

Short-Term Growth Hormone Administration Mediates Hepatic Fatty Acid Uptake and De Novo Lipogenesis Gene Expression in Obese Rats
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
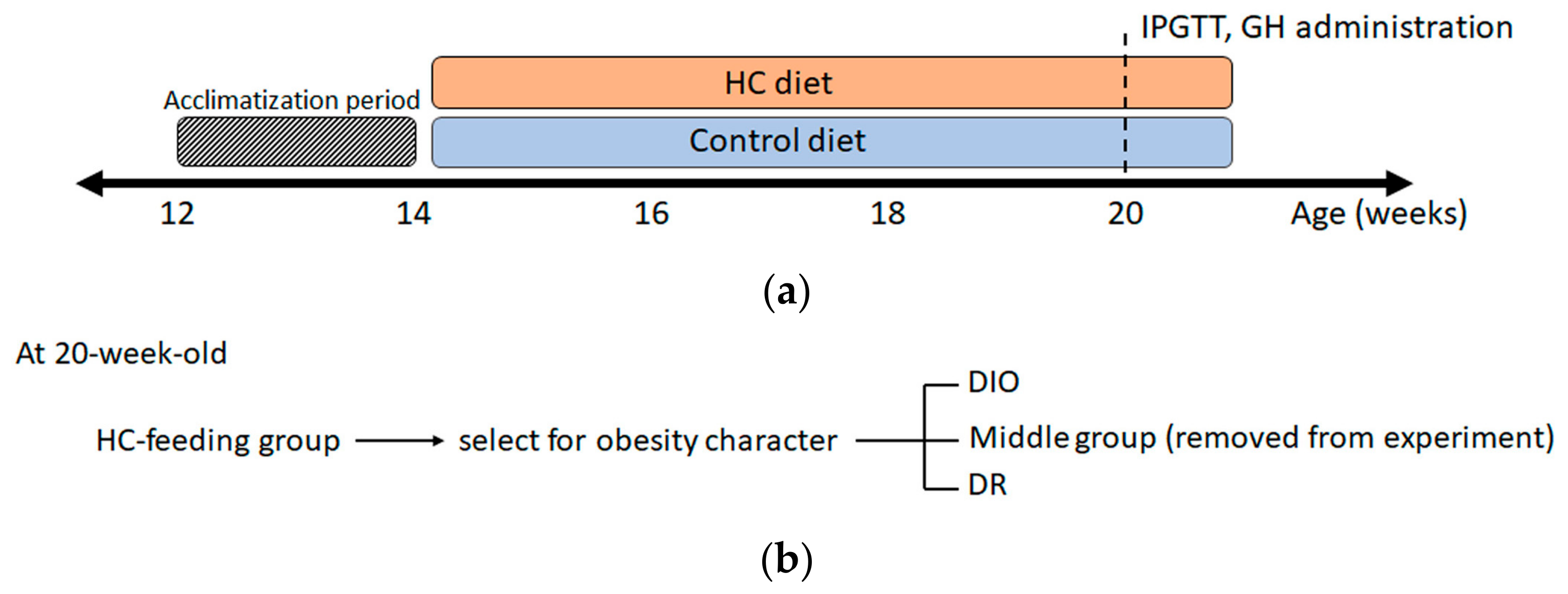
2.1. Animals and Diet
2.2. Intraperitoneal Glucose Tolerance Test
2.3. Calculation of Homeostasis Model of Insulin Resistance (HOMA-IR)
2.4. Measurement of Circulating Free Fatty Acids, Total Cholesterol and Triglyceride Levels
2.5. Liver Triglyceride (TG) Measurements
2.6. RNA Isolation and Quantitative RT-PCR
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
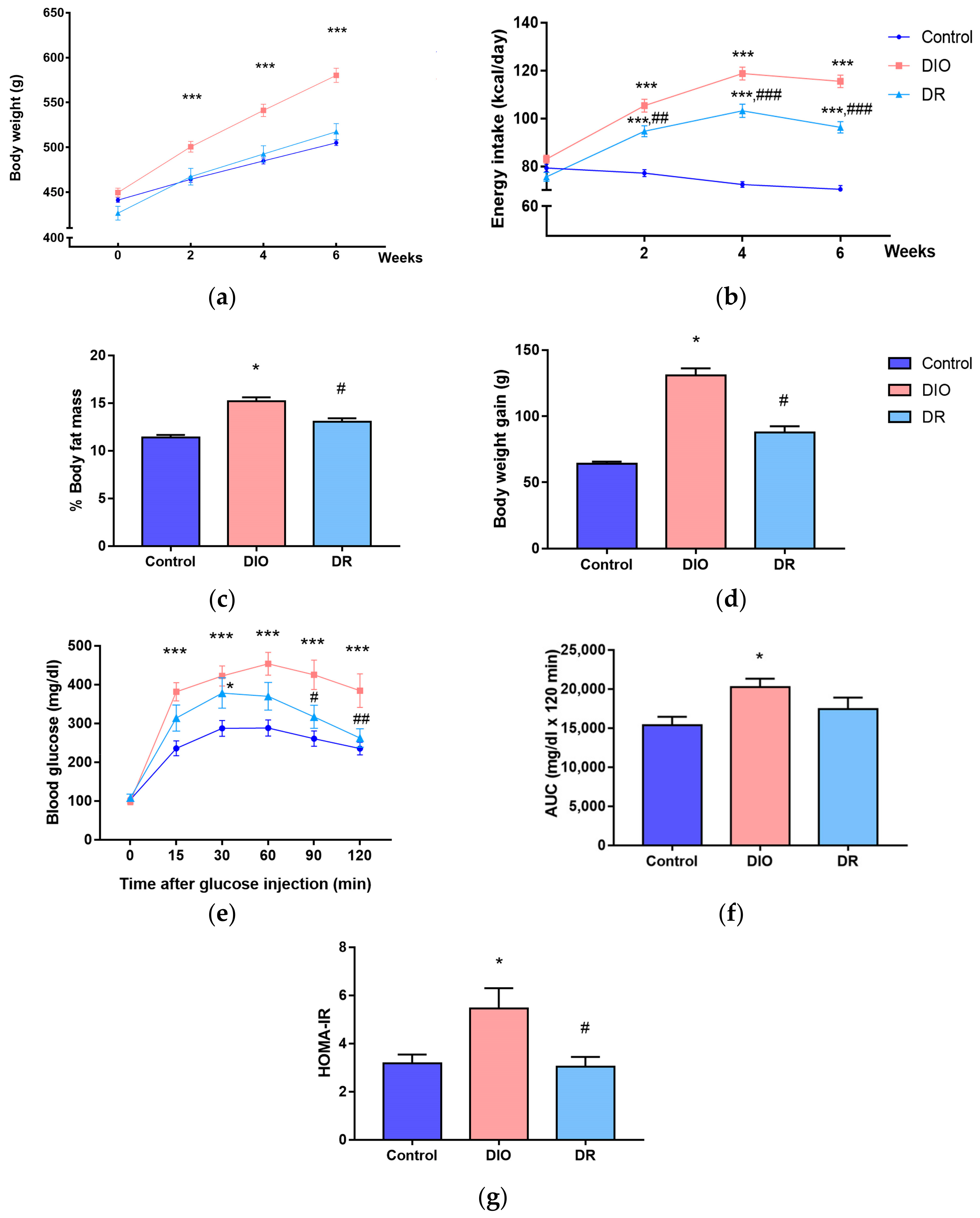
3.1. Characteristics of Diet-Induced Obesity (DIO) and Diet-Resistant (DR) Rats
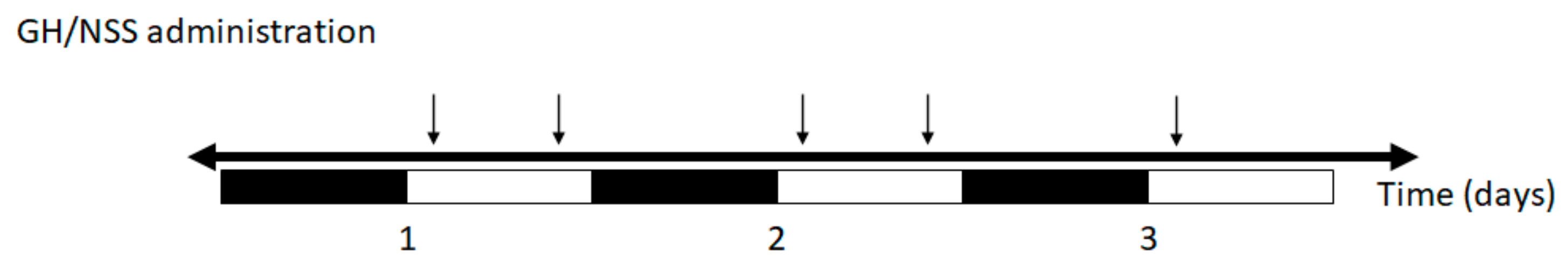
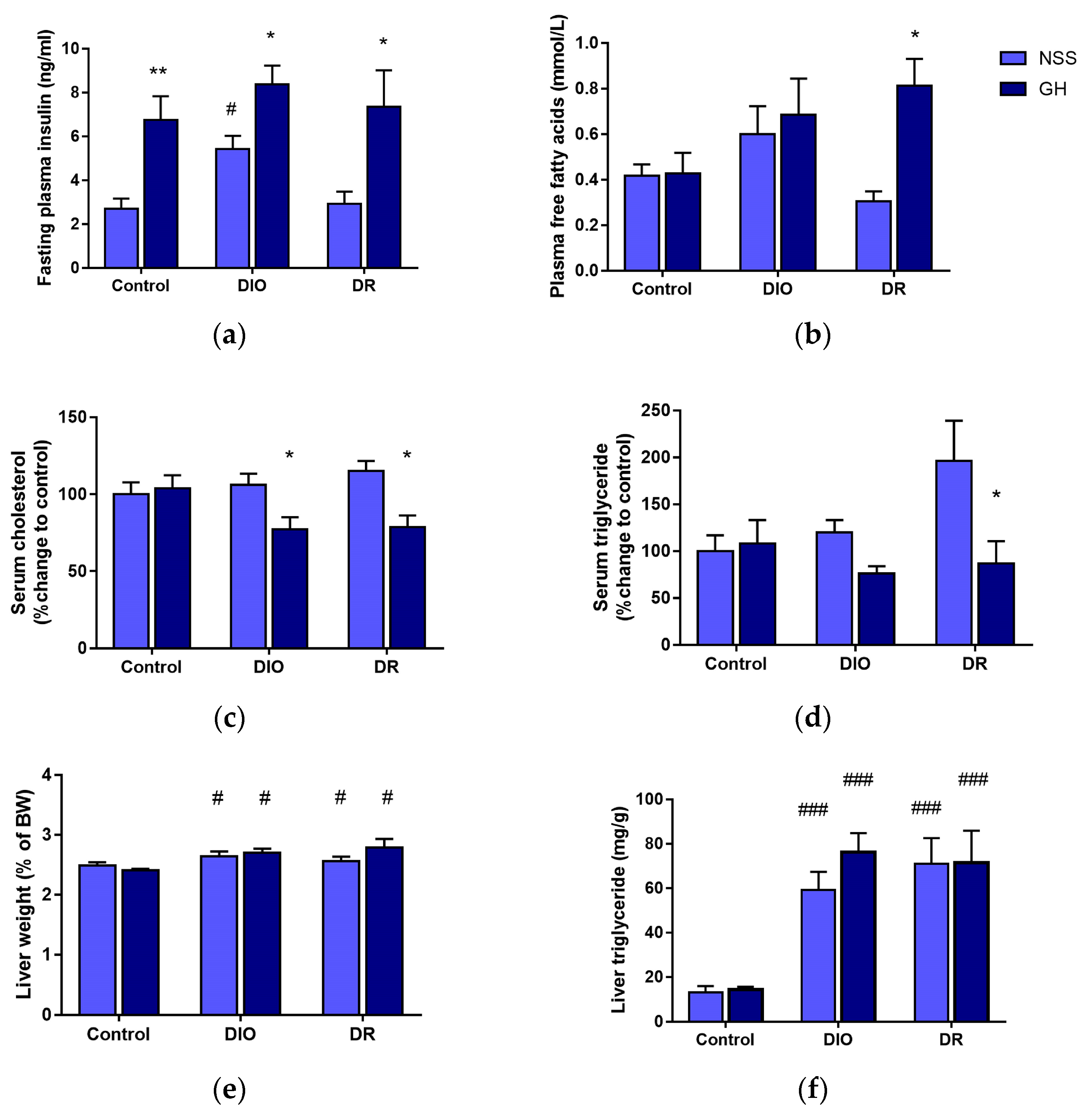
3.2. Effects of Short-Term GH Administration on the Circulating Insulin, FFAs, Cholesterol, TG and Liver TG Levels
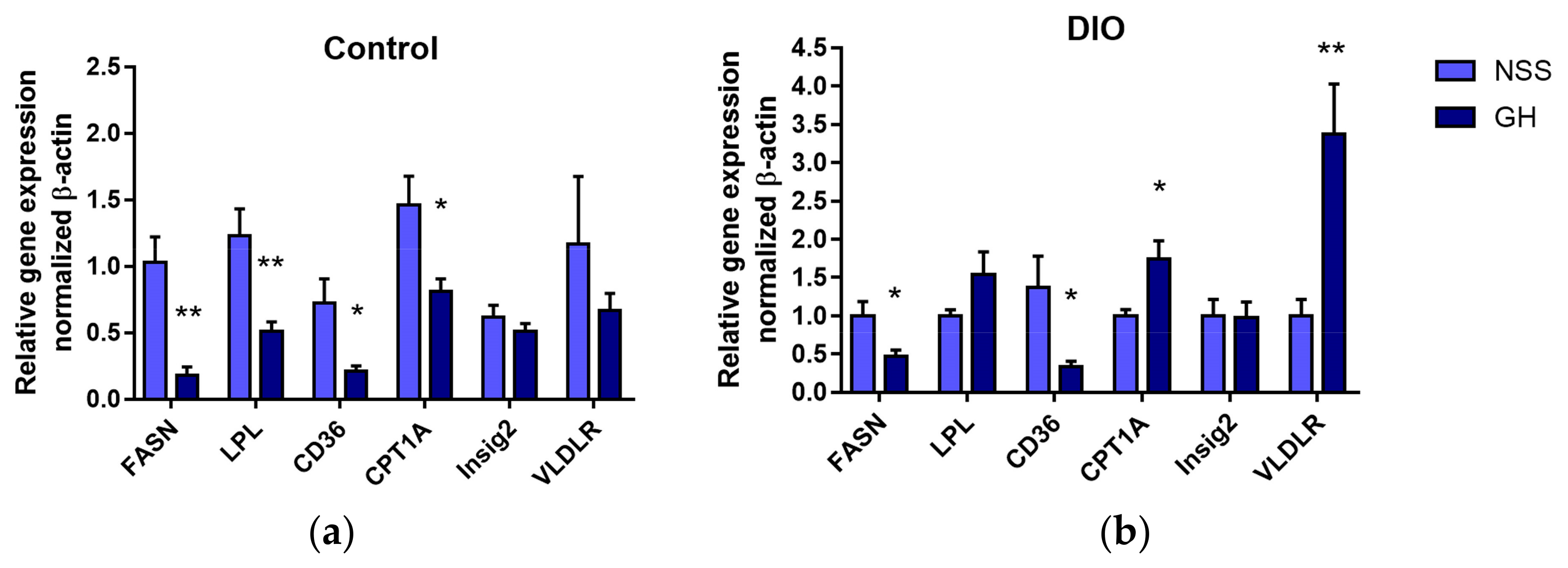
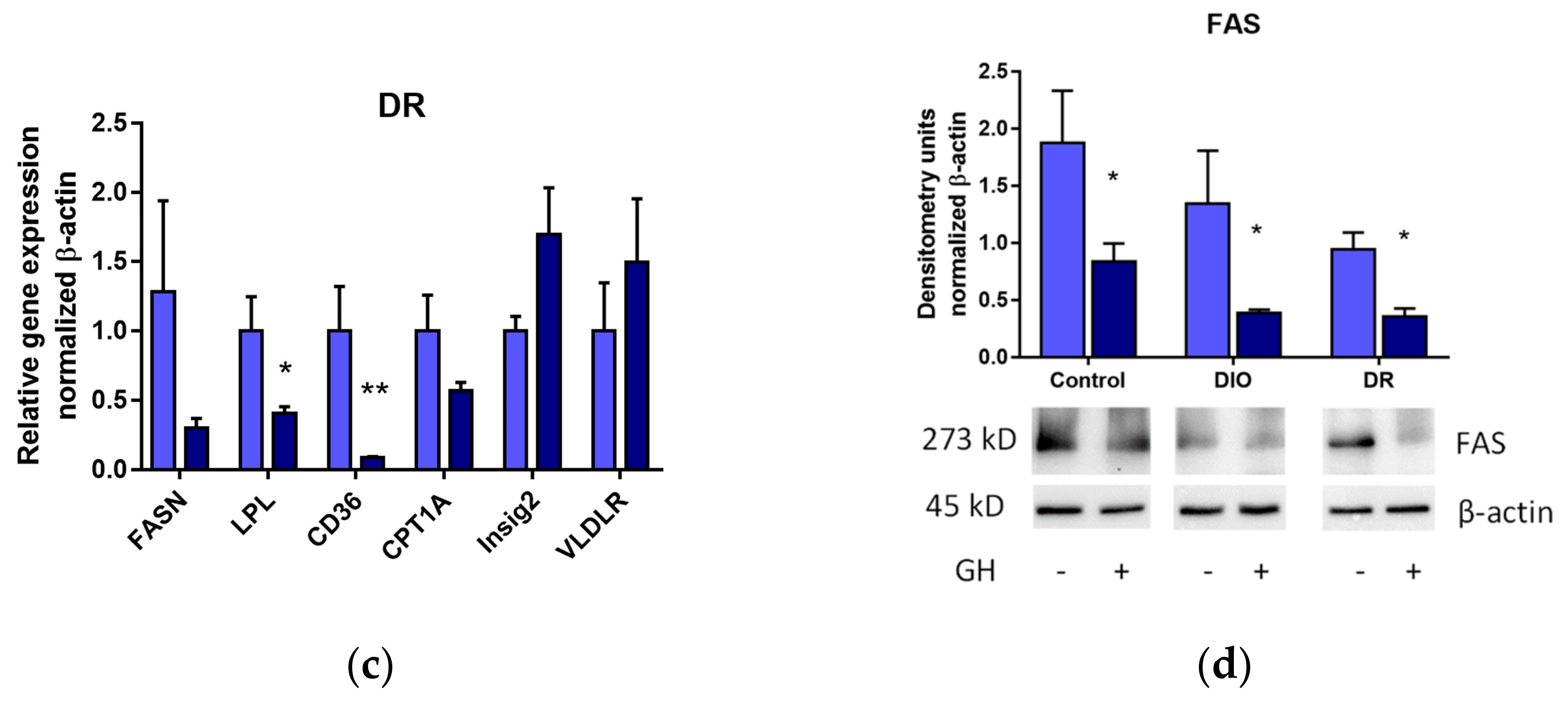
3.3. Effects of Short-Term GH Administration on mRNA Expression of Genes Related to Hepatic Lipid Metabolism and the Hepatic FAS Protein Levels
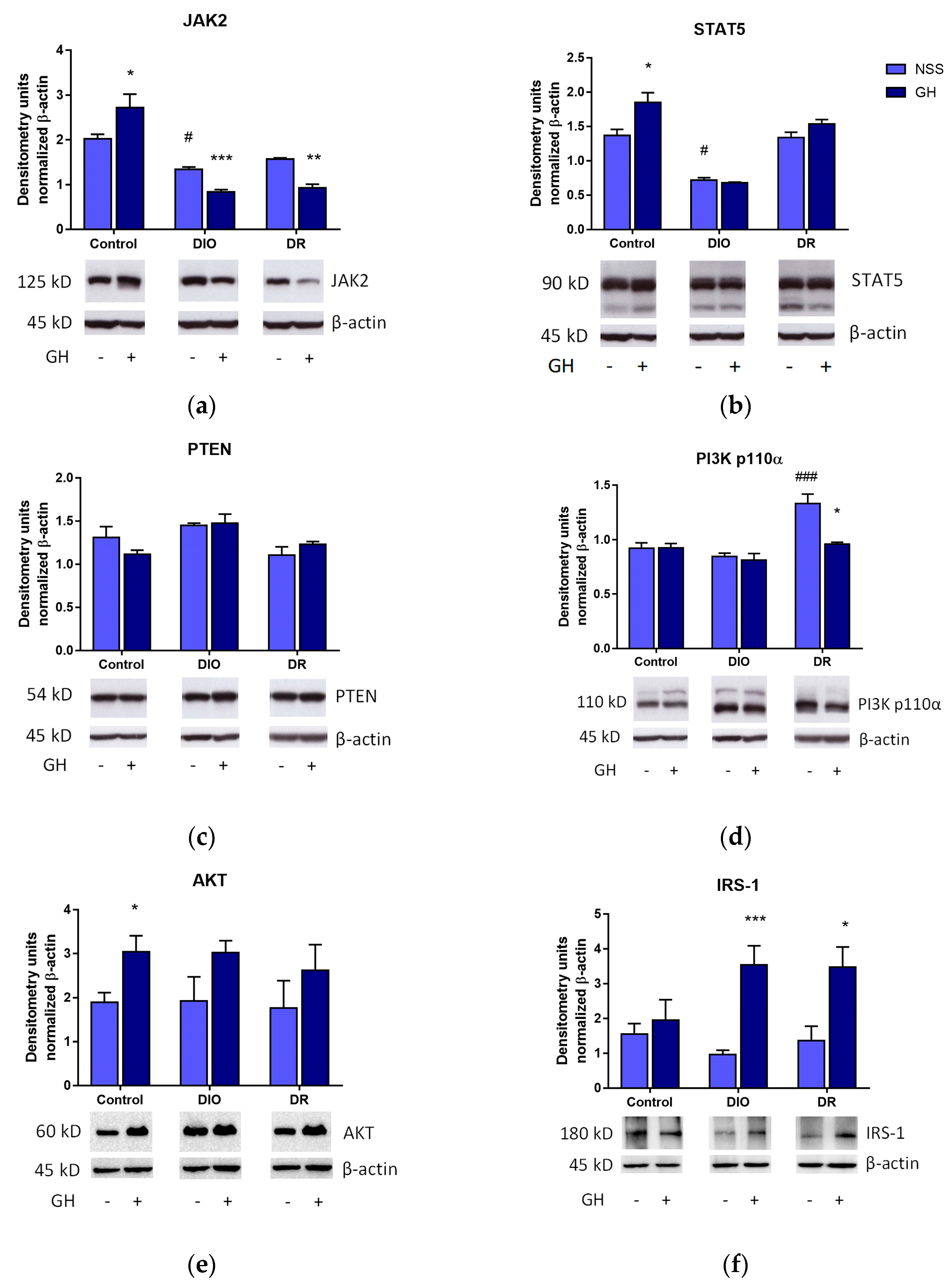
3.4. Effects of Short-Term GH Treatment on the Expression of Effectors Involved in the GH and Insulin Receptor Signaling Pathways in the Liver
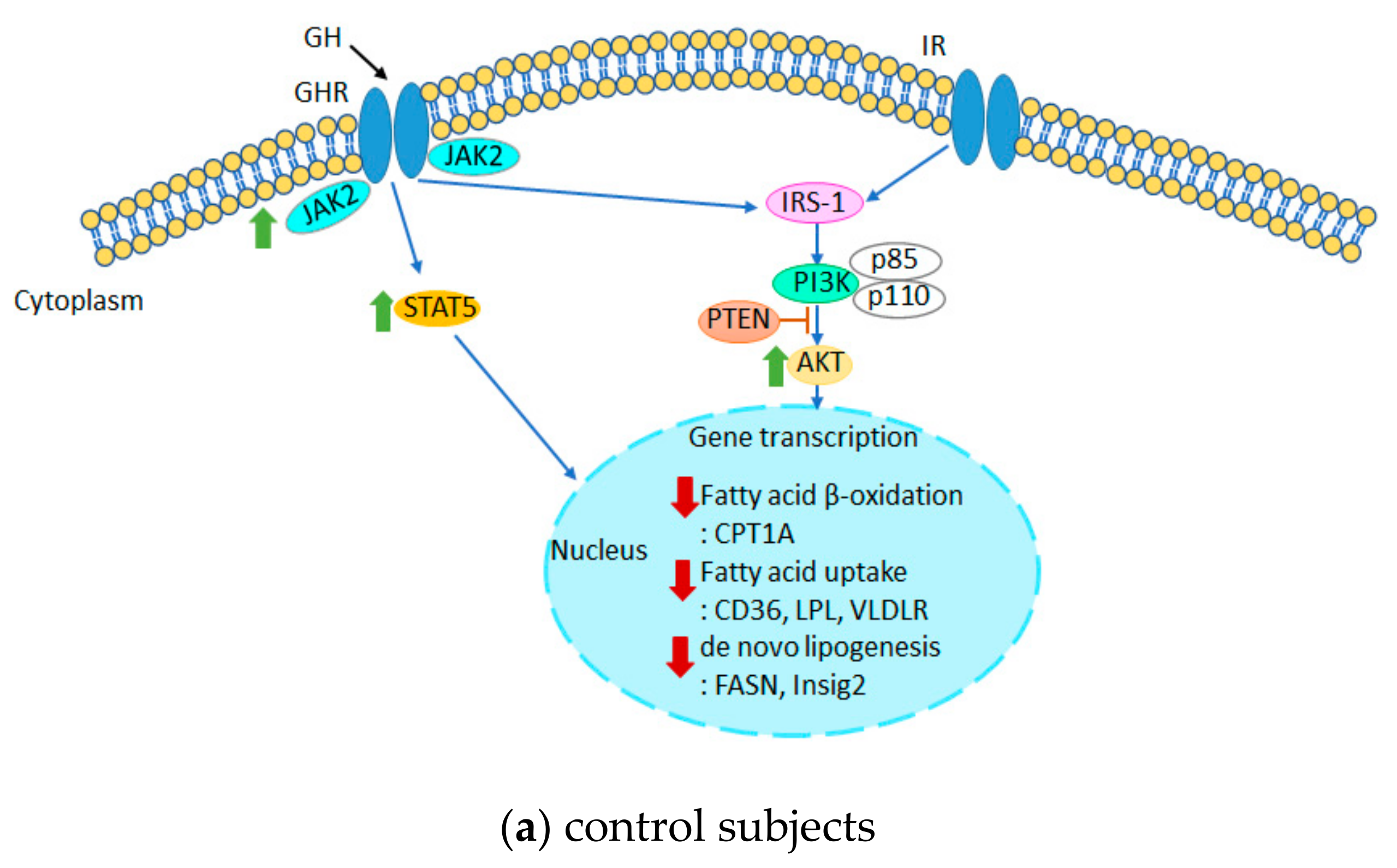
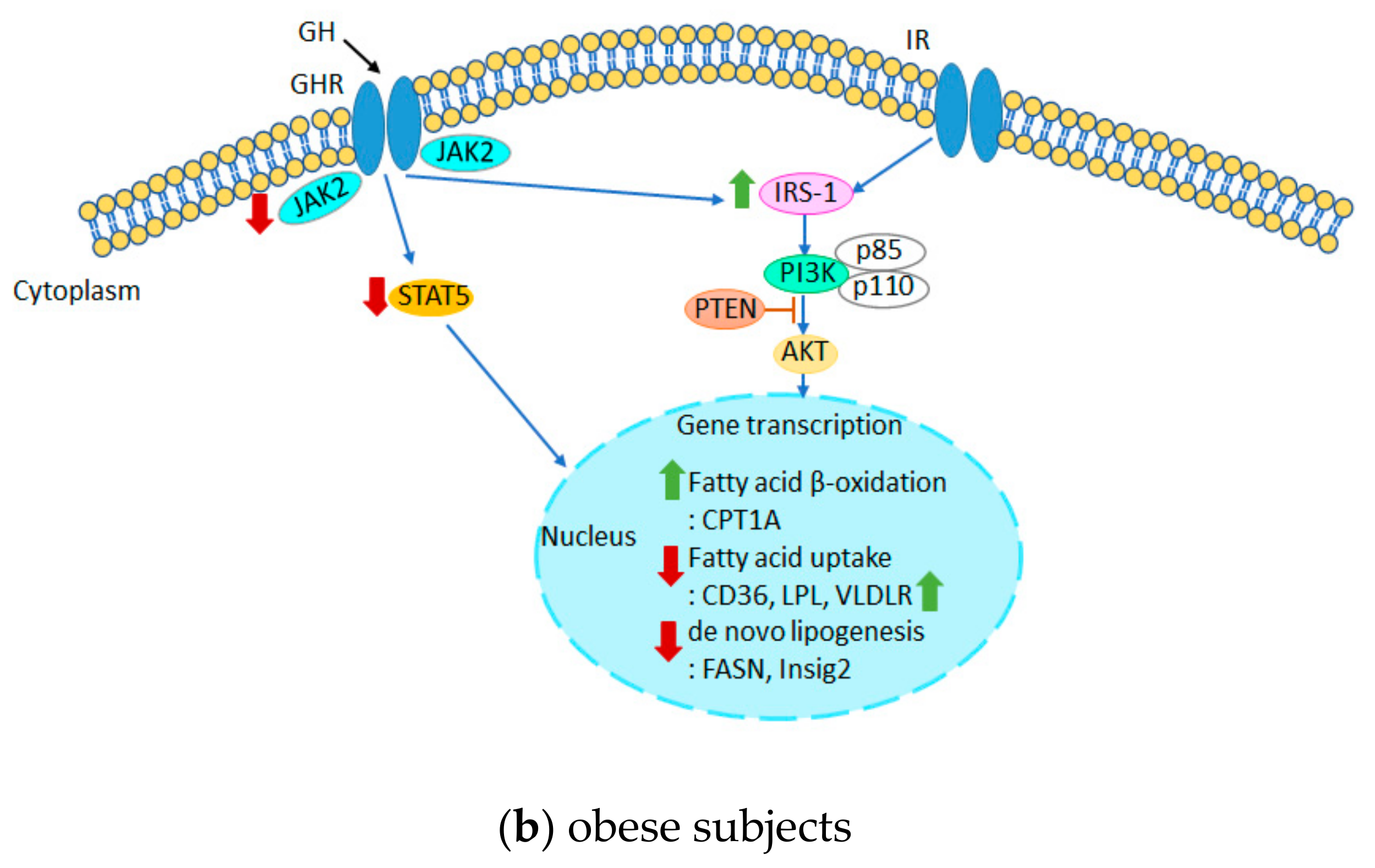
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aekplakorn, W.; Inthawong, R.; Kessomboon, P.; Sangthong, R.; Chariyalertsak, S.; Putwatana, P.; Taneepanichskul, S. Prevalence and trends of obesity and association with socioeconomic status in Thai adults: National Health Examination Surveys, 1991–2009. J. Obes. 2014, 2014, 410259. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Beydoun, M.A.; Min, J.; Xue, H.; Kaminsky, L.A.; Cheskin, L.J. Has the prevalence of overweight, obesity and central obesity levelled off in the United States? Trends, patterns, disparities, and future projections for the obesity epidemic. Int. J. Epidemiol. 