1. Introduction
Staphylococcus aureus, a Gram-positive, cocci-shaped, and immotile bacterium of the Firmicutes family, is associated with a wide range of infections in mammalian skin, various mucosal membranes, and the nostrils. This human pathogen is a significant source of infections contracted in the community or hospital, with antibiotic resistance making it difficult to treat these infections effectively [
1]. Due to its metabolic adaptability and pharmacy resistance,
Staphylococcus aureus can thrive in a wide range of conditions, with an estimated 25–30% of healthy individuals harboring the bacteria on their skin and nasopharyngeal membranes as a natural component of the human microbiome (Raafat et al., 2020).
Staphylococcus aureus is responsible for tens of thousands of infections annually and is a leading cause of pneumonia and some respiratory tract infections, cardiovascular infections, nosocomial infections, and prosthetic joints. Studies have shown that more deaths occur because of
Staphylococcus aureus bacteremia than from AIDS, viral hepatitis, tuberculosis, and other diseases combined [
2]. Furthermore, this bacterium is also associated with abscesses, wound infections, and furuncles, which can cause substantial morbidity and sickness but are rarely life-threatening.
However, when
Staphylococcus aureus enters the circulation or internal tissues, it causes a wide range of severe illnesses. The emergence of antibiotic-resistant strains of bacteria, particularly methicillin-resistant
Staphylococcus aureus (MRSA), has become a major public health issue due to the wide range of diseases it can cause, from mild skin infections to life-threatening conditions [
3].
Staphylococcus aureus infections are typically treated with antibiotics as the primary means of defense. However, the impulsive use of antibiotics, which initially had a high success rate, led to the emergence and expansion of antibiotic-resistant bacteria. For instance, MRSA and vancomycin-resistant
staphylococcus aureus (VRSA) were both discovered within a short period after the clinical application of these antibiotics [
4]). The low affinity of penicillin-binding protein 2a (PBP2a) encoded by the
mecA gene of
Staphylococcus aureus for β-lactam medications, combined with the high production of the lysyl-phosphatidylglycerol enzyme, has caused methicillin-resistant
Staphylococcus aureus to be resistant to β-lactam antibiotics. Additionally,
Staphylococcus aureus produces the antibiotic-inactivating enzyme β-lactamase and, without the auxiliary gene regulator (Agr), β-lactam antibiotics are rendered ineffective [
5]).
Staphylococcus aureus infections can be particularly severe due to their limited virulence factors, making it difficult for them to be fought by the body’s immune system. These virulence factors, mostly found on the surface of the bacteria, include proteins such as protein A, clumping factor, and binding proteins, as well as polysaccharide intercellular adhesins and toxins that function as superantigens. Enzymes, including coagulase, staphylokinase, and protease, aid in immune evasion and host tissue penetration. The production of
Staphylococcal protein A and surface proteins by virtually all
Staphylococcus aureus isolates serves as a superantigen by binding to immunoglobulins, inhibiting opsonization, and phagocytosis [
6]. Adhesion and evasion of the immune response depend on clumping factors A and B, which bind to fibrinogen facilitating the invasion of host tissues. These proteins can also adhere to the extracellular matrix, promoting the invasion of host tissues through binding to fibronectin and elastin [
7].
Staphylococcus aureus releases toxins and virulent genes associated with toxic shock syndrome, food poisoning, Staphylococcal scalded skin syndrome, and Staphylococcal scarlet fever. Moreover, these genes encode diverse virulent factors that play a crucial role in disease pathology. Factors such as toxic shock syndrome toxin-1, delta-hemolysin (hld), and Staphylococcal protein A (Spa) are all involved in the pathogenesis of bacterial infections. To combat the prevalent multidrug resistance of Staphylococcus aureus, this study aimed to identify virulent factors of the bacterium and construct an mRNA vaccine utilizing an immunoinformatic approach and various online bioinformatics tools. Additional tests utilizing both in vitro and in vivo studies on mRNA vaccines will be required to assess the efficacy of an mRNA vaccine in the future.
