
Identification of a NACC1-Regulated Gene Signature Implicated in the Features of Triple-Negative Breast Cancer
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
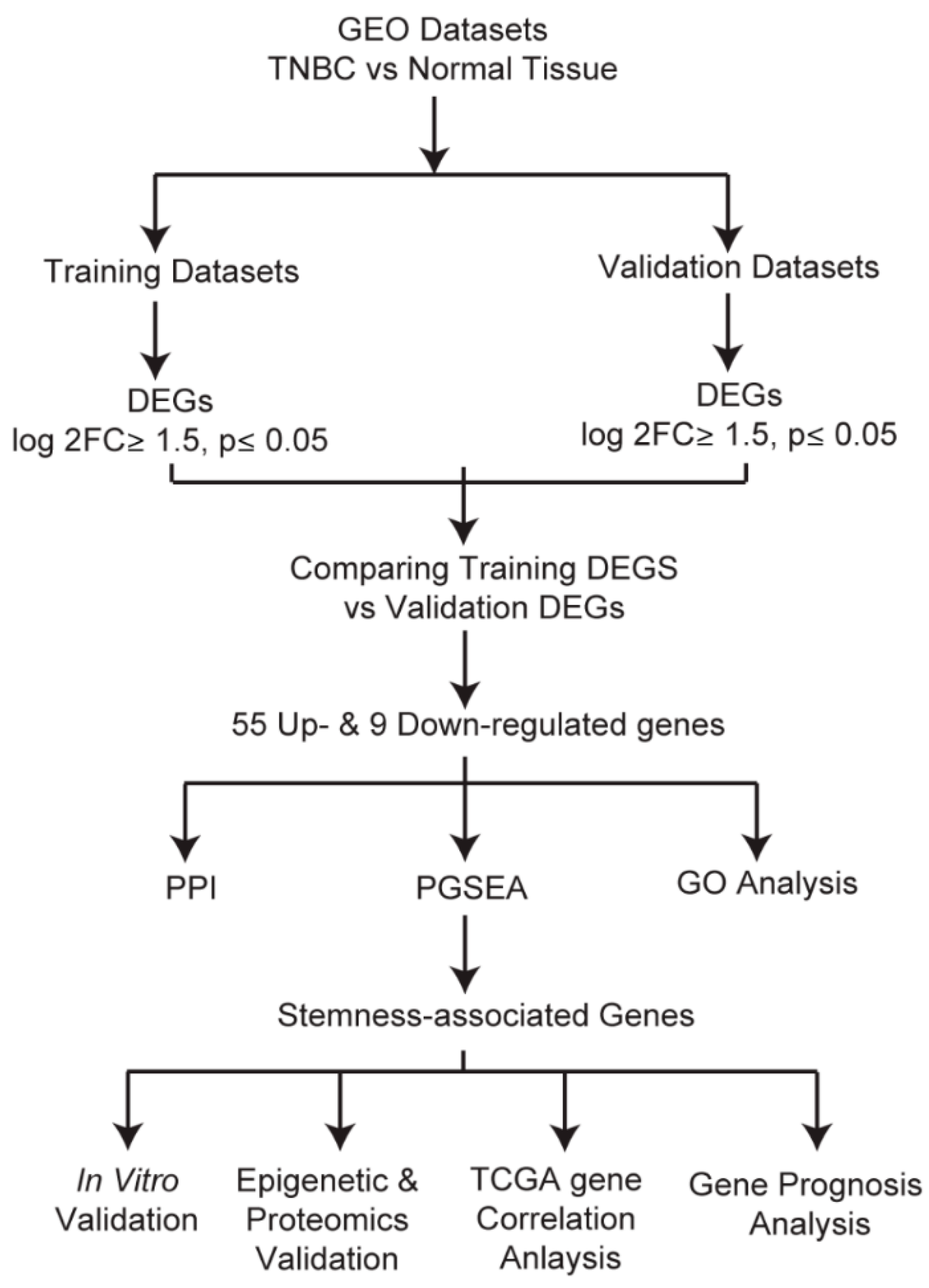
2.1. Identification and Evaluation of Differentially Expressed Genes
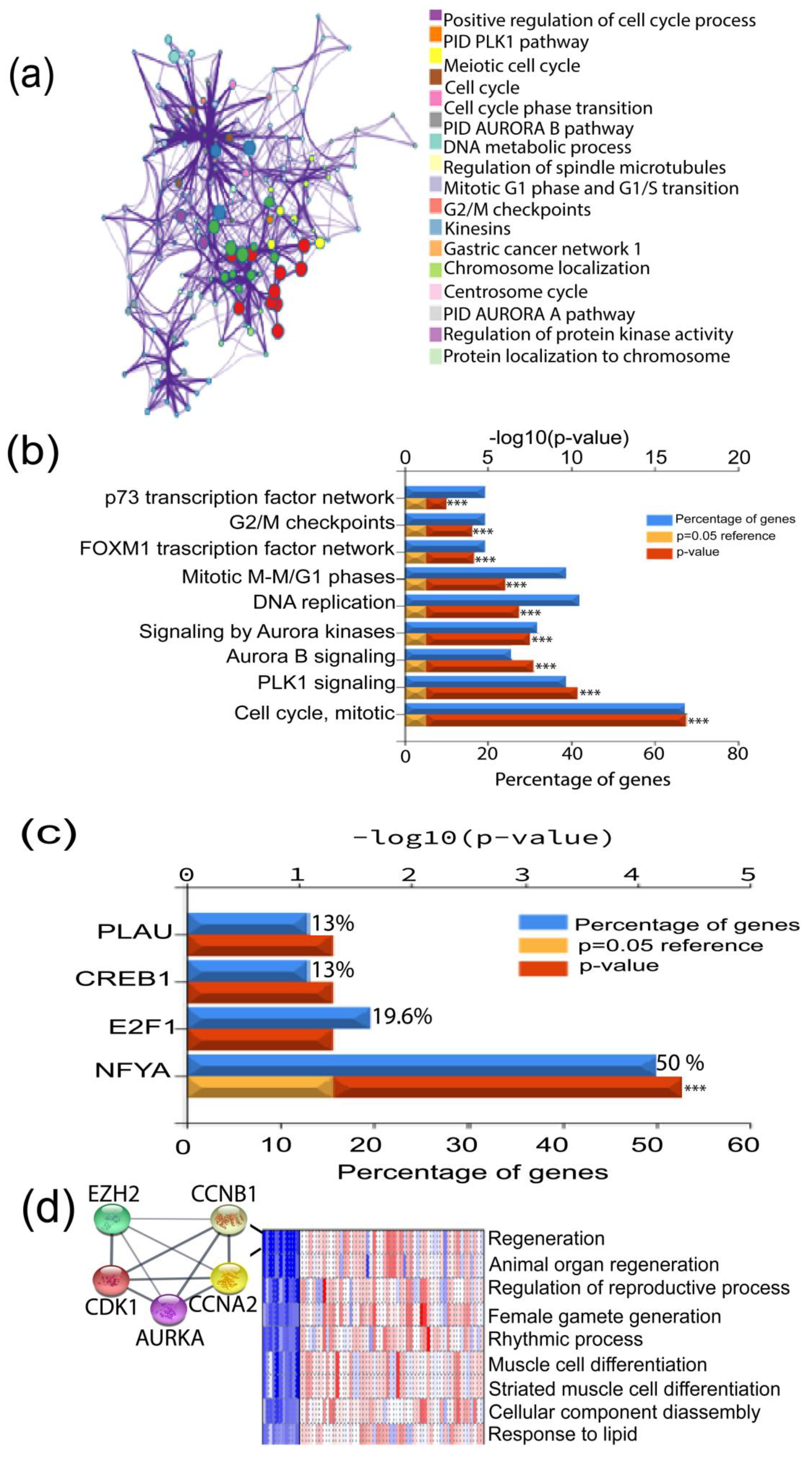
2.2. GO Analysis
2.3. Protein–Protein Interaction (PPI) Network Analysis and Hub Gene Confirmation
2.4. Analysis of the Identified Signature Genes Clinical Relevance
2.5. In Vitro Expression of Signature Genes in TNBC Cell Lines
2.6. Western Blot Analysis
3. Results
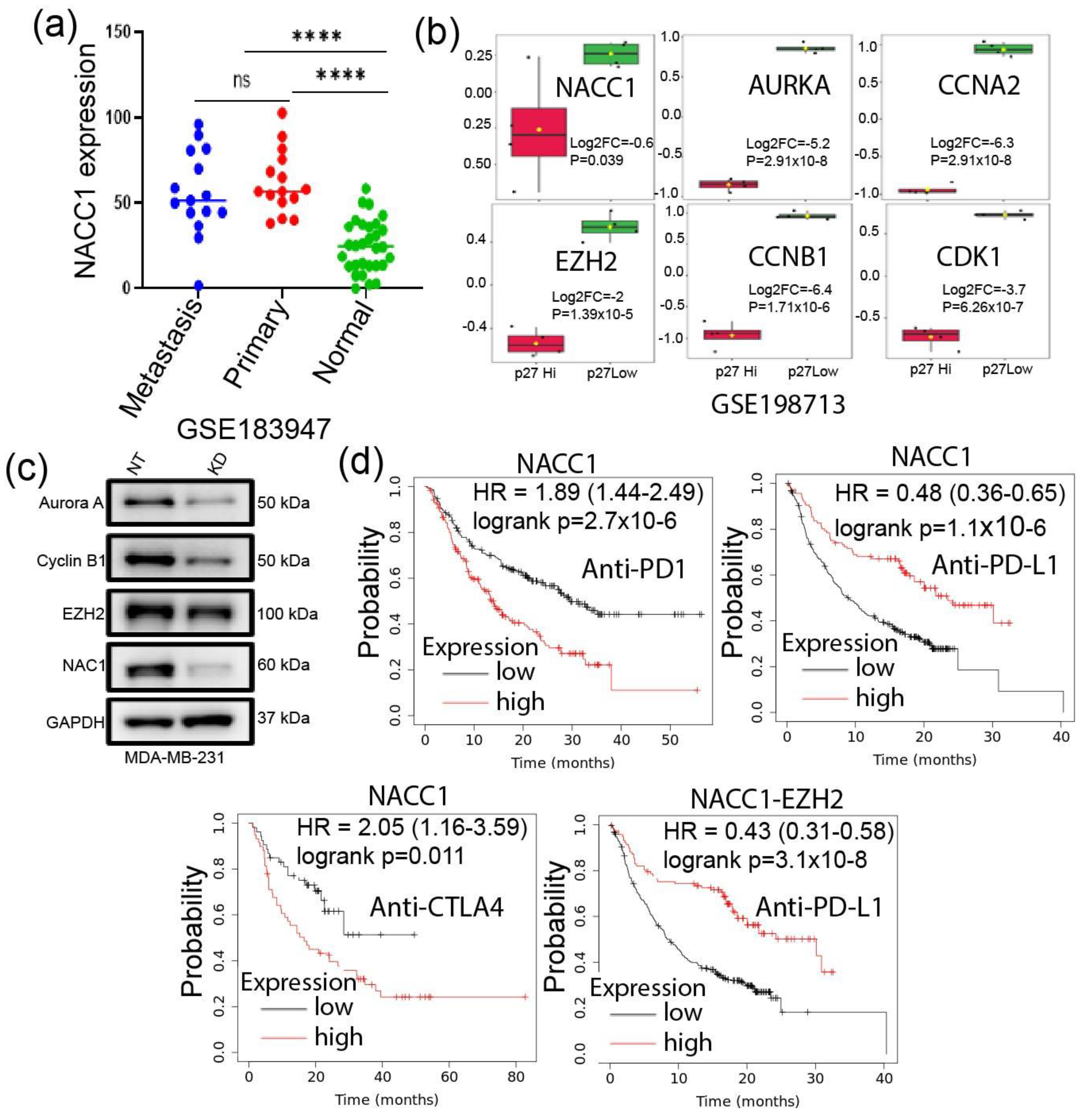
3.1. Identification of Highly Altered Genes in TNBC
3.2. Correlation of the Highly Expressed Genes with Cancer Progression
3.3. PGSEA Analysis of the TNBC Altered Genes Identifies a Five Gene-Signature with a Putative Role in Tumor Stemness
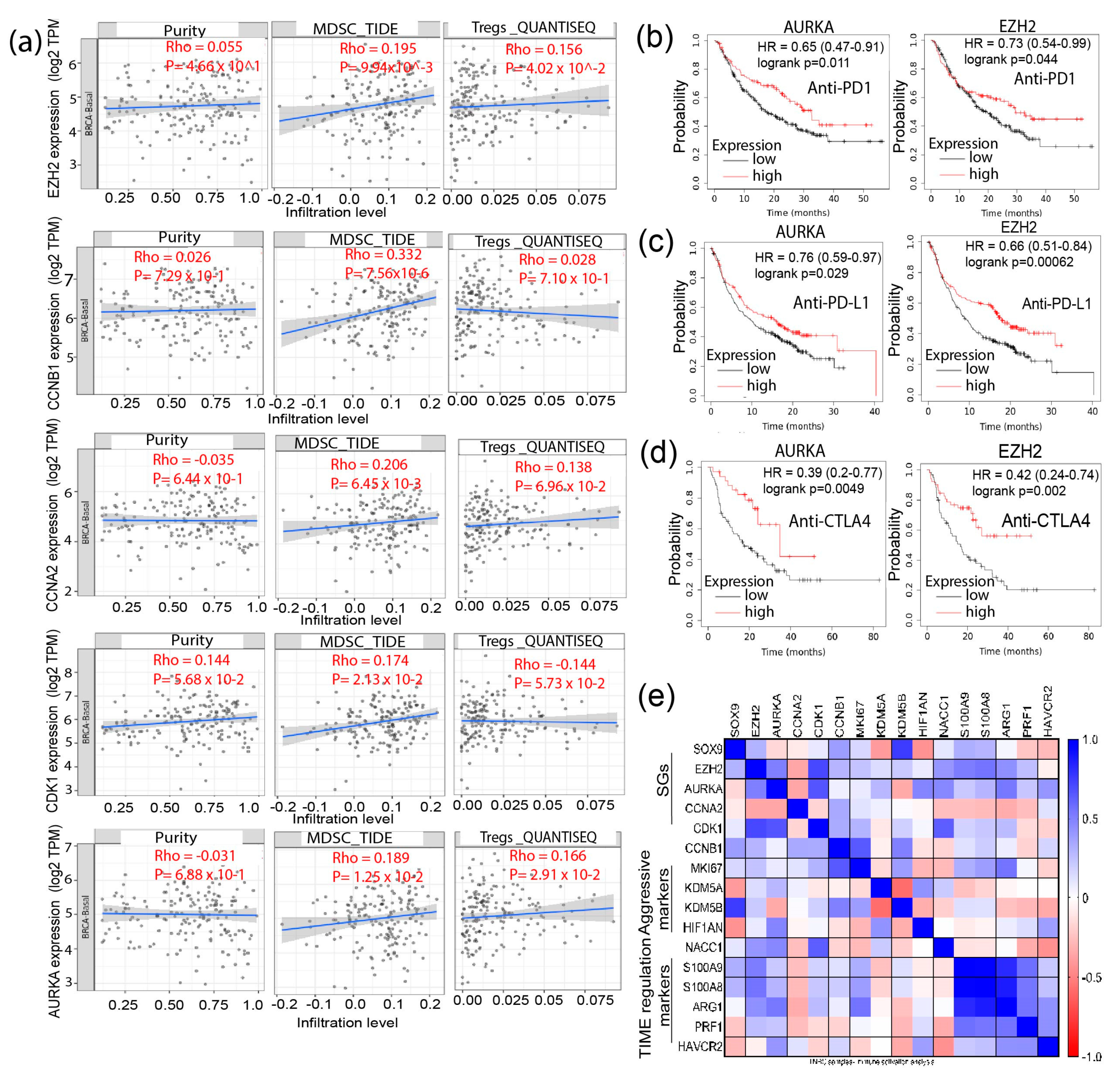
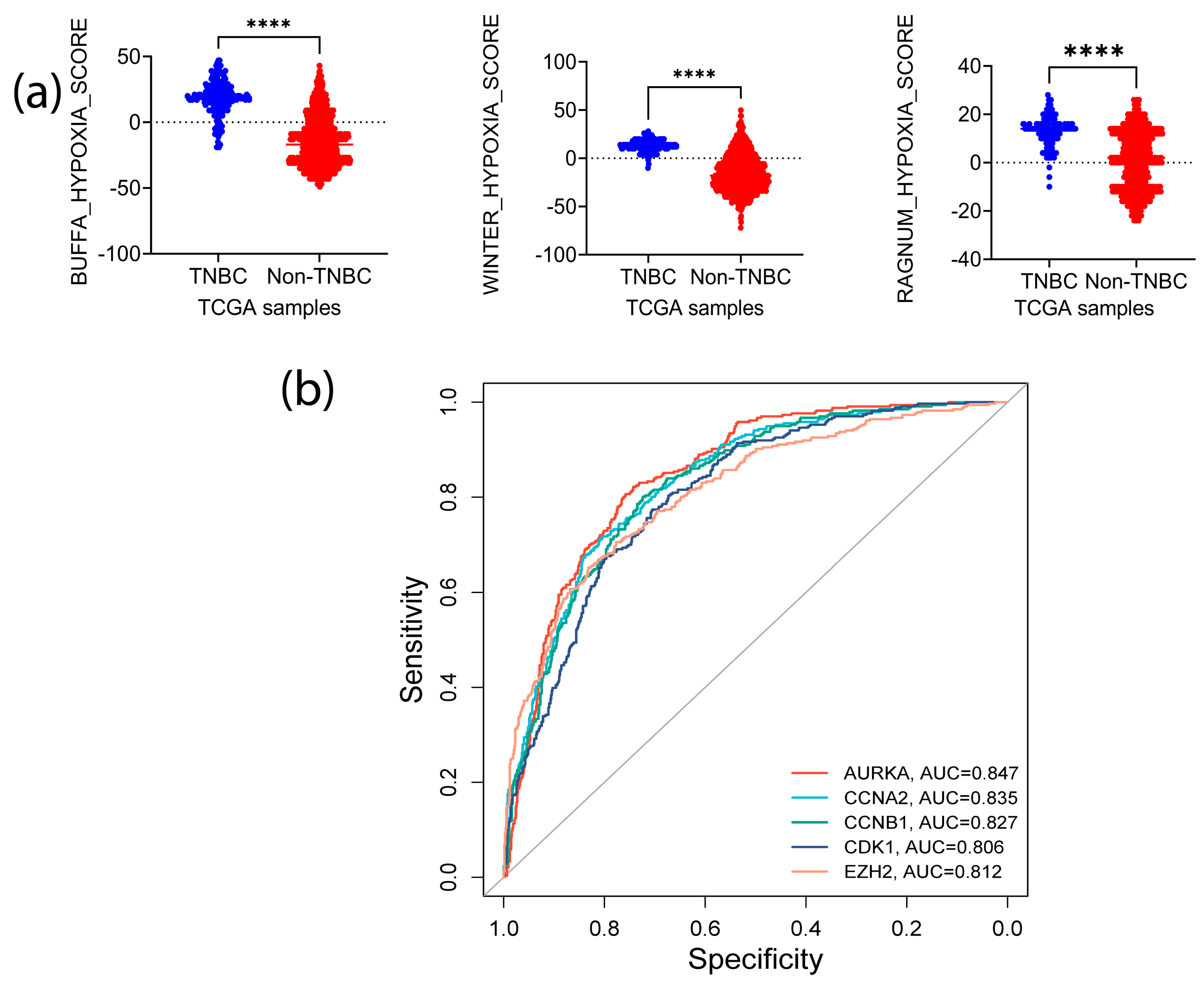
3.4. Signature Genes Expression Is Positively Associated with Immunosuppressive Cells Infiltration and Hypoxia Status in TNBC
3.5. Depletion of NACC1 Reduces the Signature Genes Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masood, S. Breast cancer subtypes: Morphologic and biologic characterization. Womens Health 2016, 12, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, R.; Ordonez-Moran, P.; Allegrucci, C. Challenges for triple negative breast cancer treatment: Defeating heterogeneity and cancer stemness. Cancers 2022, 14, 4280. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Q.; Russo, J. Eralpha-negative and triple negative breast cancer: Molecular features and potential therapeutic approaches. Biochim. Biophys. Acta 2009, 1796, 162–175. [Google Scholar] [PubMed]
