1. Introduction
Chronic myelogenous leukemia (CML) is caused by a chromosomal translocation t(9;22)(q34;q11.2), which consequences in the BCR-ABL1 chimeric gene as the carcinogenic trigger of (Ph+) leukemia or CML [
1]. This fusion gene is a clinical biomarker for CML in addition to a viable treatment approach. In the case of children (CML), it makes up 15% of all instances of myeloid leukemia. Its prevalence rises with age, reaching 1.2 instances per million annually in teenagers [
2]. The Philadelphia chromosome (Ph) is what distinguishes CML from other myeloproliferative neoplasms; however, albeit rarely, the Ph+ may also be seen in MPN other than CML [
3]. CML can manifest in one of three stages—chronic, accelerated, or blast—and is typically identified in the chronic stage in developed nations.
Asciminib is an allosteric inhibitor that binds to a myristoyl region on the BCR-ABL1 protein. Both natural and altered BCR-ABL1, including the intermediary T315I mutant, are targeted by asciminib [
4]. This mechanism of asciminib is different from that of all other ABL kinase inhibitors, as it locks the BCR-ABL1 into an inactive conformation. It exhibits low activity against unmutated BCR-ABL1 and all clinically identified ATP-site mutations, including T315I, though it has significant selectivity for only ABL1 and, presumably, ABL2 kinases. This is due to the unique shape of the myristoyl pocket [
5].
Existing ABL inhibitors can be divided into those that target the active conformation of the kinase domain and those that target the inactive kinase domain. These inhibitors compete at the ATP binding sites of these proteins. Since asciminib is distinctive in that it functions as an allosteric inhibitor, attaching to the BCR-ABL1 protein’s myristoyl pocket and immobilizing it in an inactive conformation, it is widely administered for the treatment of (Ph+)leukemia [
6]. The health risks of an overdose are likely to coincide with asciminib’s adverse effect profile; therefore, these might include serious hematological abnormalities and/or gastrointestinal side effects, among other concerns.
This study intends to overcome the existing toxicity that prevails in the already administered drugs for (Ph+)leukemia by the utilization of the natural vitamin E compound gamma-tocotrienol as a BCR-ABL1 inhibitor. The artificial intelligence deep learning algorithm application for the de novo drug design of tocotrienol was implemented, and a further toxicity comparison study was performed with asciminib. The AIGT was docked, and furthermore, analysis of AIGT has been proved vital in this study.
4. Discussion
Chronic myelogenous leukemia (CML), a slow-growing malignant hematological illness, is a result of 15% of instances of leukemia that are caused [
16]. The Philadelphia chromosome, which is formed by a reciprocal translocation that results in a prolonged chromosome 9 and a shorter chromosome 22, is the cause of this illness. The dysregulated BCR-ABL1 fusion carcinogen protein is developed as a result of the translocation, which contributes to the uncontrolled proliferating of white blood cells [
17]. Multiple processes involved in cell growth and division, including receptor endocytosis, autophagy, remodeling of the cytoskeleton and actin, cell motility and adhesion, and cell adhesion, depend on ABL1. Additionally, ABL1 translocates into the nucleus, where it assists in apoptosis, the response to DNA damage, and DNA binding activities [
4].
The mechanisms of action and toxicity of the medications used to treat CML differ. In a study conducted by Oliver Henke et al. [
18]: it was observed that Imatinib’s utilization in the management of CML radically altered the way this disease was addressed and spurred the advent of additional potent targeted protein kinase inhibitors. FDA-approved drugs for initial therapy comprise imatinib as an initial treatment. Furthermore, dasatinib binds to the kinases and prevents them from stimulating growth and is also administered as a treatment. Dasatinib and bosutinib are both regarded as the second line in therapy [
19]. Additionally, nilotinib treatment is linked to the transitory increase in serum aminotransferase levels and few incidences of clinically evident acute liver damage [
20]. Whereas, although the clinical manifestations of hepatotoxicity are still being precisely defined, occurrences of clinically evident liver problems, progressing hepatic failure, and fatality have been reported in ponatinib clinical studies [
21]. Despite hepatoxicity and other health hazardous effects of these drugs, they have been approved by the FDA, and each of these medications is orally ingested to treat (Ph+)leukemia or CML.