2020, 49, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Renier, G.; Gaudreau, P.; Hajjad, H.; Deslauriers, N.; Houde-Nadeau, M.; Brazeau, P. Decreased pituitary growth hormone response to growth hormone-releasing factor in cafeteria-fed rats: Dietary and obesity effects. Neuroendocrinology 1990, 52, 284–290. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, J.A.; Smitz, J.P.; Zhou, X.L.; Louis, O.; Velkeniers, B.E.; Vanhaelst, L. Cafeteria diet-induced obesity is associated with a low spontaneous growth hormone secretion and normal plasma insulin-like growth factor-I concentrations. Growth Horm. IGF Res. 1998, 8, 397–401. [Google Scholar] [CrossRef]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Ferrannini, E.; Natali, A.; Capaldo, B.; Lehtovirta, M.; Jacob, S.; Yki-Järvinen, H.; for the European Group for the Study of Insulin Resistance (EGIR). Insulin resistance, hyperinsulinemia, and blood pressure: Role of age and obesity. Hypertension 1997, 30, 1144–1149. [Google Scholar] [CrossRef]
- Møller, N.; Jørgensen, J.O.; Abildgård, N.; Orskov, L.; Schmitz, O.; Christiansen, J.S. Effects of growth hormone on glucose metabolism. Horm. Res. 1991, 36 (Suppl. S1), 32–35. [Google Scholar]
- Malmlöf, K.; Din, N.; Johansen, T.; Pedersen, S.B. Growth hormone affects both adiposity and voluntary food intake in old and obese female rats. Eur. J. Endocrinol. 2002, 146, 121–128. [Google Scholar] [CrossRef]
- Malmlöf, K.; Johansen, T. Growth hormone-mediated breakdown of body fat: Insulin and leptin responses to GH are modulated by diet composition and caloric intake in old rats. Horm. Metab. Res. 2003, 35, 236–242. [Google Scholar] [PubMed]
- Davidson, M.B. Effect of Growth Hormone on Carbohydrate and Lipid Metabolism. Endocr. Rev. 1987, 8, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Park, M.-J. Effects of growth hormone on glucose metabolism and insulin resistance in human. Ann. Pediatr. Endocrinol. Metab. 2017, 22, 145–152. [Google Scholar] [CrossRef]
- Malmlöf, K.; Fledelius, C.; Johansen, T.; Theodorsson, E. The anorectic response to growth hormone in obese rats is associated with an increased rate of lipid oxidation and decreased hypothalamic galanin. Physiol. Behav. 2011, 102, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Likitnukul, S.; Kalandakanond-Thongsong, S.; Thammacharoen, S. Evidence of growth hormone effect on plasma leptin in diet-induced obesity and diet-resistant rats. Asian Biomed. 2018, 12, 219–228. [Google Scholar] [CrossRef]
