Cancer Res. 1999, 59, 5989–5994. [Google Scholar]
- Feldser, D.; Agani, F.; Iyer, N.V.; Pak, B.; Ferreira, G.; Semenza, G.L. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999, 59, 3915–3918. [Google Scholar]
- Scalia, P.; Giordano, A.; Martini, C.; Williams, S.J. Isoform- and Paralog-Switching in IR-Signaling: When Diabetes Opens the Gates to Cancer. Biomolecules 2020, 10, 1617. [Google Scholar] [CrossRef]
- Zheng, Q.F.; Xu, B.; Wang, H.M.; Ding, L.H.; Liu, J.Y.; Zhu, L.Y.; Qiu, H.; Zhang, L.; Ni, G.Y.; Ye, J.; et al. Epigenetic alterations contribute to promoter activity of imprinting gene IGF2. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 117–124. [Google Scholar] [CrossRef]
- Voz, M.L.; Agten, N.S.; Van de Ven, W.J.; Kas, K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000, 60, 106–113. [Google Scholar]
- Hensen, K.; Braem, C.; Declercq, J.; Van Dyck, F.; Dewerchin, M.; Fiette, L.; Denef, C.; Van de Ven, W.J. Targeted disruption of the murine Plag1 proto-oncogene causes growth retardation and reduced fertility. Dev. Growth Differ. 2004, 46, 459–470. [Google Scholar] [CrossRef]
- Zatkova, A.; Rouillard, J.M.; Hartmann, W.; Lamb, B.J.; Kuick, R.; Eckart, M.; von Schweinitz, D.; Koch, A.; Fonatsch, C.; Pietsch, T.; et al. Amplification and overexpression of the IGF2 regulator PLAG1 in hepatoblastoma. Genes Chromosomes Cancer 2004, 39, 126–137. [Google Scholar] [CrossRef]
- Akhtar, M.; Holmgren, C.; Gondor, A.; Vesterlund, M.; Kanduri, C.; Larsson, C.; Ekstrom, T.J. Cell type and context-specific function of PLAG1 for IGF2 P3 promoter activity. Int. J. Oncol. 2012, 41, 1959–1966. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Kashanchi, F.; Zhan, Q.; Zhan, S.; Brady, J.N.; Fornace, A.J.; Seth, P.; Helman, L.J. Regulation of insulin-like growth factor II P3 promotor by p53: A potential mechanism for tumorigenesis. Cancer Res. 1996, 56, 1367–1373. [Google Scholar]
- Haley, V.L.; Barnes, D.J.; Sandovici, I.; Constancia, M.; Graham, C.F.; Pezzella, F.; Buhnemann, C.; Carter, E.J.; Hassan, A.B. Igf2 pathway dependency of the Trp53 developmental and tumour phenotypes. EMBO Mol. Med. 2012, 4, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Dynkevich, Y.; Rother, K.I.; Whitford, I.; Qureshi, S.; Galiveeti, S.; Szulc, A.L.; Danoff, A.; Breen, T.L.; Kaviani, N.; Shanik, M.H.; et al. Tumors, IGF-2, and hypoglycemia: Insights from the clinic, the laboratory, and the historical archive. Endocr. Rev. 2013, 34, 798–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalia, P.; Williams, S.J.; Fujita-Yamaguchi, Y.; Giordano, A. Cell cycle control by the insulin-like growth factor signal: At the crossroad between cell growth and mitotic regulation. Cell Cycle 2022, 22, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, A.S.; Nguyen, M.; Braunschweig, M.; Nezer, C.; Collette, C.; Moreau, L.; Archibald, A.L.; Haley, C.S.; Buys, N.; Tally, M.; et al. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 2003, 425, 832–836. [Google Scholar] [CrossRef]