- Fultang, N.; Chakraborty, M.; Peethambaran, B. Regulation of cancer stem cells in triple negative breast cancer. Cancer Drug Resist. 2021, 4, 321–342. [Google Scholar] [CrossRef]
- Baldominos, P.; Barbera-Mourelle, A.; Barreiro, O.; Huang, Y.; Wight, A.; Cho, J.W.; Zhao, X.; Estivill, G.; Adam, I.; Sanchez, X.; et al. Quiescent cancer cells resist t cell attack by forming an immunosuppressive niche. Cell 2022, 185, 1694–1708.e19. [Google Scholar] [CrossRef]
- He, L.; Wick, N.; Germans, S.K.; Peng, Y. The role of breast cancer stem cells in chemoresistance and metastasis in triple-negative breast cancer. Cancers 2021, 13, 6209. [Google Scholar] [CrossRef]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef]
- Nakayama, K.; Nakayama, N.; Davidson, B.; Sheu, J.J.C.; Jinawath, N.; Santillan, A.; Salani, R.; Bristow, R.E.; Morin, P.J.; Kurman, R.J.; et al. A btb/poz protein, nac-1, is related to tumor recurrence and is essential for tumor growth and survival. Proc. Natl. Acad. Sci. USA 2006, 103, 18739–18744. [Google Scholar] [CrossRef]
- Perez-Torrado, R.; Yamada, D.; Defossez, P.A. Born to bind: The btb protein-protein interaction domain. Bioessays 2006, 28, 1194–1202. [Google Scholar] [CrossRef]
- Sean, D.; Meltzer, P.S. Geoquery: A bridge between the gene expression omnibus (geo) and bioconductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.J.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. Funrich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
- Ge, X.J. Idep web application for rna-seq data analysis. Methods Mol. Biol. 2021, 2284, 417–443. [Google Scholar]