- Weltman, A.; Wideman, L.; Weltman, J.Y.; Veldhuis, J.D. Neuroendocrine control of GH release during acute aerobic exercise. J. Endocrinol. Investig. 2003, 26, 843–850. [Google Scholar] [CrossRef]
- Thomas, G.A.; Kraemer, W.J.; Comstock, B.A.; Dunn-Lewis, C.; Maresh, C.M.; Volek, J.S. Obesity, Growth Hormone and Exercise. Sport. Med. 2013, 43, 839–849. [Google Scholar] [CrossRef]
- Bisker-Kassif, O.; Kauli, R.; Lilos, P.; Laron, Z. Biphasic response of subscapular skinfold thickness to hGH or IGF-1 administration to patients with congenital IGHD, congenital MPHD and Laron syndrome. Obes. Res. Clin. Pract. 2014, 8, e55–e62. [Google Scholar] [CrossRef]
- Fox, K.R.; Hillsdon, M. Physical activity and obesity. Obes. Rev. 2007, 8 (Suppl. S1), 115–121. [Google Scholar] [CrossRef]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Carlsson, L.; Nilsson, I.; Oscarsson, J. Hormonal regulation of liver fatty acid-binding protein in vivo and in vitro: Effects of growth hormone and insulin. Endocrinology 1998, 139, 2699–2709. [Google Scholar] [CrossRef]
- Latasa, M.-J.; Moon, Y.S.; Kim, K.-H.; Sul, H.S. Nutritional regulation of the fatty acid synthase promoter in vivo: Sterol regulatory element binding protein functions through an upstream region containing a sterol regulatory element. Proc. Natl. Acad. Sci. USA 2000, 97, 10619. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Menon, R.K.; Cohen, P.; Hwang, D.; Clemens, T.; DiGirolamo, D.J.; Kopchick, J.J.; Le Roith, D.; Trucco, M.; Sperling, M.A. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J. Biol. Chem. 2009, 284, 19937–19944. [Google Scholar] [CrossRef] [PubMed]
- Barclay, J.; Nelson, C.N.; Ishikawa, M.; Murray, L.A.; Kerr, L.M.; McPhee, T.R.; Powell, E.; Waters, M.J. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology 2011, 152, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Cordoba-Chacon, J.; Majumdar, N.; List, E.O.; Diaz-Ruiz, A.; Frank, S.J.; Manzano, A.; Bartrons, R.; Puchowicz, M.; Kopchick, J.J.; Kineman, R.D. Growth Hormone Inhibits Hepatic De Novo Lipogenesis in Adult Mice. Diabetes 2015, 64, 3093–3103. [Google Scholar] [CrossRef]
