
The First Large Deletion of ATL3 Identified in a Patient Presenting with a Sensory Polyneuropathy
 , and
, and 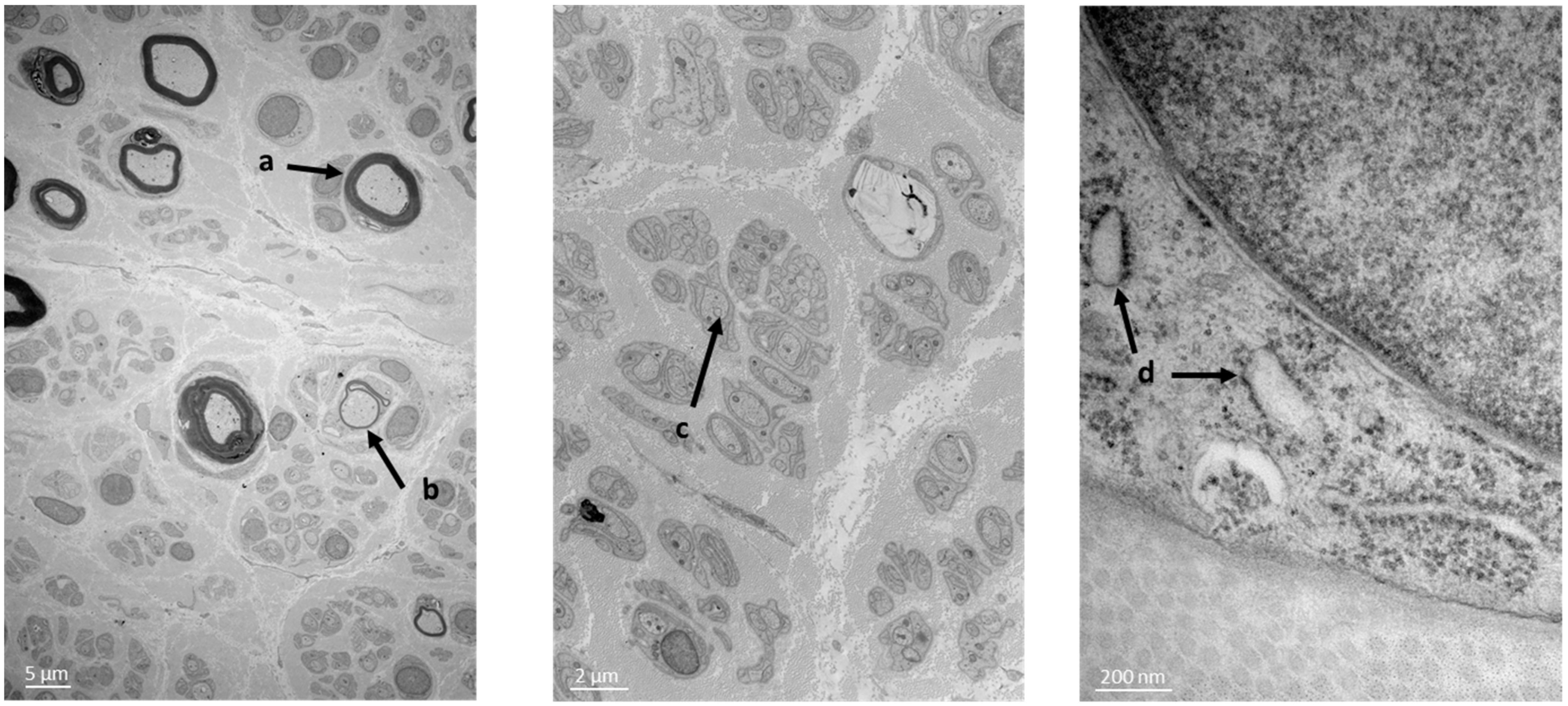{kind=link}
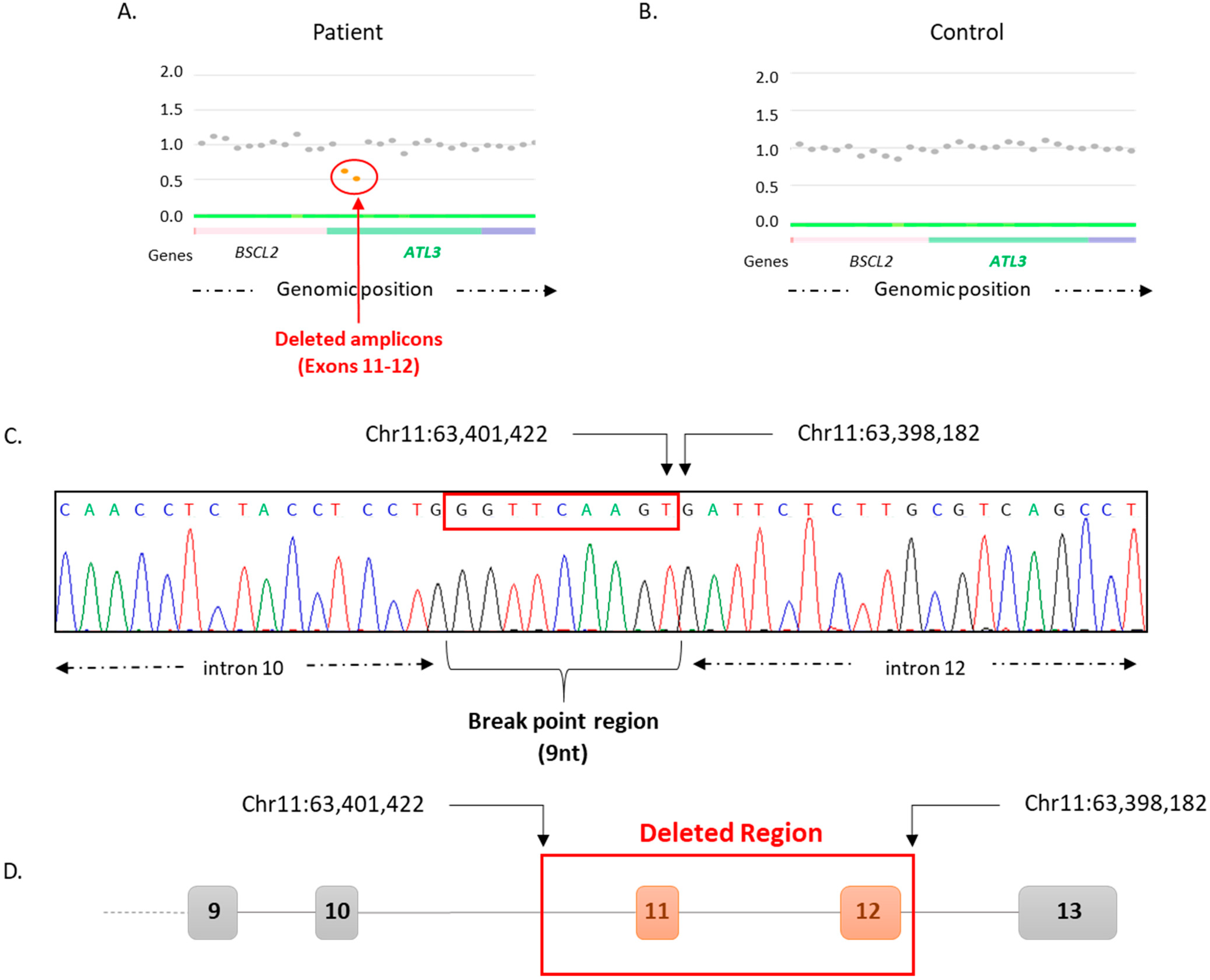{kind=link}
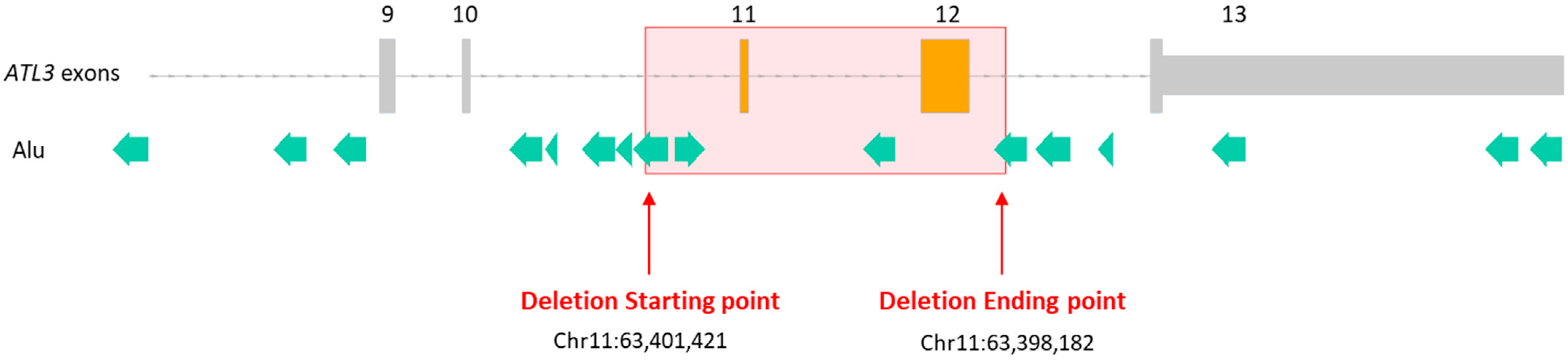{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient’s Samples
2.2. Electronic Microscopy
2.3. Next Generation Sequencing (NGS) and Bioinformatics Analysis
2.4. Cartography
3. Results
3.1. Patient’s Phenotype
3.2. Structural Variant Detection
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Auer-Grumbach, M. Hereditary sensory and autonomic neuropathies. Handb. Clin. Neurol. 2013, 115, 893–906. [Google Scholar] [CrossRef]
- Dyck, P.J.; Chance, P.F.; Lebo, R.; Carney, J.A. Neuronal Atrophy and Degeneration Predominantly Affecting Peripheral Sensory and Autonomic Neurons. In Hereditary Motor and Sensory Neuropathy, 3rd ed.; Dyck, P.J., Thomas, P.K., Eds.; WB Saunders Company: Philadelphia, PA, USA, 1993; Volume 2, pp. 1065–1093. [Google Scholar]
- Dyck, P.J.; Chance, P.F.; Lebo, R.; Carney, J.A. Hereditary Motor and Sensory Neuropathy, 3rd ed.; Dyck, P.J., Thomas, P.K., Eds.; WB Saunders Company: Philadelphia, PA, USA, 1993; Volume 2, pp. 1094–1136. [Google Scholar]
- Zhu, P.P.; Patterson, A.; Lavoie, B.; Stadler, J.; Shoeb, M.; Patel, R.; Blackstone, C. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J. Biol. Chem. 2003, 278, 49063–49071. [Google Scholar] [CrossRef]
- Kornak, U.; Mademan, I.; Schinke, M.; Voigt, M.; Krawitz, P.; Hecht, J.; Barvencik, F.; Schinke, T.; Gießelmann, S.; Beil, F.T.; et al. Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain 2014, 137, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Rismanchi, N.; Soderblom, C.; Stadler, J.; Zhu, P.P.; Blackstone, C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet. 2008, 17, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Shibata, Y.; Zhu, P.P.; Voss, C.; Rismanchi, N.; Prinz, W.A.; Rapoport, T.A.; Blackstone, C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 2009, 138, 549–561. [Google Scholar] [CrossRef]
