
The Effect of the Sodium—Glucose Cotransporter Inhibitor on Cognition and Metabolic Parameters in a Rat Model of Sporadic Alzheimer’s Disease
 ,
,  , ,
, ,  , and
, and 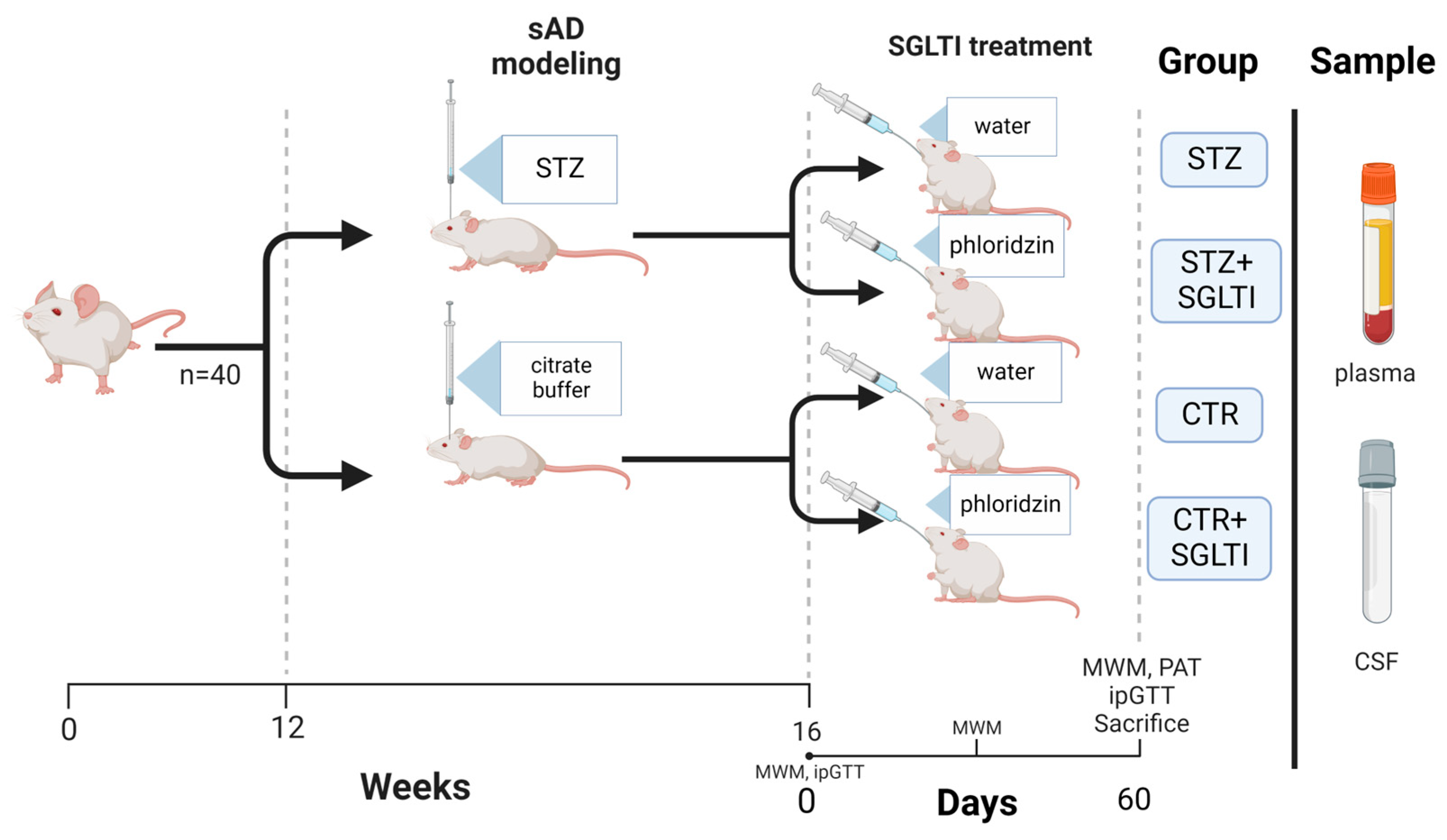{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Materials
2.3. Drug Treatments
2.4. Cognitive Testing
2.5. Intraperitoneal Glucose Tolerance Test (ipGTT)
2.6. Biochemical Analyses
2.7. Ethics
2.8. Statistics
3. Results
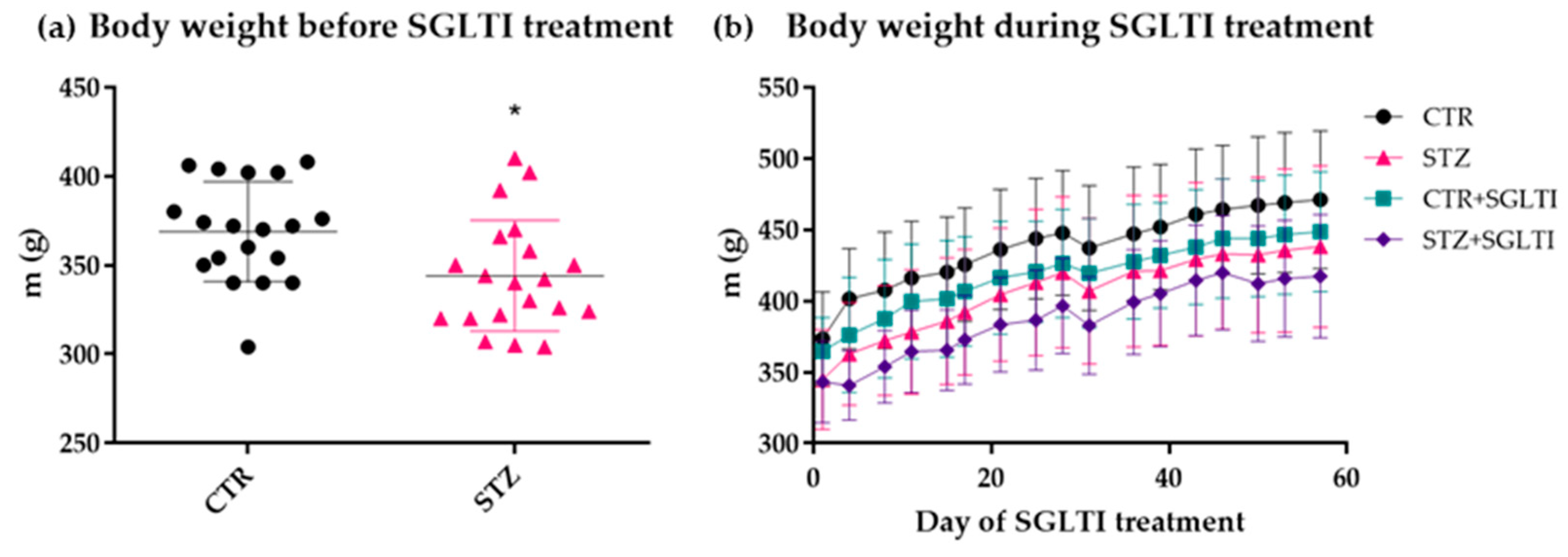
3.1. Body Weight
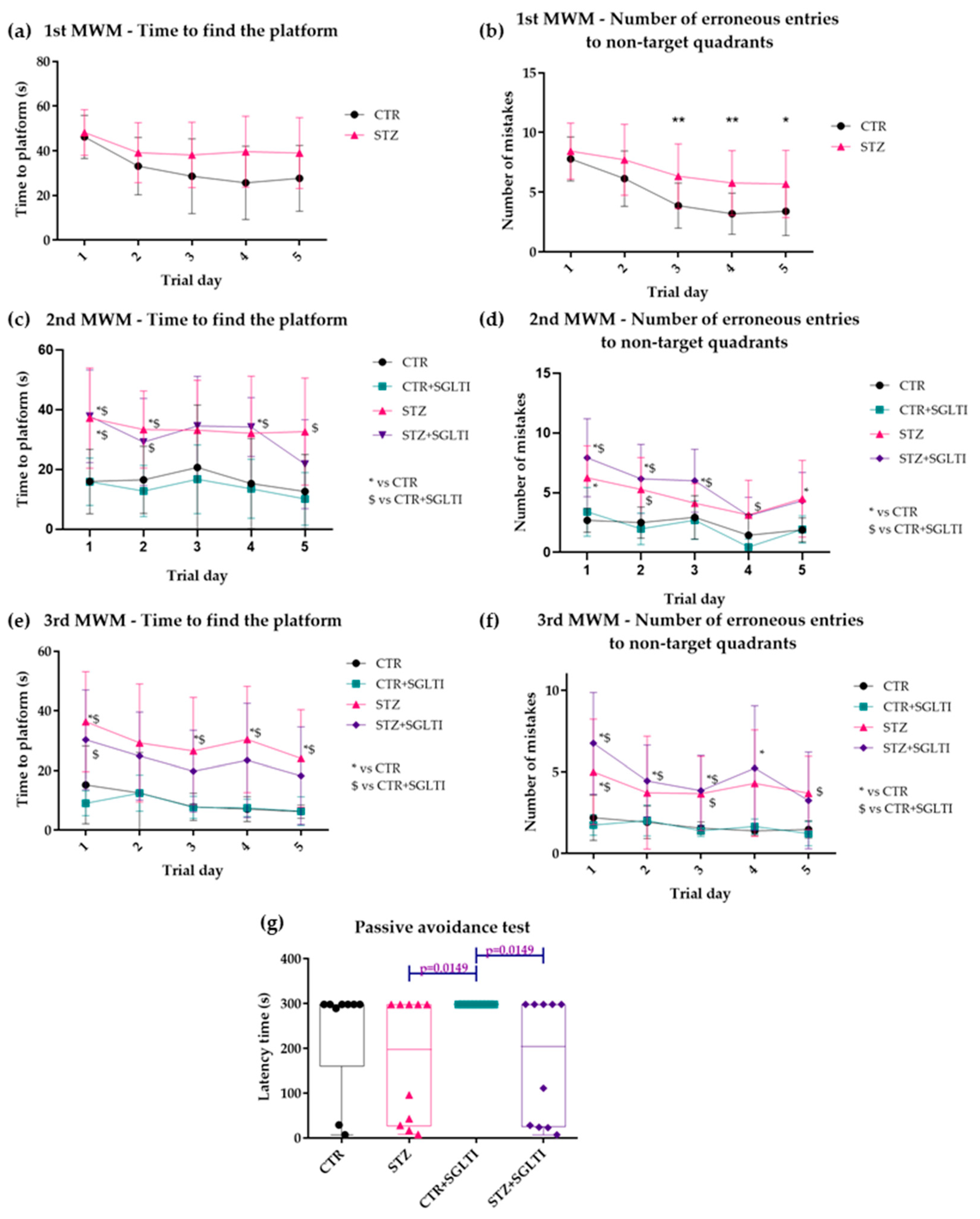
3.2. Cognitive Assessment
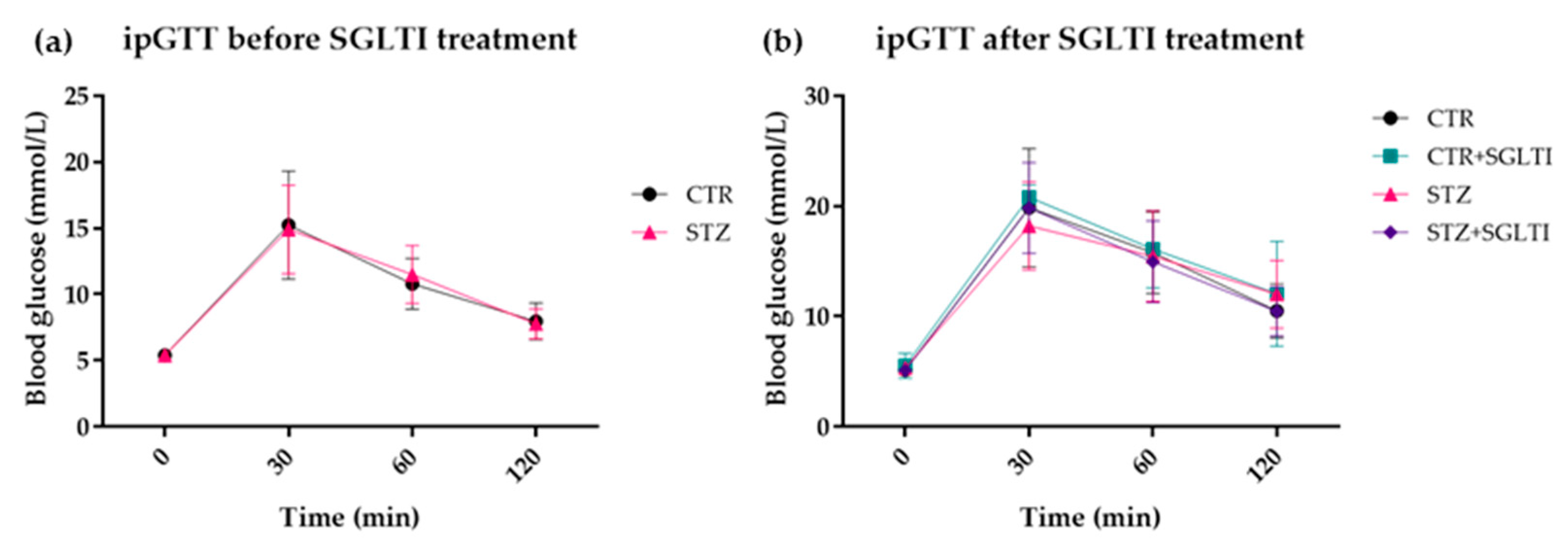
3.3. ipGTT
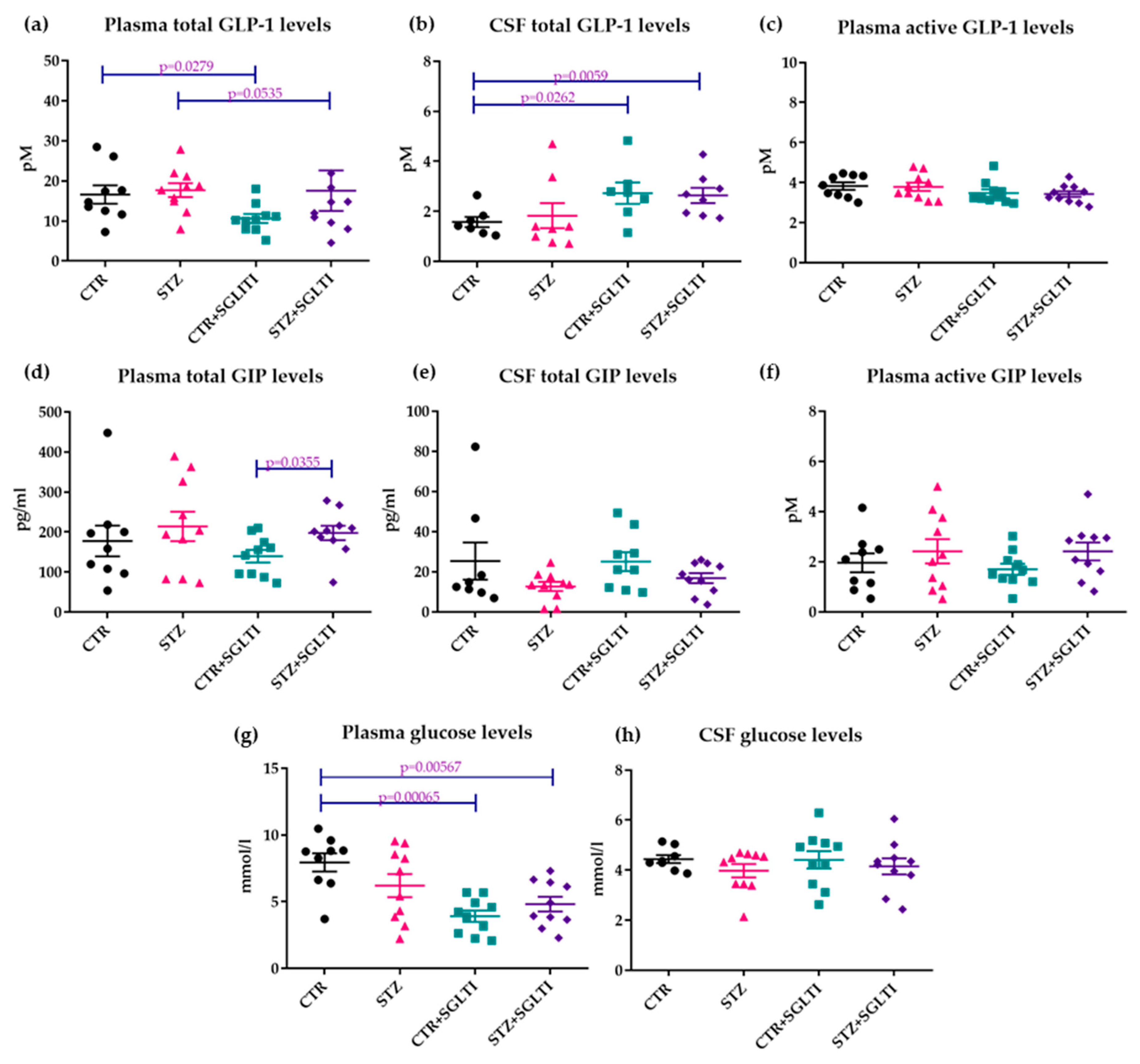
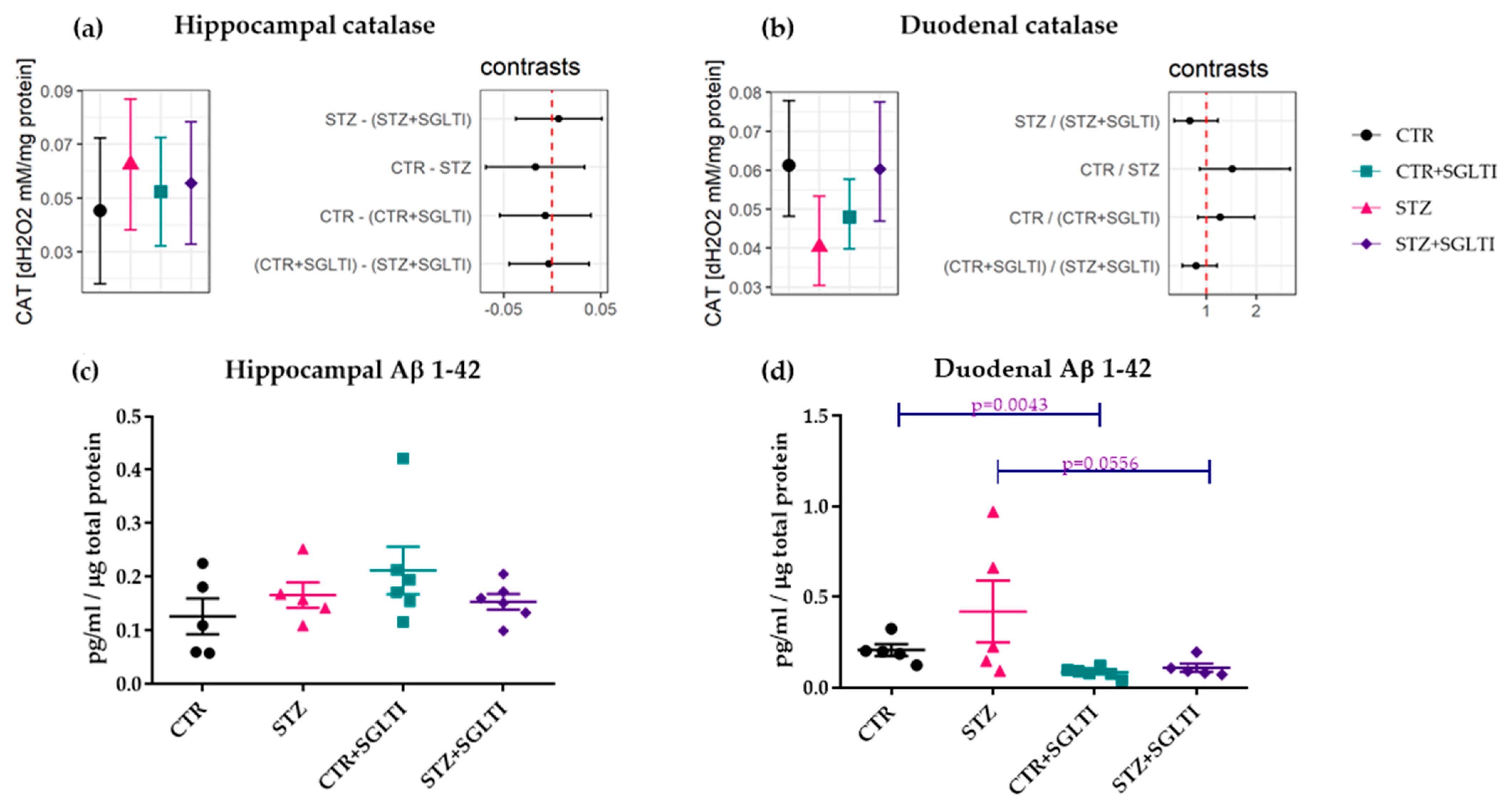
3.4. Biochemical Analyses
4. Discussion
4.1. Chronic Effect of Low Dose of Phloridzin/Phloretin on Cognition
4.2. Chronic Effect of Low Dose of Phloridzin/Phloretin on Glucose and GLP-1 Levels and Body Weight
4.3. Chronic Effect of Low Dose of Phloridzin/Phloretin on Aβ 1-42 and Catalase Interplay in the Hippocampus and Duodenum
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Błaszczyk, J.W. Energy Metabolism Decline in the Aging Brain—Pathogenesis of Neurodegenerative Disorders. Metabolites 2020, 10, 450. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin Resistance and Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults with Prediabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Michailidis, M.; Tata, D.A.; Moraitou, D.; Kavvadas, D.; Karachrysafi, S.; Papamitsou, T.; Vareltzis, P.; Papaliagkas, V. Antidiabetic Drugs in the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 4641. [Google Scholar] [CrossRef] [PubMed]