- Younis, S.; Schonke, M.; Massart, J.; Hjortebjerg, R.; Sundstrom, E.; Gustafson, U.; Bjornholm, M.; Krook, A.; Frystyk, J.; Zierath, J.R.; et al. The ZBED6-IGF2 axis has a major effect on growth of skeletal muscle and internal organs in placental mammals. Proc. Natl. Acad. Sci. USA 2018, 115, E2048–E2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Call, K.M.; Glaser, T.; Ito, C.Y.; Buckler, A.J.; Pelletier, J.; Haber, D.A.; Rose, E.A.; Kral, A.; Yeger, H.; Lewis, W.H.; et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990, 60, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Drummond, I.A.; Madden, S.L.; Rohwer-Nutter, P.; Bell, G.I.; Sukhatme, V.P.; Rauscher, F.J., 3rd. Repression of the insulin-like growth factor II gene by the Wilms tumor suppressor WT1. Science 1992, 257, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, F.J.; Morris, J.F.; Tournay, O.E.; Cook, D.M.; Curran, T. Binding of the Wilms’ Tumor Locus Zinc Finger Protein to the EGR-1 Consensus Sequence. Science 1990, 250, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Pooler, J.A.; Miyagawa, K.; Duarte, A.; Hastie, N.D.; Caricasole, A. Repression of promoters for the mouse insulin-like growth factor II-encoding gene (Igf-2) by products of the Wilms’ tumour suppressor gene wt1. Gene 1995, 167, 239–243. [Google Scholar] [CrossRef]
- Lee, Y.I.; Kim, S.J. Transcriptional repression of human insulin-like growth factor-II P4 promoter by Wilms’ tumor suppressor WT1. DNA Cell Biol. 1996, 15, 99–104. [Google Scholar] [CrossRef]
- Caricasole, A.; Duarte, A.; Larsson, S.H.; Hastie, N.D.; Little, M.; Holmes, G.; Todorov, I.; Ward, A. RNA binding by the Wilms tumor suppressor zinc finger proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 7562–7566. [Google Scholar] [CrossRef] [Green Version]
- Scharnhorst, V.; Menke, A.L.; Attema, J.; Haneveld, J.K.; Riteco, N.; van Steenbrugge, G.J.; van der Eb, A.J.; Jochemsen, A.G. EGR-1 enhances tumor growth and modulates the effect of the Wilms’ tumor 1 gene products on tumorigenicity. Oncogene 2000, 19, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Haber, D.A.; Sohn, R.L.; Buckler, A.J.; Pelletier, J.; Call, K.M.; Housman, D.E. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc. Natl. Acad. Sci. USA 1991, 88, 9618–9622. [Google Scholar] [CrossRef] [Green Version]
- Bickmore, W.A.; Oghene, K.; Little, M.H.; Seawright, A.; van Heyningen, V.; Hastie, N.D. Modulation of DNA binding specificity by alternative splicing of the Wilms tumor wt1 gene transcript. Science 1992, 257, 235–237. [Google Scholar] [CrossRef]
- Duarte, A.; Caricasole, A.; Graham, C.F.; Ward, A. Wilms’ tumour-suppressor protein isoforms have opposite effects on Igf2 expression in primary embryonic cells, independently of p53 genotype. Br. J. Cancer 1998, 77, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, K.; Jinno, Y.; Sohda, T.; Niikawa, N.; Ikeda, T. Promoter-specific insulin-like growth factor 2 gene imprinting in human fetal liver and hepatoblastoma. J. Pathol. 1998, 185, 91–98. [Google Scholar] [CrossRef]
- van Dijk, M.A.; Holthuizen, P.E.; Sussenbach, J.S. Elements required for activation of the major promoter of the human insulin-like growth factor II gene. Mol. Cell. Endocrinol. 1992, 88, 175–185. [Google Scholar] [CrossRef]