- Ge, S.X.; Son, E.W.; Yao, R.N. Idep: An integrated web application for differential expression and pathway analysis of rna-seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]
- Kim, S.Y.; Volsky, D.J. Page: Parametric analysis of gene set enrichment. BMC Bioinform. 2005, 6, 144. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. Correction to ‘the string database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets’. Nucleic Acids Res. 2021, 49, 10800. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.H.; Kumar, S.; Manne, U.; et al. Ualcan: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. Ualcan: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Bartha, A.; Gyorffy, B. Tnmplot.Com: A web tool for the comparison of gene expression in normal, tumor and metastatic tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Gao, J.J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.C.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Li, T.W.; Fu, J.X.; Zeng, Z.X.; Cohen, D.; Li, J.; Chen, Q.M.; Li, B.; Liu, X.S. Timer 2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.T.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Lanczky, A.; Gyorffy, B. Web-based survival analysis tool tailored for medical research (kmplot): Development and implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Mathews, J.C.; Nadeem, S.; Levine, A.J.; Pouryahya, M.; Deasy, J.O.; Tannenbaum, A. Robust and interpretable pam50 reclassification exhibits survival advantage for myoepithelial and immune phenotypes. NPJ Breast Cancer 2019, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hsu, W.H.; Han, J.; Xia, Y.; DePinho, R.A. Cancer stemness meets immunity: From mechanism to therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef]