Asciminib works as a therapeutic agent by blocking an oncogenic protein that promotes the growth of CML. It functions to inhibit both the wild-type as well as some mutation forms of BCR-ABL1, along with the T315I mutation [
17]. Upon its administration, it recognizes and attaches to the myristoyl pocket of the BCR-ABL1 fusion protein, which is distinctive from the ATP-binding domain. An overdose’s side effects are likely to correspond to asciminib’s adverse effect profile; therefore, they may furthermore include severe gastrointestinal problems and hematological abnormalities mainly. Asciminib’s drug-induced death of the proliferating cells was defined by a power model. Additionally, the computational analysis of hepatotoxicity along with other toxicities was computed in this study that validates the administration of asciminib as highly risky. ProTox-II and VNN-Admet simultaneously justify asciminib as crucial for its role in liver damage.
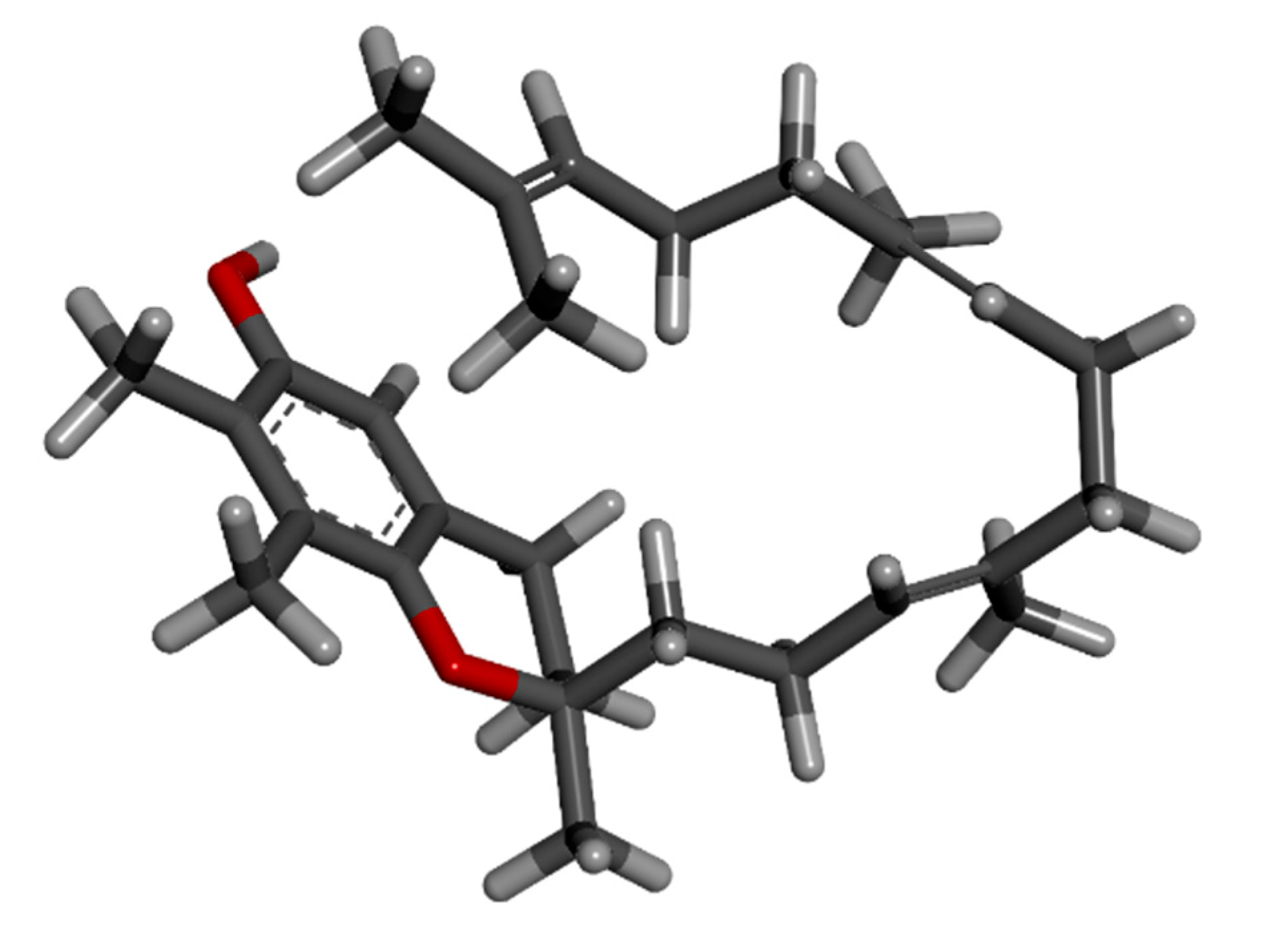
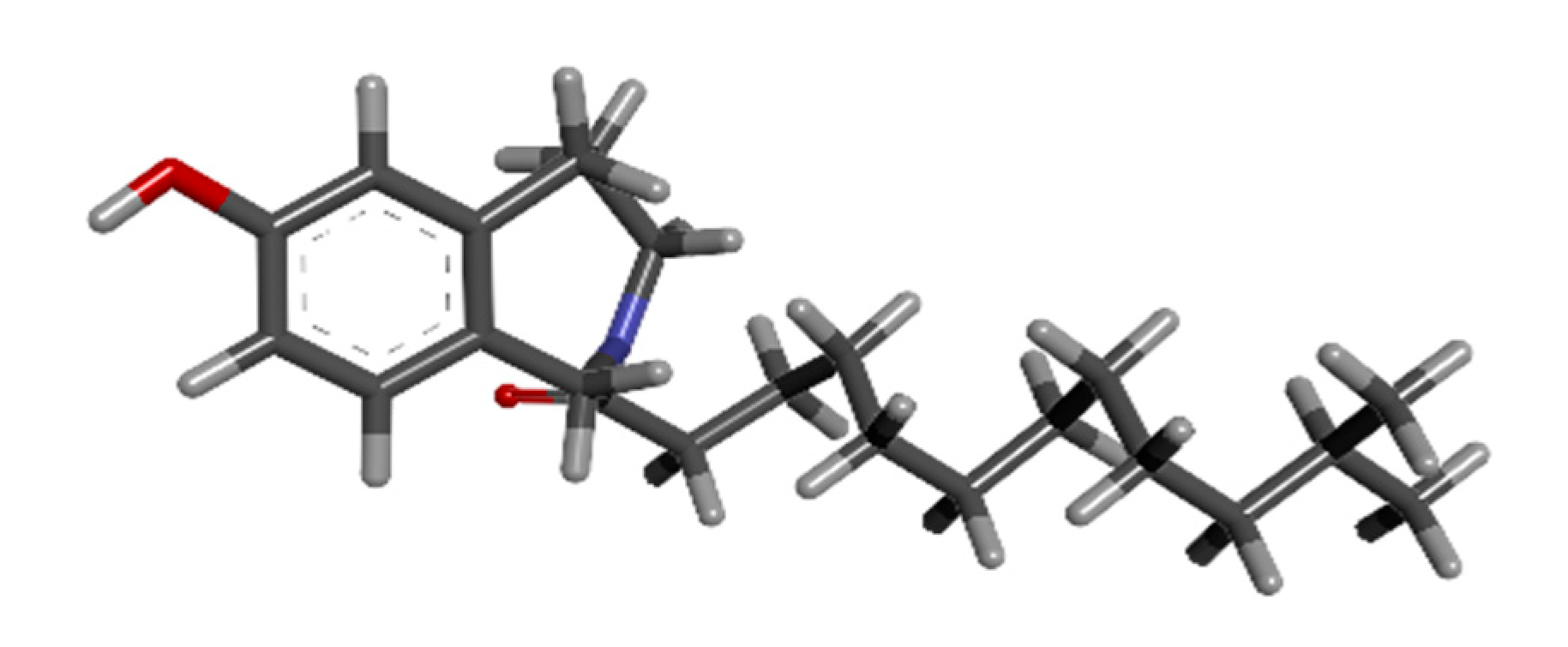
The vitamin E has eight members: four tocopherols, namely α-, β-, δ- and γ-tocopherol, and four tocotrienols in the form of α-, β-, δ- and γ-tocotrienols. Tocotrienols are an underappreciated isomer of vitamin E that has unmatched health advantages [
22]. Tocotrienols were rarely used in vitamin E studies until recently, despite their relative superiority to tocopherol and their widespread occurrence in palm oil. This study specifically focuses on the utilization of gamma-tocotrienol since it functions as an apoptosis inducer, a radiation protective agent, a plant metabolite, an antioxidant, an antineoplastic agent, and a hepatoprotective agent [
23].