- Rodenburg, R.J.; Teertstra, W.; Holthuizen, P.E.; Sussenbach, J.S. Postnatal liver-specific expression of human insulin-like growth factor-II is highly stimulated by the transcriptional activators liver-enriched activating protein and CCAAT/enhancer binding protein-alpha. Mol. Endocrinol. 1995, 9, 424–434. [Google Scholar] [CrossRef]
- Tiwari, N.; Meyer-Schaller, N.; Arnold, P.; Antoniadis, H.; Pachkov, M.; van Nimwegen, E.; Christofori, G. Klf4 is a transcriptional regulator of genes critical for EMT, including Jnk1 (Mapk8). PLoS ONE 2013, 8, e57329. [Google Scholar] [CrossRef]
- Rowland, B.D.; Bernards, R.; Peeper, D.S. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat. Cell Biol. 2005, 7, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Schagdarsurengin, U.; Lammert, A.; Schunk, N.; Sheridan, D.; Gattenloehner, S.; Steger, K.; Wagenlehner, F.; Dansranjavin, T. Impairment of IGF2 gene expression in prostate cancer is triggered by epigenetic dysregulation of IGF2-DMR0 and its interaction with KLF4. Cell Commun. Signal. 2017, 15, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Higashimoto, K.; Miyake, N.; Morita, S.; Horii, T.; Kimura, M.; Suzuki, T.; Maeda, T.; Hidaka, H.; Aoki, S.; et al. DNA methylation analysis of multiple imprinted DMRs in Sotos syndrome reveals IGF2-DMR0 as a DNA methylation-dependent, P0 promoter-specific enhancer. FASEB J. 2020, 34, 960–973. [Google Scholar] [CrossRef] [Green Version]
- Rodenburg, R.J.; Holthuizen, P.E.; Sussenbach, J.S. A functional Sp1 binding site is essential for the activity of the adult liver-specific human insulin-like growth factor II promoter. Mol. Endocrinol. 1997, 11, 237–250. [Google Scholar] [CrossRef]
- Yang, P.; Wang, Y.; Hoang, D.; Tinkham, M.; Patel, A.; Sun, M.A.; Wolf, G.; Baker, M.; Chien, H.C.; Lai, K.N.; et al. A placental growth factor is silenced in mouse embryos by the zinc finger protein ZFP568. Science 2017, 356, 757–759. [Google Scholar] [CrossRef] [Green Version]
- Marasek, P.; Dzijak, R.; Studenyak, I.; Fiserova, J.; Ulicna, L.; Novak, P.; Hozak, P. Paxillin-dependent regulation of IGF2 and H19 gene cluster expression. J. Cell Sci. 2015, 128, 3106–3116. [Google Scholar] [CrossRef] [Green Version]
- Blattler, A.; Farnham, P.J. Cross-talk between site-specific transcription factors and DNA methylation states. J. Biol. Chem. 2013, 288, 34287–34294. [Google Scholar] [CrossRef] [Green Version]
- Borensztein, M.; Monnier, P.; Court, F.; Louault, Y.; Ripoche, M.A.; Tiret, L.; Yao, Z.; Tapscott, S.J.; Forne, T.; Montarras, D.; et al. Myod and H19-Igf2 locus interactions are required for diaphragm formation in the mouse. Development 2013, 140, 1231–1239. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.L.; Wächter, K.; Mühleck, B.; Pazaitis, N.; Köhn, M.; Lederer, M.; Hüttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of cancer progression? Cell. Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, H.T.; Sorrell, A.M.; Siddle, K. Two isoforms of the mRNA binding protein IGF2BP2 are generated by alternative translational initiation. PLoS ONE 2012, 7, e33140. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, K.E.; Noubissi, F.K.; Katti, P.S.; Hahn, C.M.; Davey, S.R.; Lundsmith, E.T.; Klein-Szanto, A.J.; Rhim, A.D.; Spiegelman, V.S.; Rustgi, A.K. IMP1 promotes tumor growth, dissemination and a tumor-initiating cell phenotype in colorectal cancer cell xenografts. Carcinogenesis 2013, 34, 2647–2654. [Google Scholar] [CrossRef] [Green Version]