- Gillies, C.L.; Abrams, K.R.; Lambert, P.C.; Cooper, N.J.; Sutton, A.J.; Hsu, R.T.; Khunti, K. Pharmacological and Lifestyle Interventions to Prevent or Delay Type 2 Diabetes in People with Impaired Glucose Tolerance: Systematic Review and Meta-Analysis. BMJ 2007, 334, 299. [Google Scholar] [CrossRef] [Green Version]
- Bendlin, B.B. Antidiabetic Therapies and Alzheimer Disease. Dialogues Clin. Neurosci. 2019, 21, 83–91. [Google Scholar] [CrossRef]
- Cefalo, C.M.A.; Cinti, F.; Moffa, S.; Impronta, F.; Sorice, G.P.; Mezza, T.; Pontecorvi, A.; Giaccari, A. Sotagliflozin, the First Dual SGLT Inhibitor: Current Outlook and Perspectives. Cardiovasc. Diabetol. 2019, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, M.R.; Di Meo, I.; Polito, R.; Auriemma, M.C.; Gambardella, A.; di Mauro, G.; Capuano, A.; Paolisso, G. Cognitive Impairment and Type 2 Diabetes Mellitus: Focus of SGLT2 Inhibitors Treatment. Pharmacol. Res. 2022, 176, 106062. [Google Scholar] [CrossRef] [PubMed]
- Pawlos, A.; Broncel, M.; Woźniak, E.; Gorzelak-Pabiś, P. Neuroprotective Effect of SGLT2 Inhibitors. Molecules 2021, 26, 7213. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Shinozaki, Y.; Ohta, T. Sodium–glucose Cotransporters: Functional Properties and Pharmaceutical Potential. J. Diabetes Investig. 2020, 11, 770. [Google Scholar] [CrossRef] [Green Version]