- Moon, Y.S.; Latasa, M.-J.; Kim, K.-H.; Wang, D.; Sul, H.S. Two 5’-regions are required for nutritional and insulin regulation of the fatty-acid synthase promoter in transgenic mice. J. Biol. Chem. 2000, 275, 10121–10127. [Google Scholar] [CrossRef]
- Alam, S.; Mustafa, G.; Alam, M.; Ahmad, N. Insulin resistance in development and progression of nonalcoholic fatty liver disease. World J. Gastrointest. Pathophysiol. 2016, 7, 211–217. [Google Scholar] [CrossRef]
- Bugianesi, E.; McCullough, A.J.; Marchesini, G. Insulin resistance: A metabolic pathway to chronic liver disease. Hepatology 2005, 42, 987–1000. [Google Scholar] [CrossRef]
- Qin, Y.; Tian, Y. Preventive effects of chronic exogenous growth hormone levels on diet-induced hepatic steatosis in rats. Lipids Health Dis. 2010, 9, 78. [Google Scholar] [CrossRef]
- Huang, L.; Steyn, F.J.; Tan, H.Y.; Xie, T.Y.; Veldhuis, J.D.; Ngo, S.T.; Chen, C. The Decline in Pulsatile GH Secretion throughout Early Adulthood in Mice Is Exacerbated by Dietary-Induced Weight Gain. Endocrinology 2012, 153, 4380–4388. [Google Scholar] [CrossRef]
- Steyn, F.J.; Xie, T.Y.; Huang, L.; Ngo, S.T.; Veldhuis, J.D.; Waters, M.J.; Chen, C. Increased adiposity and insulin correlates with the progressive suppression of pulsatile GH secretion during weight gain. J. Endocrinol. 2013, 218, 233–244. [Google Scholar] [CrossRef]
- Yaligar, J.; Gopalan, V.; Kiat, O.W.; Sugii, S.; Shui, G.; Lam, B.D.; Henry, C.J.; Wenk, M.R.; Tai, E.S.; Velan, S.S. Evaluation of Dietary Effects on Hepatic Lipids in High Fat and Placebo Diet Fed Rats by In Vivo MRS and LC-MS Techniques. PLoS ONE 2014, 9, e91436. [Google Scholar] [CrossRef]
- Recena Aydos, L.; Aparecida do Amaral, L.; Serafim de Souza, R.; Jacobowski, A.C.; Freitas Dos Santos, E.; Rodrigues Macedo, M.L. Nonalcoholic Fatty Liver Disease Induced by High-Fat Diet in C57bl/6 Models. Nutrients 2019, 11, 3067. [Google Scholar] [CrossRef] [PubMed]
- Sang, J.; Qu, H.; Gu, R.; Chen, D.; Chen, X.; Yin, B.; Huang, Y.; Xi, W.; Wang, C.; Huang, Y. Proteomics study of the effect of high-fat diet on rat liver. Br. J. Nutr. 2019, 122, 1062–1072. [Google Scholar] [CrossRef]