- Wang, S.; Tukachinsky, H.; Romano, F.B.; Rapoport, T.A. Cooperation of the ER-shaping proteins atlastin, lunapark, and reticulons to generate a tubular membrane network. Elife 2016, 5, e18605. [Google Scholar] [CrossRef]
- Krols, M.; Detry, S.; Asselbergh, B.; Almeida-Souza, L.; Kremer, A.; Lippens, S.; De Rycke, R.; De Winter, V.; Müller, F.J.; Kurth, I.; et al. Sensory-Neuropathy-Causing Mutations in ATL3 Cause Aberrant ER Membrane Tethering. Cell Rep. 2018, 23, 2026–2038. [Google Scholar] [CrossRef]
- Zhu, P.P.; Soderblom, C.; Tao-Cheng, J.H.; Stadler, J.; Blackstone, C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet. 2006, 15, 1343–1353. [Google Scholar] [CrossRef]
- Sulek, A.; Elert, E.; Rajkiewicz, M.; Zdzienicka, E.; Stepniak, I.; Krysa, W.; Zaremba, J. Screening for the hereditary spastic paraplaegias SPG4 and SPG3A with the multiplex ligation-dependent probe amplification technique in a large population of affected individuals. Neurol. Sci. 2013, 34, 239–242. [Google Scholar] [CrossRef]
- Fischer, D.; Schabhüttl, M.; Wieland, T.; Windhager, R.; Strom, T.M.; Auer-Grumbach, M. A novel missense mutation confirms ATL3 as a gene for hereditary sensory neuropathy type 1. Brain 2014, 137, e286. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, C.; Cao, L.; Song, J.; Xu, X.; Zhang, B.; Chen, B.; Zhao, G. ATL3 gene mutation in a Chinese family with hereditary sensory neuropathy type 1F. J. Peripher. Nerv. Syst. 2019, 24, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Cintra, V.P.; Dohrn, M.F.; Tomaselli, P.J.; Figueiredo, F.B.; Marques, S.E.; Camargos, S.T.; Barbosa, L.S.M.; Rebelo, A.; Abreu, L.; Danzi, M.; et al. Rare mutations in ATL3, SPTLC2 and SCN9A explaining hereditary sensory neuropathy and congenital insensitivity to pain in a Brazilian cohort. J. Neurol. Sci. 2021, 427, 117498. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Jafari Khamirani, H.; Baneshi, M.; Kamal, N.; Manoocheri, J.; Saffar, M.; Dianatpour, M.; Tabei, S.M.B.; Dastgheib, S.A. A novel nonsense variant in the ATL3 gene is associated with disturbed pain sensitivity, numbness of distal limbs and muscle weakness. Ann. Hum. Genet. 2023. Available online: https://onlinelibrary.wiley.com/doi/epdf/10.1111/ahg.12501 (accessed on 9 May 2023). [CrossRef]
- Derouault, P.; Chauzeix, J.; Rizzo, D.; Miressi, F.; Magdelaine, C.; Bourthoumieu, S.; Durand, K.; Dzugan, H.; Feuillard, J.; Sturtz, F.; et al. CovCopCan: An Efficient Tool to Detect Copy Number Variation from Amplicon Sequencing Data in Inherited Diseases and Cancer. PLoS Comput. Biol. 2020, 16, e1007503. [Google Scholar] [CrossRef] [PubMed]
- Miressi, F.; Faye, P.A.; Pyromali, I.; Bourthoumieu, S.; Derouault, P.; Husson, M.; Favreau, F.; Sturtz, F.; Magdelaine, C.; Lia, A.S. A mutation can hide another one: Think Structural Variants! Comput. Struct. Biotechnol. J. 2020, 18, 2095–2099. [Google Scholar] [CrossRef]
- Hahne, F.; Ivanek, R. Statistical Genomics: Methods and Protocols. In Visualizing Genomic Data Using Gviz and Bioconductor; Mathé, E., Davis, S., Eds.; Springer: New York, NY, USA, 2016; pp. 335–351. ISBN 978-1-4939-3578-9. [Google Scholar] [CrossRef]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef]
- Budczies, J.; Pfarr, N.; Stenzinger, A.; Treue, D.; Endris, V.; Ismaeel, F.; Bangemann, N.; Blohmer, J.U.; Dietel, M.; Loibl, S.; et al. Ioncopy: A novel method for calling copy number alterations in amplicon sequencing data including significance assessment. Oncotarget 2016, 7, 13236–13247. [Google Scholar] [CrossRef]
- Derouault, P.; Parfait, B.; Moulinas, R.; Barrot, C.C.; Sturtz, F.; Merillou, S.; Lia, A.S. ‘COV’COP’ allows to detect CNVs responsible for inherited diseases among amplicons sequencing data. Bioinformatics 2017, 33, 1586–1588. [Google Scholar] [CrossRef]