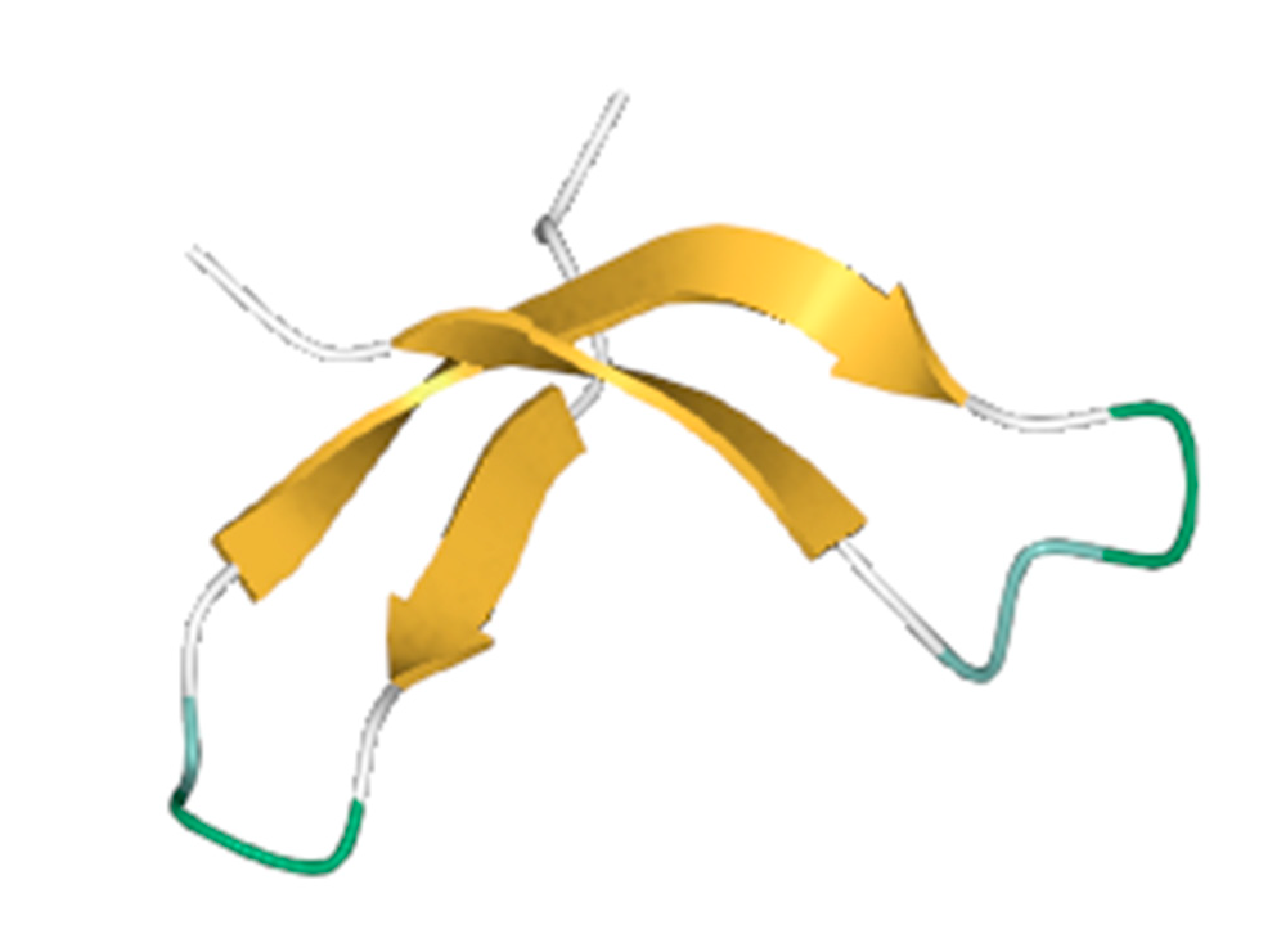
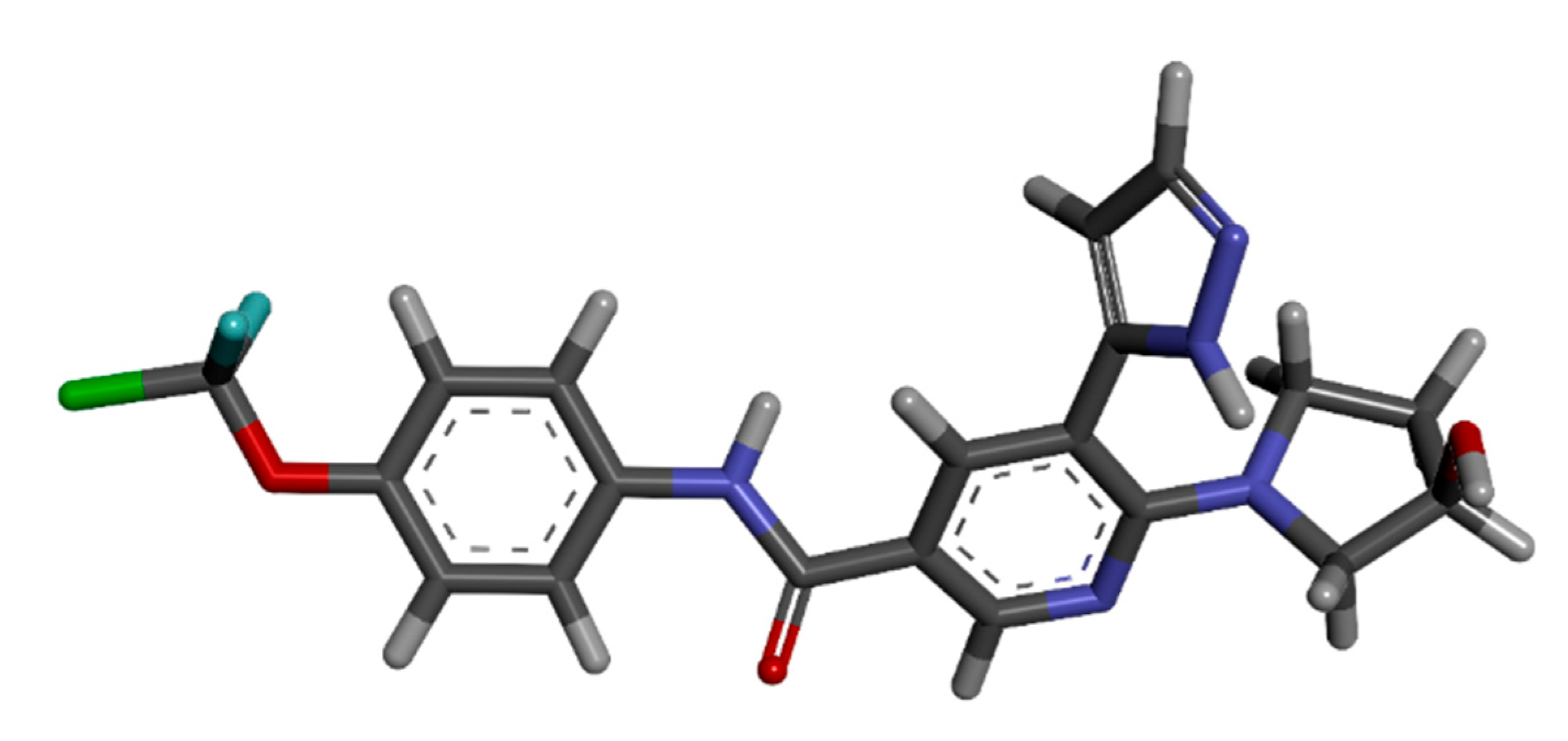
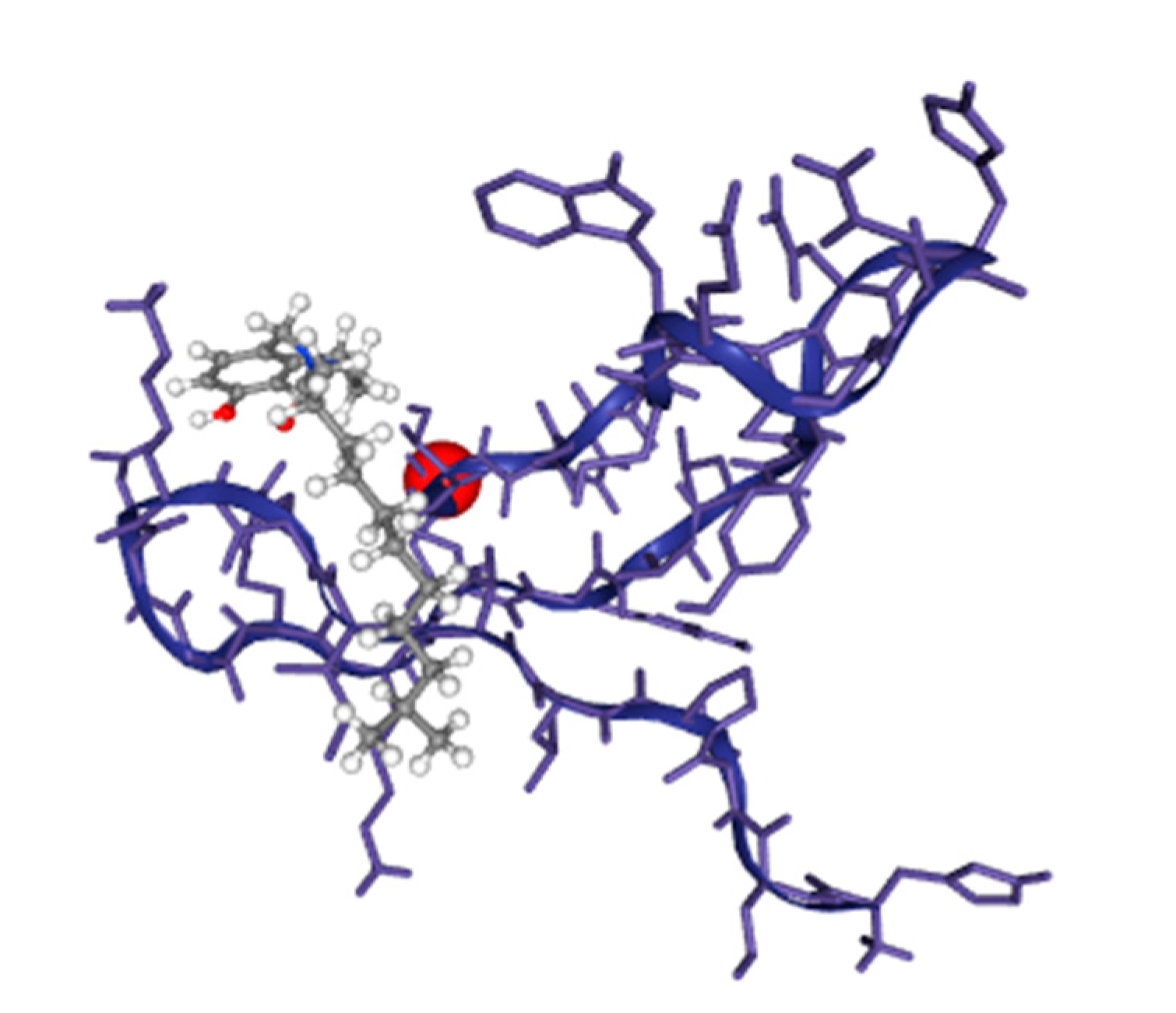
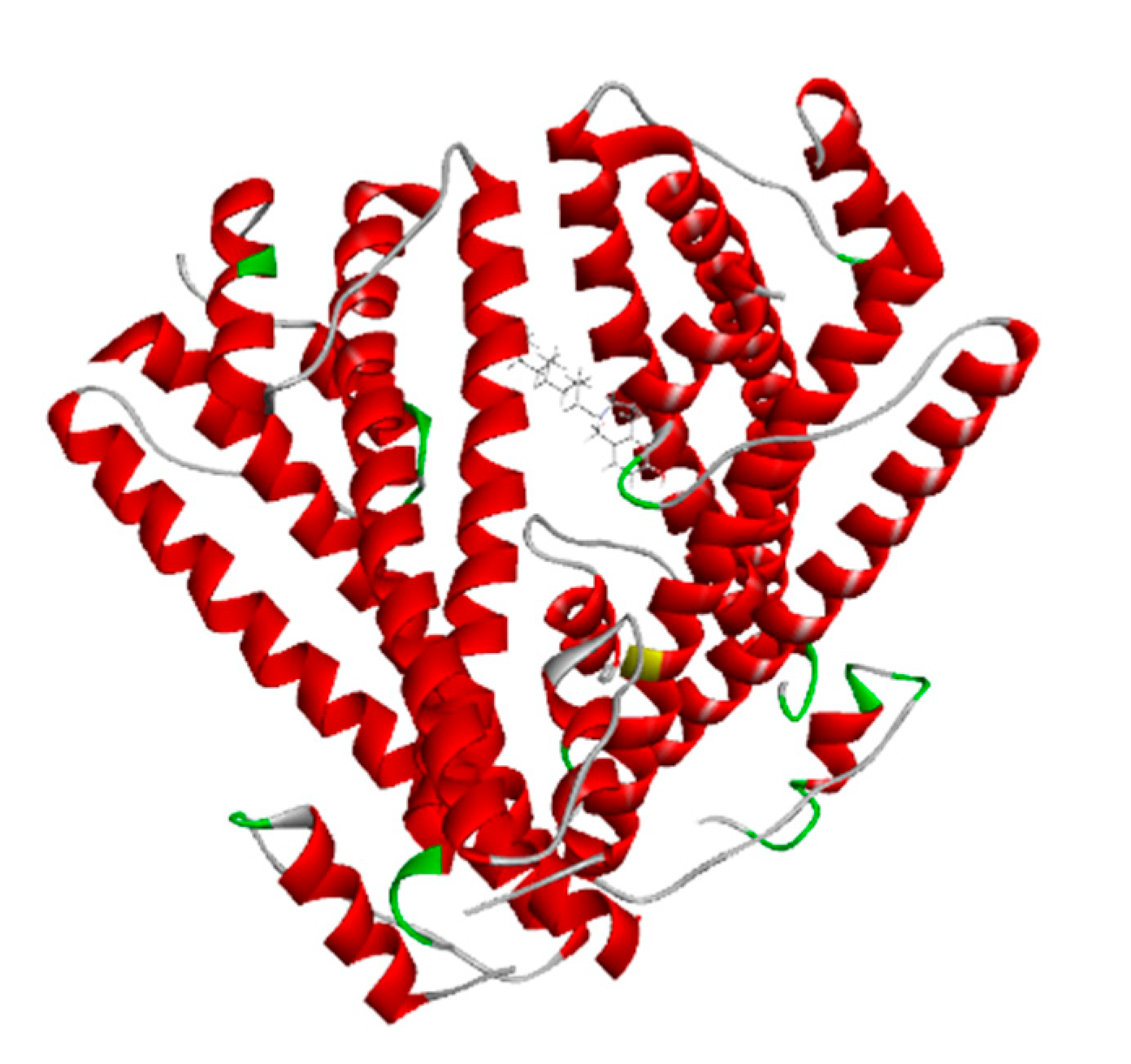
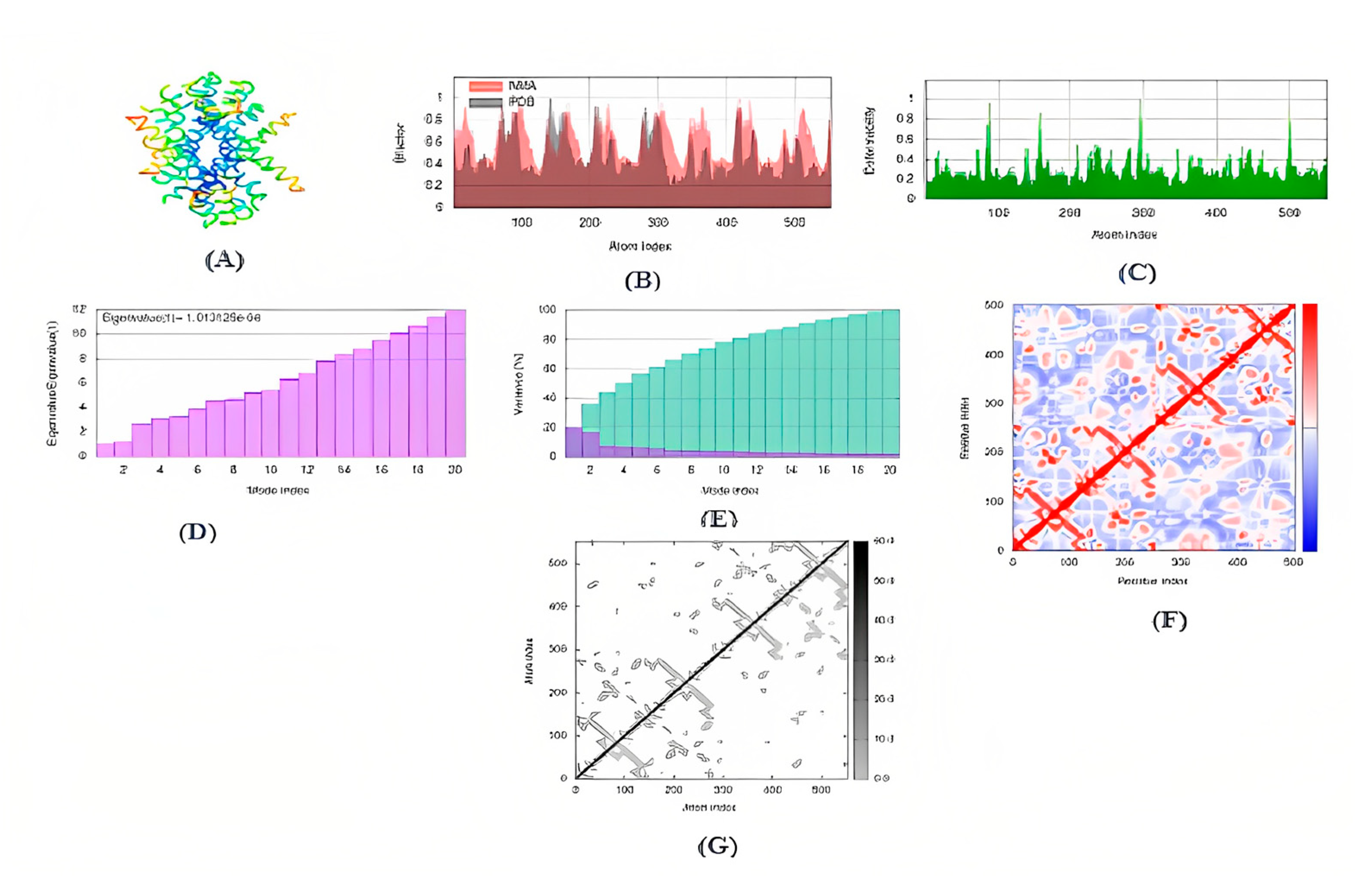
The advanced technique of revolutionary artificial intelligence for drug designing through deep learning algorithms was implemented by assessing the WADDAICA online server. Docking is a valuable tool for screening because it allows for the virtual prediction of how small molecules, such