- Sos, B.C.; Harris, C.; Nordstrom, S.M.; Tran, J.L.; Balázs, M.; Caplazi, P.; Febbraio, M.; Applegate, M.A.B.; Wagner, K.-U.; Weiss, E.J. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte-specific deletion of JAK2. J. Clin. Investig. 2011, 121, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Han, L.; Zhu, L.; Yu, Y. Free fatty acids, not triglycerides, are associated with non-alcoholic liver injury progression in high fat diet induced obese rats. Lipids Health Dis. 2016, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Householder, L.A.; Lubbers, E.R.; List, E.O.; Troike, K.; Vesel, C.; Duran-Ortiz, S.; Kopchick, J.J.; Berryman, D.E. Growth Hormone Receptor Antagonist Transgenic Mice Are Protected from Hyperinsulinemia and Glucose Intolerance Despite Obesity When Placed on a HF Diet. Endocrinology 2015, 156, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Cacho, J.; Sevillano, J.; de Castro, J.; Herrera, E.; Ramos, M.P. Validation of simple indexes to assess insulin sensitivity during pregnancy in Wistar and Sprague-Dawley rats. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1269–E1276. [Google Scholar] [CrossRef]
- Huang, K.-W.; Reebye, V.; Czysz, K.; Ciriello, S.; Dorman, S.; Reccia, I.; Lai, H.-S.; Peng, L.; Kostomitsopoulos, N.; Nicholls, J.; et al. Liver Activation of Hepatocellular Nuclear Factor-4α by Small Activating RNA Rescues Dyslipidemia and Improves Metabolic Profile. Mol. Nucleic Acids 2020, 19, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, C.; Sun, H.; Vijayakumar, A.; Giglou, P.R.; Qiao, R.; Oppenheimer, J.; Yakar, S.; Leroith, D. Growth hormone receptor regulates β cell hyperplasia and glucose-stimulated insulin secretion in obese mice. J. Clin. Investig. 2011, 121, 2422–2426. [Google Scholar] [CrossRef]
- Huang, Y.; Chang, Y. Chapter Ten—Regulation of Pancreatic Islet Beta-Cell Mass by Growth Factor and Hormone Signaling. In Progress in Molecular Biology and Translational Science; Tao, Y.-X., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 121, pp. 321–349. [Google Scholar]