- Yi, L.; Huang, P.; Zou, X.; Guo, L.; Gu, Y.; Wen, C.; Wu, G. Integrative stemness characteristics associated with prognosis and the immune microenvironment in esophageal cancer. Pharmacol. Res. 2020, 161, 105144. [Google Scholar] [CrossRef]
- Bayik, D.; Lathia, J.D. Cancer stem cell-immune cell crosstalk in tumour progression. Nat. Rev. Cancer 2021, 21, 526–536. [Google Scholar] [CrossRef]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019, 35, I436–I445. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.Q.; Pan, D.; Fu, J.X.; Sahu, A.; Hu, X.H.; Li, Z.Y.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of t-cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Hu, J.Y.; Li, X.Y.; Yang, L.; Li, H.Y. Hypoxia, a key factor in the immune microenvironment. Biomed. Pharmacother. 2022, 151, 113068. [Google Scholar] [CrossRef]
- Winter, S.C.; Buffa, F.M.; Silva, P.; Miller, C.; Valentine, H.R.; Turley, H.; Shah, K.A.; Cox, G.J.; Corbridge, R.J.; Homer, J.J.; et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 2007, 67, 3441–3449. [Google Scholar] [CrossRef]
- Ragnum, H.B.; Vlatkovic, L.; Lie, A.K.; Axcrona, K.; Julin, C.H.; Frikstad, K.M.; Hole, K.H.; Seierstad, T.; Lyng, H. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br. J. Cancer 2015, 112, 382–390. [Google Scholar] [CrossRef]
- Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 2010, 102, 428–435. [Google Scholar] [CrossRef]