as drugs, will interact with proteins, such as receptors or enzymes. This simulation can provide valuable information about the binding affinity, orientation and energetics of the protein-ligand interaction without the need for experimental techniques. It’s important to note that different scoring functions may have different strengths and weaknesses, and the choice of scoring function depends on the specific problem and the desired trade-off between accuracy and speed. While DockThor Vina may work well for a particular use case, other scoring functions may be better suited for other situations. It’s always a good idea to test and compare multiple scoring functions to determine which is the best fit for a given problem. Post-refining schemes are computational methods that are used to improve the accuracy of protein-ligand docking predictions. Two commonly used post-refining methods are MM/PBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) and MM/GBSA (Molecular Mechanics/Generalized Born Surface Area). The gamma-tocotrienol was employed for drug design by AI tool for the retrieval of three competent de novo drug molecules against the BCR-ABL1 fusion protein. The AIGT was selected as the target candidate based on its drug-likeliness analysis amongst all three. The comparison study of the toxicity of asciminib and AIGT proves that AIGT is not only more efficacious but hepatoprotective as well. Moreover, the ADMET analysis comparison further justifies the findings, The docking of BCR-ABL1-1 with AIGT resulted in the binding affinity of −7.486 kcal/mol hence proving that AIGT is a potential drug candidate. The execution of docking with Patchdock to assure the shape complementarity of the protein and ligand substantially reinforced the docking findings. Thus, the molecular dynamic stimulation also enhanced the reliability of the efficiency of the results.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}