- Gutschner, T.; Hämmerle, M.; Pazaitis, N.; Bley, N.; Fiskin, E.; Uckelmann, H.; Heim, A.; Groβ, M.; Hofmann, N.; Geffers, R.; et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology 2014, 59, 1900–1911. [Google Scholar] [CrossRef]
- Müller, S.; Bley, N.; Glaß, M.; Busch, B.; Rousseau, V.; Misiak, D.; Fuchs, T.; Lederer, M.; Hüttelmaier, S. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018, 46, 6285–6303. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Ji, F.; Wright, J.; Minichiello, L.; Sadreyev, R.; Avruch, J. IGF2 mRNA binding protein-2 is a tumor promoter that drives cancer proliferation through its client mRNAs IGF2 and HMGA1. eLife 2017, 6, e27155. [Google Scholar] [CrossRef]
- Ennajdaoui, H.; Howard, J.M.; Sterne-Weiler, T.; Jahanbani, F.; Coyne, D.J.; Uren, P.J.; Dargyte, M.; Katzman, S.; Draper, J.M.; Wallace, A.; et al. IGF2BP3 Modulates the Interaction of Invasion-Associated Transcripts with RISC. Cell Rep. 2016, 15, 1876–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, N.; Rapley, J.; Angel, M.; Yanik, M.F.; Blower, M.D.; Avruch, J. mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 2011, 25, 1159–1172. [Google Scholar] [CrossRef] [Green Version]
- Liao, B.; Hu, Y.; Herrick, D.J.; Brewer, G. The RNA-binding protein IMP-3 is a translational activator of insulin-like growth factor II leader-3 mRNA during proliferation of human K562 leukemia cells. J. Biol. Chem. 2005, 280, 18517–18524. [Google Scholar] [CrossRef] [Green Version]
- Hüttelmaier, S.; Zenklusen, D.; Lederer, M.; Dictenberg, J.; Lorenz, M.; Meng, X.; Bassell, G.J.; Condeelis, J.; Singer, R.H. Spatial regulation of beta-actin translation by Src-dependent phosphorylation of ZBP1. Nature 2005, 438, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.Y.; Zhu, Z.M.; Pei, D.S. The biological function of IGF2BPs and their role in tumorigenesis. Investig. New Drugs 2021, 39, 1682–1693. [Google Scholar] [CrossRef]
- Eun, B.; Sampley, M.L.; Good, A.L.; Gebert, C.M.; Pfeifer, K. Promoter cross-talk via a shared enhancer explains paternally biased expression of Nctc1 at the Igf2/H19/Nctc1 imprinted locus. Nucleic Acids Res. 2013, 41, 817–826. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Li, J.; Singh, J.; Alif, R.; Vazquez-Padron, R.I.; Gomes, S.A.; Hare, J.M.; Shehadeh, L.A. miR-30e targets IGF2-regulated osteogenesis in bone marrow-derived mesenchymal stem cells, aortic smooth muscle cells, and ApoE−/− mice. Cardiovasc. Res. 2015, 106, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Sun, Y.; Chen, J. IGF-II is regulated by microRNA-125b in skeletal myogenesis. J. Cell Biol. 2011, 192, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Seol, H.S.; Akiyama, Y.; Lee, S.E.; Shimada, S.; Jang, S.J. Loss of miR-100 and miR-125b results in cancer stem cell properties through IGF2 upregulation in hepatocellular carcinoma. Sci. Rep. 2020, 10, 21412. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Dong, J.; He, X.; Shen, L.; Long, C.; Liu, F.; Liu, X.; Lin, T.; He, D.; Wei, G. MiR-155-5p exerts tumor-suppressing functions in Wilms tumor by targeting IGF2 via the PI3K signaling pathway. Biomed. Pharmacother. 2020, 125, 109880. [Google Scholar] [CrossRef]