- Ren, Y.J.; Wang, X.H.; Ji, C.; Guan, Y.D.; Lu, X.J.; Liu, X.R.; Zhang, H.H.; Guo, L.C.; Xu, Q.H.; Zhu, W.D.; et al. Silencing of nac1 expression induces cancer cells oxidative stress in hypoxia and potentiates the therapeutic activity of elesclomol. Front. Pharmacol. 2017, 8, 804. [Google Scholar] [CrossRef]
- Ren, Y.J.; Kumar, A.; Das, J.K.; Peng, H.Y.; Wang, L.Q.; Balllard, D.; Xiong, X.F.; Ren, X.C.; Zhang, Y.; Yang, J.M.; et al. Tumorous expression of nac1 restrains antitumor immunity through the ldha-mediated immune evasion. J. ImmunoTherapy Cancer 2022, 10, e004856. [Google Scholar] [CrossRef]
- Zhang, Y.; Tong, G.H.; Wei, X.X.; Chen, H.Y.; Liang, T.; Tang, H.P.; Wu, C.A.; Wen, G.M.; Yang, W.K.; Liang, L.; et al. Identification of five cytotoxicity-related genes involved in the progression of triple-negative breast cancer. Front. Genet. 2022, 12, 723477. [Google Scholar] [CrossRef]
- De Santis, F.; Romero-Cordoba, S.L.; Castagnoli, L.; Volpari, T.; Faraci, S.; Fuca, G.; Tagliabue, E.; De Braud, F.; Pupa, S.M.; Di Nicola, M. Bcl6 and the notch pathway: A signaling axis leading to a novel druggable biotarget in triple negative breast cancer. Cell. Oncol. 2022, 45, 257–274. [Google Scholar] [CrossRef]
- Das, P.K.; Pillai, S.; Rakib, M.A.; Khanam, J.A.; Gopalan, V.; Lam, A.K.Y.; Islam, F. Plasticity of cancer stem cell: Origin and role in disease progression and therapy resistance. Stem Cell Rev. Rep. 2020, 16, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Ameri, M.; Salimi, H.; Eskandari, S.; Nezafat, N. Identification of potential biomarkers in hepatocellular carcinoma: A network-based approach. Inform. Med. Unlocked 2022, 28, 100864. [Google Scholar] [CrossRef]