- Rani, R.; Kumar, A.; Jaggi, A.S.; Singh, N. Pharmacological Investigations on Efficacy of Phlorizin a Sodium-Glucose Co-Transporter (SGLT) Inhibitor in Mouse Model of Intracerebroventricular Streptozotocin Induced Dementia of AD Type. J. Basic Clin. Physiol. Pharmacol. 2021, 32, 1057–1064. [Google Scholar] [CrossRef]
- Tian, Y.; Lu, W.; Deng, H.; Yang, F.; Guo, Y.; Gao, L.; Xu, Y. Phlorizin Administration Ameliorates Cognitive Deficits by Reducing Oxidative Stress, Tau Hyper-Phosphorylation, and Neuroinflammation in a Rat Model of Alzheimer’s Disease. J. Food Biochem. 2018, 42, e12644. [Google Scholar] [CrossRef]
- Lannert, H.; Hoyer, S. Intracerebroventricular Administration of Streptozotocin Causes Long-Term Diminutions in Learning and Memory Abilities and in Cerebral Energy Metabolism in Adult Rats. Behav. Neurosci. 1998, 112, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Osmanovic Barilar, J.; Knezovic, A.; Grünblatt, E.; Riederer, P.; Salkovic-Petrisic, M. Nine-Month Follow-up of the Insulin Receptor Signalling Cascade in the Brain of Streptozotocin Rat Model of Sporadic Alzheimer’s Disease. J. Neural Transm. 2015, 122, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Gupta, Y.K. Intracerebroventricular Injection of Streptozotocin in Rats Produces Both Oxidative Stress in the Brain and Cognitive Impairment. Life Sci. 2001, 68, 1021–1029. [Google Scholar] [CrossRef]