- Gao, Y.; Shen, W.; Lu, B.; Zhang, Q.; Hu, Y.; Chen, Y. Upregulation of hepatic VLDLR via PPARα is required for the triglyceride-lowering effect of fenofibrate. J. Lipid Res. 2014, 55, 1622–1633. [Google Scholar] [CrossRef]
- Mesotten, D.; Berghe, G.V.D.; Liddle, C.; Coulter, S.; McDougall, F.; Baxter, R.; Delhanty, P.J.D. Growth hormone modulation of the rat hepatic bile transporter system in endotoxin-induced cholestasis. Endocrinology 2003, 144, 4008–4017. [Google Scholar] [CrossRef]
- Xue, J.; Liang, S.; Ma, J.; Xiao, Y. Effect of growth hormone therapy on liver enzyme and other cardiometabolic risk factors in boys with obesity and nonalcoholic fatty liver disease. BMC Endocr. Disord. 2022, 22, 49. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-J.; Fan, J.-G.; Ding, X.-D.; Qiao, L.; Wang, G.-L. Characterization of High-Fat, Diet-Induced, Non-alcoholic Steatohepatitis with Fibrosis in Rats. Dig. Dis. Sci. 2010, 55, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Kučera, O.; Garnol, T.; Lotková, H.; Staňková, P.; Mazurová, Y.; Hroch, M.; Bolehovská, R.; Roušar, T.; Červinková, Z. The effect of rat strain, diet composition and feeding period on the development of a nutritional model of non-alcoholic fatty liver disease in rats. Physiol. Res. 2011, 60, 317–328. [Google Scholar] [CrossRef]
- Donkin, S.S.; McNall, A.D.; Swencki, B.S.; Peters, J.L.; Etherton, T.D. The growth hormone-dependent decrease in hepatic fatty acid synthase mRNA is the result of a decrease in gene transcription. J. Mol. Endocrinol. 1996, 16, 151–158. [Google Scholar] [CrossRef]
- Cabrera, D.; Cabello-Verrugio, C.; Solís, N.; Martín, D.S.; Cofré, C.; Pizarro, M.; Arab, J.P.; Abrigo, J.; Campos, F.; Irigoyen, B.; et al. Somatotropic Axis Dysfunction in Non-Alcoholic Fatty Liver Disease: Beneficial Hepatic and Systemic Effects of Hormone Supplementation. Int. J. Mol. Sci. 2018, 19, 1339. [Google Scholar] [CrossRef] [PubMed]
- Kaltenecker, D.; Themanns, M.; Mueller, K.M.; Spirk, K.; Suske, T.; Merkel, O.; Kenner, L.; Luís, A.; Kozlov, A.; Haybaeck, J.; et al. Hepatic growth hormone—JAK2—STAT5 signalling: Metabolic function, non-alcoholic fatty liver disease and hepatocellular carcinoma progression. Cytokine 2019, 124, 154569. [Google Scholar] [CrossRef]