- Dolfini, D.; Andrioletti, V.; Mantovani, R. Overexpression and alternative splicing of nf-ya in breast cancer. Sci. Rep. 2019, 9, 12955. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Zhao, Y.; Zhang, Z.; Li, H.C.; Xing, J.; Guo, S.L.; Qiu, X.T.; Zhang, S.T.; Min, L.; Zhu, S.T. Gene regulatory network construction identified nfya as a diffuse subtype-specific prognostic factor in gastric cancer. Int. J. Oncol. 2018, 53, 1857–1868. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, X.; Chen, H.; Chen, Z.; Hu, K.; Yin, D. Transcription factor nfya promotes g1/s cell cycle transition and cell proliferation by transactivating cyclin d1 and cdk4 in clear cell renal cell carcinoma. Am. J. Cancer Res. 2020, 10, 2446–2463. [Google Scholar]
- Huang, Z.; Shen, G.; Gao, J. Cdk1 promotes the stemness of lung cancer cells through interacting with sox2. Clin. Transl. Oncol. 2021, 23, 1743–1751. [Google Scholar] [CrossRef]
- Chen, G.F.; Liu, B.Y.; Yin, S.S.; Li, S.D.; Guo, Y.E.; Wang, M.F.; Wang, K.; Wan, X.P. Hypoxia induces an endometrial cancer stem-like cell phenotype via hif-dependent demethylation of sox2 mrna. Oncogenesis 2020, 9, 81. [Google Scholar] [CrossRef]
- Tang, B.; Sun, R.; Wang, D.J.; Sheng, H.Y.; Wei, T.; Wang, L.G.; Zhang, J.; Ho, T.H.; Yang, L.; Wei, Q.; et al. Zmynd8 preferentially binds phosphorylated ezh2 to promote a prc2-dependent to-independent function switch in hypoxia-inducible factor-activated cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2019052118. [Google Scholar] [CrossRef]
- Kuang, Y.; Guo, W.; Ling, J.; Xu, D.; Liao, Y.; Zhao, H.; Du, X.; Wang, H.; Xu, M.; Song, H.; et al. Iron-dependent cdk1 activity promotes lung carcinogenesis via activation of the gp130/stat3 signaling pathway. Cell Death Dis. 2019, 10, 297. [Google Scholar] [CrossRef]