- Shonesy, B.C.; Thiruchelvam, K.; Parameshwaran, K.; Rahman, E.A.; Karuppagounder, S.S.; Huggins, K.W.; Pinkert, C.A.; Amin, R.; Dhanasekaran, M.; Suppiramaniam, V. Central Insulin Resistance and Synaptic Dysfunction in Intracerebroventricular-Streptozotocin Injected Rodents. Neurobiol. Aging 2012, 33, 430.e5–430.e18. [Google Scholar] [CrossRef] [PubMed]
- Prickaerts, J.; Fahrig, T.; Blokland, A. Cognitive Performance and Biochemical Markers in Septum, Hippocampus and Striatum of Rats after an i.c.v, Injection of Streptozotocin: A Correlation Analysis. Behav. Brain Res. 1999, 102, 73–88. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Knezovic, A.; Hoyer, S.; Riederer, P. What Have We Learned from the Streptozotocin-Induced Animal Model of Sporadic Alzheimer’s Disease, about the Therapeutic Strategies in Alzheimer’s Research. J. Neural Transm. 2013, 120, 233–252. [Google Scholar] [CrossRef] [Green Version]
- Knezovic, A.; Osmanovic-Barilar, J.; Curlin, M.; Hof, P.R.; Simic, G.; Riederer, P.; Salkovic-Petrisic, M. Staging of Cognitive Deficits and Neuropathological and Ultrastructural Changes in Streptozotocin-Induced Rat Model of Alzheimer’s Disease. J. Neural Transm. 2015, 122, 577–592. [Google Scholar] [CrossRef] [Green Version]
- Salkovic-Petrisic, M.; Osmanovic-Barilar, J.; Brückner, M.K.; Hoyer, S.; Arendt, T.; Riederer, P. Cerebral Amyloid Angiopathy in Streptozotocin Rat Model of Sporadic Alzheimer’s Disease: A Long-Term Follow up Study. J. Neural Transm. 2011, 118, 765–772. [Google Scholar] [CrossRef]