- Zhuang, S.T.; Cai, Y.J.; Liu, H.P.; Qin, Y.; Wen, J.F. LncRNA NEAT1/miR-185-5p/IGF2 axis regulates the invasion and migration of colon cancer. Mol. Genet. Genom. Med. 2020, 8, e1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Wang, P.; Du, X.; Wang, W.; Zhang, T.; Chen, Y. Direct repression of IGF2 is implicated in the anti-angiogenic function of microRNA-210 in human retinal endothelial cells. Angiogenesis 2018, 21, 313–323. [Google Scholar] [CrossRef]
- Li, G.; Luo, W.; Abdalla, B.A.; Ouyang, H.; Yu, J.; Hu, F.; Nie, Q.; Zhang, X. miRNA-223 upregulated by MYOD inhibits myoblast proliferation by repressing IGF2 and facilitates myoblast differentiation by inhibiting ZEB1. Cell Death Dis. 2017, 8, e3094. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Roth, A.; Yu, M.; Morris, R.; Bersani, F.; Rivera, M.N.; Lu, J.; Shioda, T.; Vasudevan, S.; Ramaswamy, S.; et al. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev. 2013, 27, 2543–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Shi, X.; Chang, W.; Li, Y.; Wang, L.; Wang, L. Epigenetic alterations of the Igf2 promoter and the effect of miR-483-5p on its target gene expression in esophageal squamous cell carcinoma. Mol. Med. Rep. 2018, 17, 2251–2256. [Google Scholar] [CrossRef]
- Gailhouste, L.; Liew, L.C.; Yasukawa, K.; Hatada, I.; Tanaka, Y.; Kato, T.; Nakagama, H.; Ochiya, T. MEG3-derived miR-493-5p overcomes the oncogenic feature of IGF2-miR-483 loss of imprinting in hepatic cancer cells. Cell Death Dis. 2019, 10, 553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Pan, D.; Zhang, S.; Wu, Q.; Zhen, L.; Liu, S.; Chen, J.; Lin, R.; Hong, Q.; Zheng, X.; et al. Exosomal miR-543 Inhibits the Proliferation of Ovarian Cancer by Targeting IGF2. J. Immunol. Res. 2022, 2022, 2003739. [Google Scholar] [CrossRef]
- Gao, W.; Gu, Y.; Li, Z.; Cai, H.; Peng, Q.; Tu, M.; Kondo, Y.; Shinjo, K.; Zhu, Y.; Zhang, J.; et al. miR-615-5p is epigenetically inactivated and functions as a tumor suppressor in pancreatic ductal adenocarcinoma. Oncogene 2015, 34, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Icli, B.; Wu, W.; Ozdemir, D.; Li, H.; Cheng, H.S.; Haemmig, S.; Liu, X.; Giatsidis, G.; Avci, S.N.; Lee, N.; et al. MicroRNA-615-5p Regulates Angiogenesis and Tissue Repair by Targeting AKT/eNOS (Protein Kinase B/Endothelial Nitric Oxide Synthase) Signaling in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1458–1474. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.S.; Stroup, E.K.; Budhipramono, A.; Rakheja, D.; Nichols-Vinueza, D.; Xu, L.; Stuart, S.H.; Shukla, A.A.; Fraire, C.; Mendell, J.T.; et al. Mutations in microRNA processing genes in Wilms tumors derepress the IGF2 regulator PLAG1. Genes Dev. 2018, 32, 996–1007. [Google Scholar] [CrossRef] [Green Version]
- Xing, S.; Tian, Z.; Zheng, W.; Yang, W.; Du, N.; Gu, Y.; Yin, J.; Liu, H.; Jia, X.; Huang, D.; et al. Hypoxia downregulated miR-4521 suppresses gastric carcinoma progression through regulation of IGF2 and FOXM1. Mol. Cancer 2021, 20, 9. [Google Scholar] [CrossRef]