4. Discussion
The bacterium
Staphylococcus aureus, widely acknowledged for its ability to cause a spectrum of illnesses, particularly in individuals with nosocomial infections, has been reported to possess natural resistance to ampicillin, macrolide antibiotics, cephalosporins, and cefotaxime. The phenomenon of antibiotic defense in bacteria is a worldwide health issue, as it affects both animals and humans [
34]. The increasing opposition to penicillin in
Staphylococcus aureus has caused more frequent use of vancomycin to treat resistant bacterial infections worldwide. However, the frequent use of vancomycin has diminished its effectiveness in combating
Staphylococcus aureus infections [
35]. Considering these challenges, this study aims to identify virulent factors of
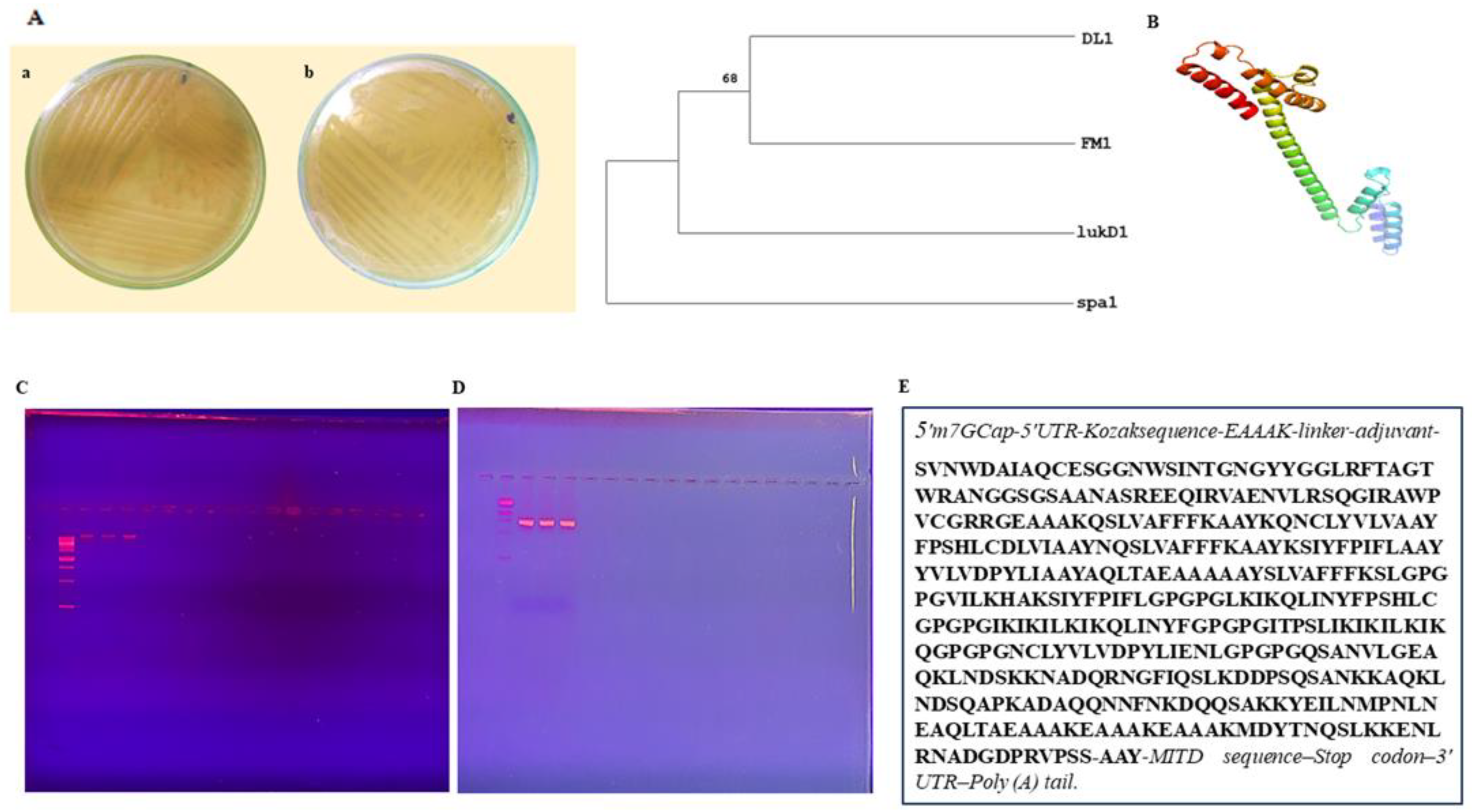
Staphylococcus aureus to design an mRNA vaccine that can combat antibiotic resistance. The process involves isolating desired bacteria through culturing, selecting three strains for molecular identification, and detecting virulent genes, such as
spa,
lukD,
fmhA, and
hld genes, by means of PCR with the product size of 293 bp, 243 bp, 345 bp and 357 bp, respectively. DNA extraction from samples of
Staphylococcus aureus obtained from the Sheikh Zayed Hospital and the PCSIR was performed utilizing the CTAB technique and confirmed by the gel doc technique. Identification of bacterial strains was accomplished by utilizing 16S rRNA and primers such as
spa,
lukD,
fmhA, and
hld for gene amplification. Sequencing was conducted at ABI, Malaysia, and the phylogenetic tree, and alignment of strains was constructed after sequencing. In silico analysis of
spa,
fmhA, and
hld genes was also carried out, including creating an antigen-specific vaccine as performed by [
36].