- Babic Perhoc, A.; Osmanovic Barilar, J.; Knezovic, A.; Farkas, V.; Bagaric, R.; Svarc, A.; Grünblatt, E.; Riederer, P.; Salkovic-Petrisic, M. Cognitive, Behavioral and Metabolic Effects of Oral Galactose Treatment in the Transgenic Tg2576 Mice. Neuropharmacology 2019, 148, 50–67. [Google Scholar] [CrossRef]
- Najafian, M.; Jahromi, M.Z.; Nowroznejhad, M.J.; Khajeaian, P.; Kargar, M.M.; Sadeghi, M.; Arasteh, A. Phloridzin Reduces Blood Glucose Levels and Improves Lipids Metabolism in Streptozotocin-Induced Diabetic Rats. Mol. Biol. Rep. 2012, 39, 5299–5306. [Google Scholar] [CrossRef] [PubMed]
- Knezovic, A.; Osmanovic Barilar, J.; Babic, A.; Bagaric, R.; Farkas, V.; Riederer, P.; Salkovic-Petrisic, M. Glucagon-like Peptide-1 Mediates Effects of Oral Galactose in Streptozotocin-Induced Rat Model of Sporadic Alzheimer’s Disease. Neuropharmacology 2018, 135, 48–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorhees, C.V.; Williams, M.T. Morris Water Maze: Procedures for Assessing Spatial and Related Forms of Learning and Memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ögren, S.O.; Stiedl, O. Passive Avoidance. In Encyclopedia of Psychopharmacology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–10. [Google Scholar]
- Walters, G.C.; Abel, E.L. Passive Avoidance Learning in Rats, Mice, Gerbils, and Hamsters. Psychon. Sci. 1971, 22, 269–270. [Google Scholar] [CrossRef] [Green Version]