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1447. [Google Scholar] [CrossRef]
- Jang, H.S.; Kim, K.; Lee, M.R.; Kim, S.H.; Choi, J.H.; Park, M.J. Treatment of growth hormone attenuates hepatic steatosis in hyperlipidemic mice via downregulation of hepatic CD36 expression. Anim. Cells Syst. 2020, 24, 151–159. [Google Scholar] [CrossRef]
- Oshio, Y.; Hattori, Y.; Kamata, H.; Ozaki-Masuzawa, Y.; Seki, A.; Tsuruta, Y.; Takenaka, A. Very low-density lipoprotein receptor increases in a liver-specific manner due to protein deficiency but does not affect fatty liver in mice. Sci. Rep. 2021, 11, 8003. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Mera, P.; Casas, J.; Salvador, J.; Rodríguez, A.; Alonso, S.; Sebastián, D.; Soler-Vázquez, M.C.; Montironi, C.; Recalde, S.; et al. Liver CPT1A gene therapy reduces diet-induced hepatic steatosis in mice and highlights potential lipid biomarkers for human NAFLD. FASEB J. 2020, 34, 11816–11837. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Guan, R.; Frank, S.J.; Messina, J.L. Insulin inhibits growth hormone signaling via the growth hormone receptor/JAK2/STAT5B pathway. J. Biol. Chem. 1999, 274, 13434–13442. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, A.; Greenhalgh, C.J.; Norstedt, G.; Rico-Bautista, E. Negative Regulation of Growth Hormone Receptor Signaling. Mol. Endocrinol. 2006, 20, 241–253. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Titchenell, P.M.; Quinn, W.J.; Lu, M.; Chu, Q.; Lu, W.; Li, C.; Chen, H.; Monks, B.R.; Chen, J.; Rabinowitz, J.D.; et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. 2016, 23, 1154–1166. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell. Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Sopasakis, V.R.; Liu, P.; Suzuki, R.; Kondo, T.; Winnay, J.; Tran, T.T.; Asano, T.; Smyth, G.; Sajan, M.P.; Farese, V.R.; et al. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010, 11, 220–230. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Selinger, E.S.; Ballou, L.M.; Lin, R.Z. Ablation of PI3K p110-α Prevents High-Fat Diet–Induced Liver Steatosis. Diabetes 2011, 60, 1483–1492. [Google Scholar] [CrossRef]