Vaccination aims to produce a sustained immune reaction in the individual, allowing the body to confront future encounters with the pathogen more effectively [
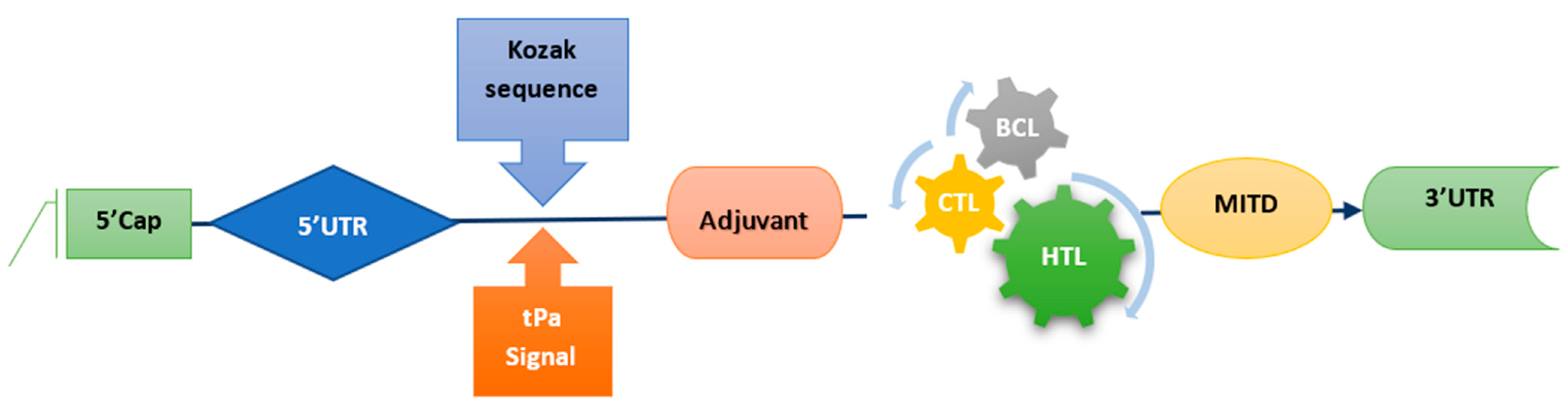
37]. In this study, virulence genes were converted into proteins, and a chimeric construct was created using various linkers. Incorporating epitopes that can activate both B and T cells into the vaccine design is essential for the vaccine to be successful [
38]. The chosen epitopes should be able to initiate the production of HTL-regulated anti-pathogen cytokines such as IFN-, IL-4, and IL-10 [
39]. After an infection is cleared, only memory cells survive, while other immune cells die. Through the presentation of processed epitopes via MHC II, B cells recognize and deliver these epitopes to T cells, allowing the T-cell receptor (TCR) to acknowledge them.