- Kang, Y.; Nam, S.H.; Park, K.S.; Kim, Y.; Kim, J.W.; Lee, E.; Ko, J.M.; Lee, K.A.; Park, I. DeviCNV: Detection and visualization of exon-level copy number variants in targeted next-generation sequencing data. BMC Bioinform. 2018, 19, 381. [Google Scholar] [CrossRef]
- Pyromali, I.; Perani, A.; Nizou, A.; Benslimane, N.; Derouault, P.; Bourthoumieu, S.; Fradin, M.; Sole, G.; Duval, F.; Gomes, C.; et al. New structural variations responsible for Charcot-Marie-Tooth disease: The first two large KIF5A deletions detected by CovCopCan software. Comput. Struct. Biotechnol. J. 2021, 19, 4265–4272. [Google Scholar] [CrossRef]
- Pyromali, I.; Benslimane, N.; Favreau, F.; Goizet, C.; Lazaro, L.; Vitry, M.; Derouault, P.; Sturtz, F.; Magdelaine, C.; Lia, A.S. From Negative to Positive Diagnosis: Structural Variation Could Be the Second Mutation You Are Looking for in a Recessive Autosomal Gene. J. Pers. Med. 2022, 12, 212. [Google Scholar] [CrossRef]
- Ando, M.; Higuchi, Y.; Yuan, J.; Yoshimura, A.; Taniguchi, T.; Kojima, F.; Noguchi, Y.; Hobara, T.; Takeuchi, M.; Takei, J.; et al. Comprehensive Genetic Analyses of Inherited Peripheral Neuropathies in Japan: Making Early Diagnosis Possible. Biomedicines 2022, 10, 1546. [Google Scholar] [CrossRef] [PubMed]
- Lia-Baldini, A.S.; Brun-Heath, I.; Carrion, C.; Simon-Bouy, B.; Serre, J.L.; Nunes, M.E.; Mornet, E. A new mechanism of dominance in hypophosphatasia: The mutated protein can disturb the cell localization of the wild-type protein. Hum. Genet. 2008, 123, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA Duplication Associated with Charcot-Marie-Tooth Disease Type 1. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, N.; Smith, B.; Ballard, L.; Lensch, M.W.; Robertson, M.; Albertsen, H.; Hanemann, C.O.; Müller, H.W.; Bird, T.D.; White, R.; et al. Peripheral Myelin Protein-22 Gene Maps in the Duplication in Chromosome 17p11.2 Associated with Charcot-Marie-Tooth 1A. Nat. Genet. 