- Ayala, J.E.; Samuel, V.T.; Morton, G.J.; Obici, S.; Croniger, C.M.; Shulman, G.I.; Wasserman, D.H.; McGuinness, O.P. NIH Mouse Metabolic Phenotyping Center Consortium Standard Operating Procedures for Describing and Performing Metabolic Tests of Glucose Homeostasis in Mice. Dis. Model. Mech. 2010, 3, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Dinger, K.; Mohr, J.; Vohlen, C.; Hirani, D.; Hucklenbruch-Rother, E.; Ensenauer, R.; Dötsch, J.; Alejandre Alcazar, M.A. Intraperitoneal Glucose Tolerance Test, Measurement of Lung Function, and Fixation of the Lung to Study the Impact of Obesity and Impaired Metabolism on Pulmonary Outcomes. J. Vis. Exp. 2018, 133, 56685. [Google Scholar] [CrossRef]
- Hadwan, M.H. Simple Spectrophotometric Assay for Measuring Catalase Activity in Biological Tissues. BMC Biochem. 2018, 19, 7. [Google Scholar] [CrossRef] [Green Version]
- Homolak, J.; Perhoc, A.B.; Knezovic, A.; Barilar, J.O.; Virag, D.; Joja, M.; Salkovic-Petrisic, M. The Effect of Acute Oral Galactose Administration on the Redox System of the Rat Small Intestine. Antioxidants 2022, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Homolak, J. The Effect of a Color Tattoo on the Local Skin Redox Regulatory Network: An N-of-1 Study. Free Radic. Res. 2021, 55, 221–229. [Google Scholar] [CrossRef]
- Homolak, J. In Vitro Analysis of Catalase and Superoxide Dismutase Mimetic Properties of Blue Tattoo Ink. Free Radic. Res. 2022, 56, 343–357. [Google Scholar] [CrossRef]
- Hall, J.L.; Reilly, R.T.; Cottrill, K.L.; Stone, W.S.; Gold, P.E. Phlorizin Enhancement of Memory in Rats and Mice. Pharmacol. Biochem. Behav. 