- Zhang, F.Q.; Duan, S.Z.; Tsai, Y.; Keng, P.C.; Chen, Y.B.; Lee, S.O.; Chen, Y. Cisplatin treatment increases stemness through upregulation of hypoxia-inducible factors by interleukin-6 in non-small cell lung cancer. Cancer Sci. 2016, 107, 746–754. [Google Scholar] [CrossRef]
- Chien, Y.C.; Liu, L.C.; Ye, H.Y.; Wu, J.Y.; Yu, Y.L. Ezh2 promotes migration and invasion of triple-negative breast cancer cells via regulating timp2-mmp-2/-9 pathway. Am. J. Cancer Res. 2018, 8, 422–434. [Google Scholar]
- Abou Khouzam, R.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor hypoxia regulates immune escape/invasion: Influence on angiogenesis and potential impact of hypoxic biomarkers on cancer therapies. Front. Immunol. 2021, 11, 613114. [Google Scholar] [CrossRef]
- Yang, J.M.; Ren, Y.J.; Kumar, A.; Xiong, X.F.; Das, J.K.; Peng, H.Y.; Wang, L.Q.; Ren, X.C.; Zhang, Y.; Ji, C.; et al. Nac1 modulates autoimmunity by suppressing regulatory t cell-mediated tolerance. Sci. Adv. 2022, 8, eabo0183. [Google Scholar] [CrossRef]
- Wang, L.Q.; Kumar, A.; Das, J.K.; Ren, Y.J.; Peng, H.Y.; Ballard, D.J.; Xiong, X.F.; Davis, J.R.; Ren, X.C.; Yang, J.M.; et al. Expression of nac1 restrains the memory formation of cd8(+) t cells during viral infection. Viruses 2022, 14, 1713. [Google Scholar] [CrossRef]
- Zou, Y.P.; Ruan, S.Y.; Jin, L.; Chen, Z.H.; Han, H.W.; Zhang, Y.P.; Jian, Z.X.; Lin, Y.; Shi, N.; Jin, H.S. Cdk1, ccnb1, and ccnb2 are prognostic biomarkers and correlated with immune infiltration in hepatocellular carcinoma. Med. Sci. Monitor 2020, 26, e925289-1–e925289-14. [Google Scholar] [CrossRef]