- Møller, N.; Jørgensen, J.O.L. Effects of Growth Hormone on Glucose, Lipid, and Protein Metabolism in Human Subjects. Endocr. Rev. 2009, 30, 152–177. [Google Scholar] [CrossRef]
- Gao, Y.; Su, P.; Wang, C.; Zhu, K.; Chen, X.; Liu, S.; He, J. The Role of PTEN in Chronic Growth Hormone-Induced Hepatic Insulin Resistance. PLoS ONE 2013, 8, e68105. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty acid synthase (FASN) | Forward (5′→3′) Reverse (5′→3′) | GCATTTCCACAACCCCAACC AACGAGTTGATGCCCACGAT |
| Lipoprotein lipase (LPL) | Forward (5′→3′) Reverse (5′→3′) | ATGGCACAGTGGCTGAAAGT CCGGCTTTCACTCGGATCTT |
| Cluster of differentiation 36 (CD36) | Forward (5′→3′) Reverse (5′→3′) | TGGACTTGTACTCTCTCCTCGG TCGTGCAGCAGAATCAAGGA |
| Carnitine palmitoyltransferase 1A (CPT1A) | Forward (5′→3′) Reverse (5′→3′) | TGCAGAGCAATAGGTCCCC ACACCCACCACCACCACGATAAG |
| Insulin induced gene 2 (Insig2) | Forward (5′→3′) Reverse (5′→3′) | GCGTGTTCCTGGCTTTAGTG CGACTTTAGCACTGGCGTGA |
| Very low-density lipoprotein receptor (VLDLR) | Forward (5′→3′) Reverse (5′→3′) | GTGATGAGCTGGACTGTGCT GCCACACTGCTCAAGAGACT |
| β-actin | Forward (5′→3′) Reverse (5′→3′) | CCACCATGTACCCAGGCATT AGGGTGTAAAACGCAGCTCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Likitnukul, S.; Thammacharoen, S.; Sriwatananukulkit, O.; Duangtha, C.; Hemstapat, R.; Sunrat, C.; Mangmool, S.; Pinthong, D. Short-Term Growth Hormone Administration Mediates Hepatic Fatty Acid Uptake and De Novo Lipogenesis Gene Expression in Obese Rats. Biomedicines 2023, 11, 1050. https://doi.org/10.3390/biomedicines11041050
Likitnukul S, Thammacharoen S, Sriwatananukulkit O, Duangtha C, Hemstapat R, Sunrat C, Mangmool S, Pinthong D. Short-Term Growth Hormone Administration Mediates Hepatic Fatty Acid Uptake and De Novo Lipogenesis Gene Expression in Obese Rats. Biomedicines. 2023; 11(4):1050. https://doi.org/10.3390/biomedicines11041050
Chicago/Turabian StyleLikitnukul, Sutharinee, Sumpun Thammacharoen, Orada Sriwatananukulkit, Chanathip Duangtha, Ruedee Hemstapat, Chotchanit Sunrat, Supachoke Mangmool, and Darawan Pinthong. 2023. "Short-Term Growth Hormone Administration Mediates Hepatic Fatty Acid Uptake and De Novo Lipogenesis Gene Expression in Obese Rats" Biomedicines 11, no. 4: 1050. https://doi.org/10.3390/biomedicines11041050