Furthermore, B-cells develop into plasma cells that create antibodies and memory cells that can be called upon in the future [
40]. This study emphasized the development of an mRNA peptide vaccine, created with antigenic proteins of
Staphylococcus aureus and utilizing only in silico methods, that was designed to induce a robust immune response. The proposed mRNA vaccine’s antigenicity, non-allergenicity, and hydrophilicity were assessed using immunoinformatic techniques; the efficacy of these tools has been discussed in the work by [
41,
42,
43]. Palladini et al. discussed that the predictions made by in silico modeling, which involves using computer simulations to predict outcomes, were accurate. However, designing and creating models of vaccines and protocols for vaccination should consider the gradual weakening of the immune system that occurs with aging and aim to enhance immune responses in older individuals [
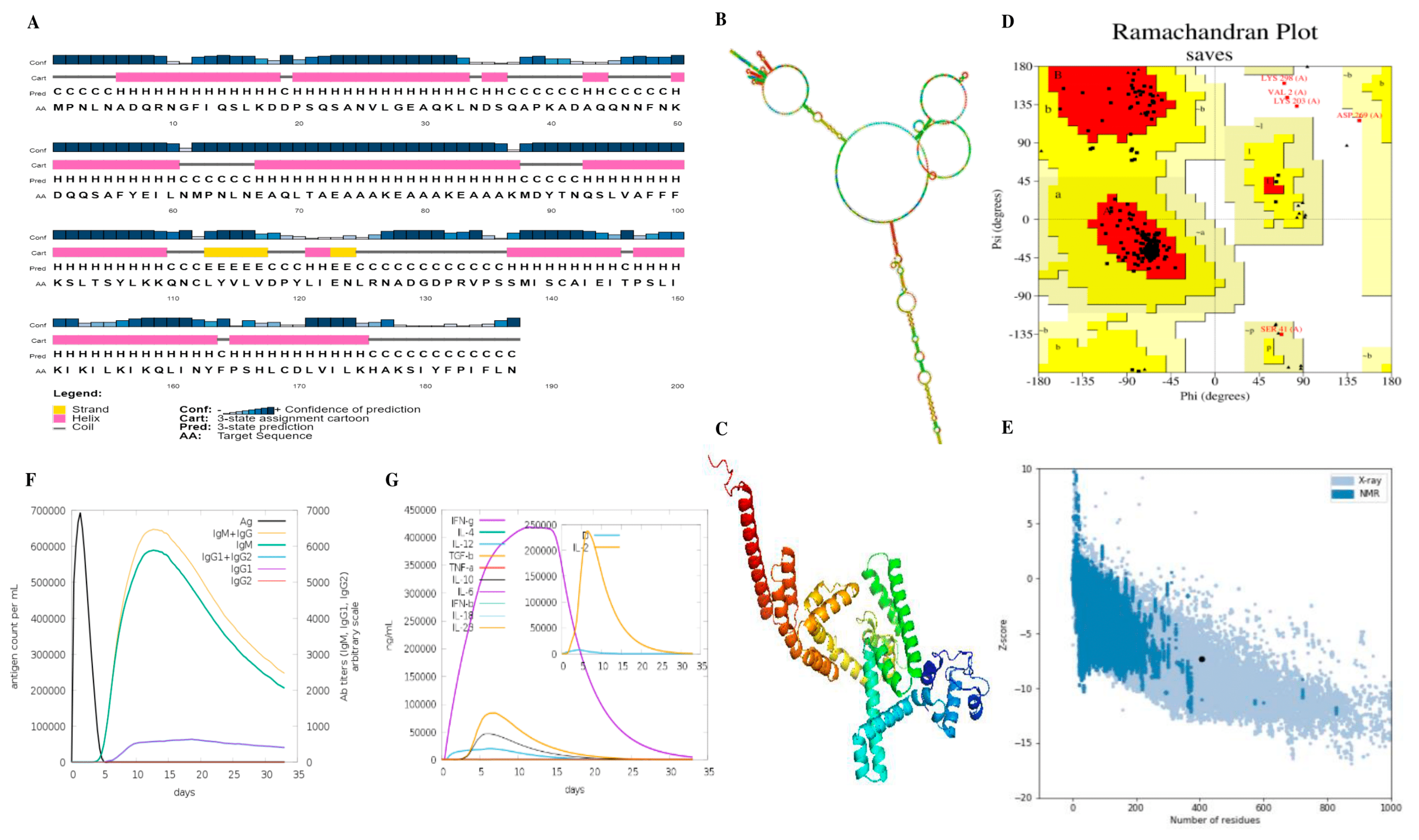
41]. To research a vaccine’s efficacy, this study used molecular simulations to determine if it induced an immune response following three injections. The simulations’ results showed that the response could generate memory cells and chemokines that stimulate B-cell and humoral reactions. The effective use of molecular dynamics simulations and the accuracy of computational tools has been confirmed previously [
44,
45,
46]. Indicators such as macrophages and dendritic cells also showed signs of memory cell formation. Ultimately, this study determined that vaccination is viable for preventing
Staphylococcus aureus infections.