- Liu, R.J.; Li, S.Y.; Liu, L.Q.; Xu, B.; Chen, M. Identification of biomarkers, pathways and potential therapeutic target for docetaxel resistant prostate cancer. Bioengineered 2021, 12, 2377–2388. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Datasets | TNBC Samples | Normal | Non-TNBC |
|---|---|---|---|
| Training datasets | |||
| GSE65194 | 55 | 11 | 98 |
| GSE36295 | 11 | 5 | 27 |
| GSE38959 | 30 | 13 | 0 |
| Sum | 96 | 29 | 125 |
| Validation datasets | |||
| GSE21653 | 75 | 29 | 162 |
| GSE20711 | 17 | 2 | 71 |
| GSE61725 | 48 | 17 | 0 |
| Sum | 140 | 48 | 233 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngule, C.M.; Hemati, H.; Ren, X.; Obaleye, O.; Akinyemi, A.O.; Oyelami, F.F.; Xiong, X.; Song, J.; Liu, X.; Yang, J.-M. Identification of a NACC1-Regulated Gene Signature Implicated in the Features of Triple-Negative Breast Cancer. Biomedicines 2023, 11, 1223. https://doi.org/10.3390/biomedicines11041223
Ngule CM, Hemati H, Ren X, Obaleye O, Akinyemi AO, Oyelami FF, Xiong X, Song J, Liu X, Yang J-M. Identification of a NACC1-Regulated Gene Signature Implicated in the Features of Triple-Negative Breast Cancer. Biomedicines. 2023; 11(4):1223. https://doi.org/10.3390/biomedicines11041223
Chicago/Turabian StyleNgule, Chrispus M., Hami Hemati, Xingcong Ren, Oluwafunminiyi Obaleye, Amos O. Akinyemi, Felix F. Oyelami, Xiaofang Xiong, Jianxun Song, Xia Liu, and Jin-Ming Yang. 2023. "Identification of a NACC1-Regulated Gene Signature Implicated in the Features of Triple-Negative Breast Cancer" Biomedicines 11, no. 4: 1223. https://doi.org/10.3390/biomedicines11041223