1992, 41, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Ghumatkar, P.J.; Patil, S.P.; Jain, P.D.; Tambe, R.M.; Sathaye, S. Nootropic, Neuroprotective and Neurotrophic Effects of Phloretin in Scopolamine Induced Amnesia in Mice. Pharmacol. Biochem. Behav. 2015, 135, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Kraml, J.; Kolínská, J.; Ellederová, D.; Hiršová, D. β-Glucosidase (Phlorizin Hydrolase) Activity of the Lactase Fraction Isolated from the Small Intestinal Mucosa of Infant Rats, and the Relationship between β-Glucosidases and β-Galactosidases. Biochim. Biophys. Acta 1972, 258, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Zhang, X.; Wang, Z.; Gao, Z.; Liu, G.; Hu, H.; Zou, L.; Li, X. Insulin Sensitivity-Enhancing Activity of Phlorizin Is Associated with Lipopolysaccharide Decrease and Gut Microbiota Changes in Obese and Type 2 Diabetes (Db/Db) Mice. J. Agric. Food Chem. 2016, 64, 7502–7511. [Google Scholar] [CrossRef]
- Lorenz-Meyer, H.; Blum, A.L.; Haemmerli, H.P.; Semenza, G. A Second Enzyme Defect in Acquired Lactase Deficiency: Lack of Small-Intestinal Phlorizin-Hydrolase. Eur. J. Clin. Investig. 1972, 2, 326–331. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, Z.; Wang, A.; Jia, L.; Zhang, X.; Fang, M.; Yi, K.; Li, Q.; Hu, H. Comparative Oral and Intravenous Pharmacokinetics of Phlorizin in Rats Having Type 2 Diabetes and in Normal Rats Based on Phase II Metabolism. Food Funct. 2019, 10, 1582–1594. [Google Scholar] [CrossRef]
- Crespy, V.; Aprikian, O.; Morand, C.; Besson, C.; Manach, C.; Demigné, C.; Rémésy, C. Bioavailability of Phloretin and Phloridzin in Rats. J. Nutr. 2001, 131, 3227–3230. [Google Scholar] [CrossRef] [Green Version]