1992, 1, 176–179. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Bolhuis, P.A.; Zorn, I.; Hoogendijk, J.E.; van den Bosch, N.; Hensels, G.W.; Jr Stanton, V.P.; Housman, D.E.; Fischbeck, K.H.; Ross, D.A.; et al. The Peripheral Myelin Gene PMP-22/GAS-3 is Duplicated in Charcot-Marie-Tooth Disease Type 1A. Nat. Genet. 1992, 1, 166–170. [Google Scholar] [CrossRef]
- Timmerman, V.; Nelis, E.; Van Hul, W.; Nieuwenhuijen, B.W.; Chen, K.L.; Wang, S.; Ben Othman, K.; Cullen, B.; Leach, R.J.; Hanemann, C.O.; et al. The Peripheral Myelin Protein Gene PMP–22 Is Contained within the Charcot–Marie–Tooth Disease Type 1A Duplication. Nat. Genet. 1992, 1, 171–175. [Google Scholar] [CrossRef]
- Behrendt, L.; Kurth, I.; Kaether, C. A disease causing ATLASTIN 3 mutation affects multiple endoplasmic reticulum-related pathways. Cell. Mol. Life Sci. 2019, 76, 1433–1445. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyromali, I.; Richard, L.; Derouault, P.; Vallat, J.-M.; Ghorab, K.; Magdelaine, C.; Sturtz, F.; Favreau, F.; Lia, A.-S. The First Large Deletion of ATL3 Identified in a Patient Presenting with a Sensory Polyneuropathy. Biomedicines 2023, 11, 1565. https://doi.org/10.3390/biomedicines11061565
Pyromali I, Richard L, Derouault P, Vallat J-M, Ghorab K, Magdelaine C, Sturtz F, Favreau F, Lia A-S. The First Large Deletion of ATL3 Identified in a Patient Presenting with a Sensory Polyneuropathy. Biomedicines. 2023; 11(6):1565. https://doi.org/10.3390/biomedicines11061565
Chicago/Turabian StylePyromali, Ioanna, Laurence Richard, Paco Derouault, Jean-Michel Vallat, Karima Ghorab, Corinne Magdelaine, Franck Sturtz, Frédéric Favreau, and Anne-Sophie Lia. 2023. "The First Large Deletion of ATL3 Identified in a Patient Presenting with a Sensory Polyneuropathy" Biomedicines 11, no. 6: 1565. https://doi.org/10.3390/biomedicines11061565