- Liu, G.; Guo, W.; Chen, G.; Li, W.; Cui, Y.; Qin, J.; Peng, J. Lnc-MCEI mediated the chemosensitivity of esophageal squamous cell carcinoma via miR-6759-5p to competitively regulate IGF2. Int. J. Biol. Sci. 2020, 16, 2938–2950. [Google Scholar] [CrossRef] [PubMed]
- JnBaptiste, C.K.; Gurtan, A.M.; Thai, K.K.; Lu, V.; Bhutkar, A.; Su, M.J.; Rotem, A.; Jacks, T.; Sharp, P.A. Dicer loss and recovery induce an oncogenic switch driven by transcriptional activation of the oncofetal Imp1-3 family. Genes Dev. 2017, 31, 674–687. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Katsaros, D.; de la Longrais, I.A.; Sochirca, O.; Yu, H. Hypermethylation of let-7a-3 in epithelial ovarian cancer is associated with low insulin-like growth factor-II expression and favorable prognosis. Cancer Res. 2007, 67, 10117–10122. [Google Scholar] [CrossRef] [Green Version]
- Fawzy, I.O.; Hamza, M.T.; Hosny, K.A.; Esmat, G.; Abdelaziz, A.I. Abrogating the interplay between IGF2BP1, 2 and 3 and IGF1R by let-7i arrests hepatocellular carcinoma growth. Growth Factors 2016, 34, 42–50. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Yu, Z.; Zhou, Y.; Tu, J.; Lou, J.; Wang, Y. Circular RNA Circ100084 functions as sponge of miR-23a-5p to regulate IGF2 expression in hepatocellular carcinoma. Mol. Med. Rep. 2020, 21, 2395–2404. [Google Scholar] [CrossRef] [Green Version]
- Veronese, A.; Lupini, L.; Consiglio, J.; Visone, R.; Ferracin, M.; Fornari, F.; Zanesi, N.; Alder, H.; D’Elia, G.; Gramantieri, L.; et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010, 70, 3140–3149. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, X.; Zeng, K.; Xu, M.; He, B.; Pan, Y.; Sun, H.; Pan, B.; Xu, X.; Xu, T.; et al. DNA-methylation-mediated silencing of miR-486-5p promotes colorectal cancer proliferation and migration through activation of PLAGL2/IGF2/β-catenin signal pathways. Cell Death Dis. 2018, 9, 1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Jia, Y.; Jia, L.; Li, T.; Yang, L.; Zhang, G. MicroRNA 615-3p Inhibits the Tumor Growth and Metastasis of NSCLC via Inhibiting IGF2. Oncol. Res. 2019, 27, 269–279. [Google Scholar] [CrossRef]
- Fawzy, I.O.; Hamza, M.T.; Hosny, K.A.; Esmat, G.; El Tayebi, H.M.; Abdelaziz, A.I. miR-1275: A single microRNA that targets the three IGF2-mRNA-binding proteins hindering tumor growth in hepatocellular carcinoma. FEBS Lett. 2015, 589, 2257–2265. [Google Scholar] [CrossRef] [Green Version]
- Henley, M.J.; Koehler, A.N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nature reviews. Drug Discov. 2021, 20, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Kaniskan, H.U.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, K.T.G.; Jaime-Figueroa, S.; Burgess, M.; Nalawansha, D.A.; Dai, K.; Hu, Z.; Bebenek, A.; Holley, S.A.; Crews, C.M. Targeted degradation of transcription factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem. Biol. 2021, 28, 648–661.e5. [Google Scholar] [CrossRef] [PubMed]



| Transcription Factor | TF Motif(s) in hIGF2 Promoter | Effect on IGF2 Gene Transcription | Reference(s) |