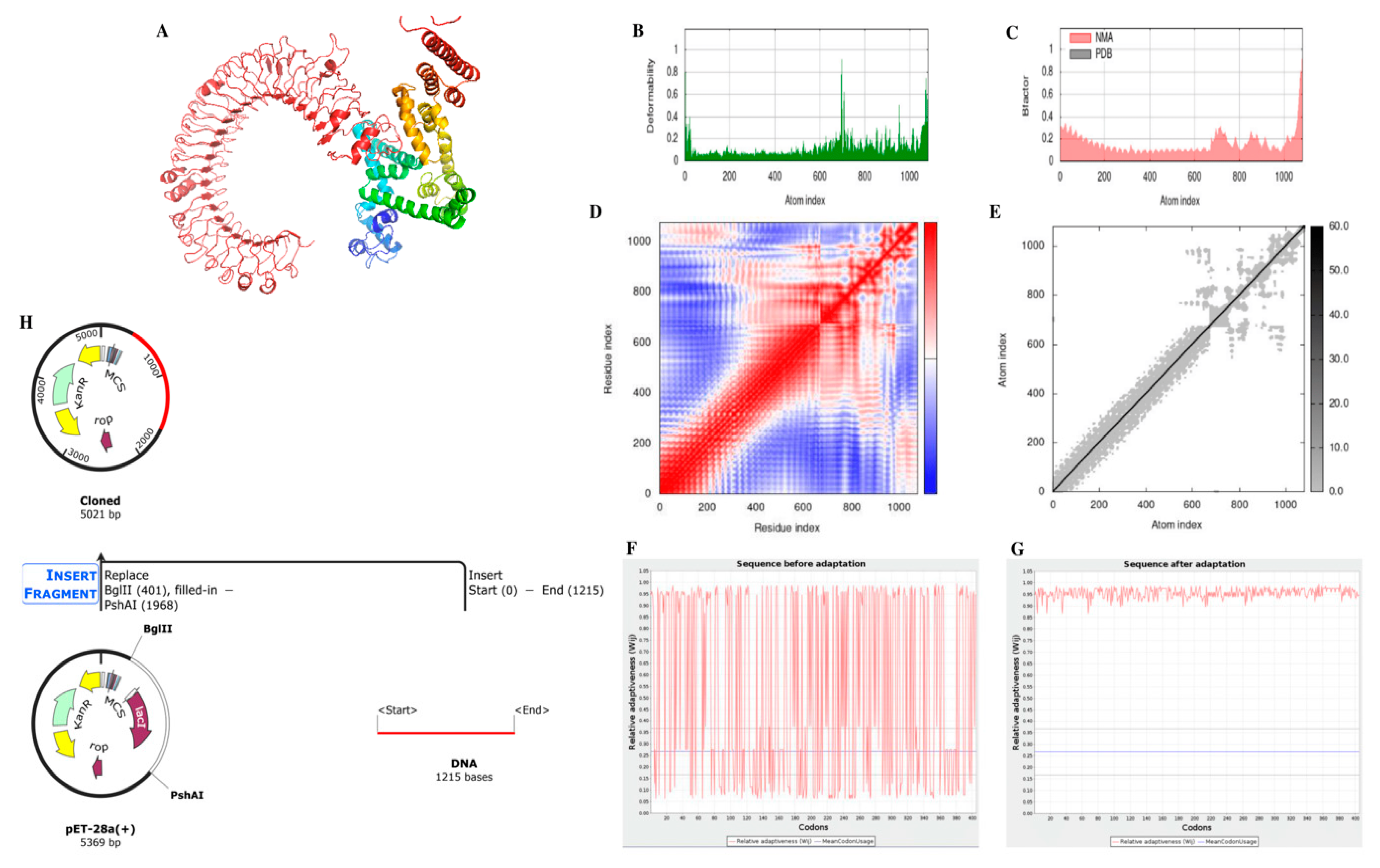
The primary advantage of using in silico tools is that they allow for the rapid and efficient identification of virulent genes and the design of an mRNA vaccine. The design can be completed in relatively short time lapses and at a lower market price compared to traditional laboratory techniques. Additionally, in silico techniques can provide a high degree of accuracy and specificity in identifying virulent genes, which is crucial for the success of the vaccination. However, there are also limitations to using only computational tools. In silico techniques are dependent on the availability and quality of data, and the validity of predictions is highly reliant on the algorithms used.
Furthermore, the results of the in silico analysis must be verified experimentally to ensure their validity. The study suggests that vaccination is a viable option for preventing Staphylococcus aureus infections, and that in silico techniques can help identify virulent genes and design an mRNA vaccine. However, it is essential to consider the limitations of using only computational tools and to verify the results experimentally.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}