|---|---|---|---|
| AP1 | p3 | Activator | [62] |
| AP2 | p3 | Activator | [64] |
| C/EBPα | p1 | Activator | [94] |
| C/EBPβ | p3 | Activator | [66] |
| E2F3 | p2 | Activator | [59] |
| Egr1(Krox24) | p3 | Activator | [69] |
| Egr2(Krox20) | p3 | Activator | [10] |
| KLF4 | p0 | Activator | [98] |
| Menin/MLL | p3 | Activator | [72] |
| NSD1 | p0 | Activator | [99] |
| PLAG1 | p3 | Activator | [76] |
| Paxillin | p3 | Activator | [102] |
| TP53 | p3 | Repressor | [72] |
| SP1 | p1 | Activator | [100] |
| ZBED6 | p1-p2 | Repressor | [82] |
| ZPF568 | p0 | Repressor | [101] |
| WT1 | p2/p4 | Repressor | [85,87] |
| miRNA | Features | References |
|---|---|---|
| Nctc1 | Coregulated with IGF2 at muscle enhancer | [116] |
| Let-7 | Suppresses IMP1-3 and their oncogenic potential | [133] |
| Let-7a/a-3 | Let7a-3 hypermethylation associated with low IGF2 in ovarian and Breast cancer | [134] |
| Let-7i | Suppresses IGF2BP2-3 in HCC | [135] |
| miR-23a-5p | Suppresses IGF2 and its inhibition by circular non-coding RNA (100084) stimulates HCC | [136] |
| miR-30e | Suppressed IGF2 in mesenchymal cells | [117] |
| miR-100 | Confers stem cell features to HCC | [119] |
| miR-125b | Suppresses IGF2 in skeletal muscle Confers stem cell features to HCC | [118] |
| miR-155-5p | Suppresses IGF2 and PI3K-AKT in WT | [120] |
| miR-185-5p | Mediates NEAT1 upregulation of IGF2 in CRC | [121] |
| miR-210 | Suppresses IGF2 in HRECs | [122] |
| miR-223 | Suppresses IGF2 and ZEB1 in myoblasts | [123] |
| miR-483-3p | Is co-regulated and over-expressed in WT, CRC, Breast ca, and HCC | [137] |
| miR-483-5p | Overexpressed in WT/ Enhances IGF2 Increased in low methylated IGF2 promoter ESCC | [124] |
| miR-486-5p | Upregulate IGF2/βCatenin axis effects in CRC by suppression of PLAG2 | [138] |
| miR-491-5p | Suppresses IGF2 in CRC | [46] |
| miR-493-5p | Suppresses miR-483-5p in HCC/ inhibits IGF2 | [126] |
| miR-543 | Suppresses IGF2 and ovarian ca cells proliferation | [127] |
| miR-4521 | Suppresses IGF2 and FOXM in gastric ca | [131] |
| miR-615-3p | Inhibits IGF2 in NSLC | [139] |
| miR-615-5p | Suppresses IGF2 in Human PDAC- Inhibits angiogenesis by targeting IGF2 in ECs | [128] |
| miR-1275 | Suppresses IGF2BP1-3 and inhibits HCC malignant growth | [140] |
| miR-6759-5p | Suppresses IGF2 and mediates the competing effects of lnc-MCEI in ESCC | [132] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalia, P.; Williams, S.J.; Fujita-Yamaguchi, Y. Human IGF2 Gene Epigenetic and Transcriptional Regulation: At the Core of Developmental Growth and Tumorigenic Behavior. Biomedicines 2023, 11, 1655. https://doi.org/10.3390/biomedicines11061655
Scalia P, Williams SJ, Fujita-Yamaguchi Y. Human IGF2 Gene Epigenetic and Transcriptional Regulation: At the Core of Developmental Growth and Tumorigenic Behavior. Biomedicines. 2023; 11(6):1655. https://doi.org/10.3390/biomedicines11061655
Chicago/Turabian StyleScalia, Pierluigi, Stephen J. Williams, and Yoko Fujita-Yamaguchi. 2023. "Human IGF2 Gene Epigenetic and Transcriptional Regulation: At the Core of Developmental Growth and Tumorigenic Behavior" Biomedicines 11, no. 6: 1655. https://doi.org/10.3390/biomedicines11061655