- Macey, R.L.; Farmer, R.E.L. Inhibition of Water and Solute Permeability in Human Red Cells. Biochim. Biophys. Acta 1970, 211, 104–106. [Google Scholar] [CrossRef]
- Miksicek, R.J. Estrogenic Flavonoids: Structural Requirements for Biological Activity. Proc. Soc. Exp. Biol. Med. 1995, 208, 44–50. [Google Scholar] [CrossRef]
- De Jonge, P.C.; Wieringa, T.; Van Putten, J.P.M.; Michiel, H.; Krans, J.; Van Dam, K. Phloretin—An Uncoupler and an Inhibitor of Mitochondrial Oxidative Phosphorylation. Biochim. Biophys. Acta-Bioenerg. 1983, 722, 219–225. [Google Scholar] [CrossRef]
- Yu, A.S.; Hirayama, B.A.; Timbol, G.; Liu, J.; Basarah, E.; Kepe, V.; Satyamurthy, N.; Huang, S.C.; Wright, E.M.; Barrio, J.R. Functional Expression of SGLTs in Rat Brain. Am. J. Physiol.-Cell Physiol. 2010, 299, 1277–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucci, S.J. Developmental Expression of GLUT1 and GLUT3 Glucose Transporters in Rat Brain. J. Neurochem. 1994, 62, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Kamdi, S.P.; Raval, A.; Nakhate, K.T. Phloridzin Attenuates Lipopolysaccharide-Induced Cognitive Impairment via Antioxidant, Anti-Inflammatory and Neuromodulatory Activities. Cytokine 2021, 139, 155048. [Google Scholar] [CrossRef]
- Kamdi, S.P.; Badwaik, H.R.; Raval, A.; Ajazuddin; Nakhate, K.T. Ameliorative Potential of Phloridzin in Type 2 Diabetes-Induced Memory Deficits in Rats. Eur. J. Pharmacol. 2021, 913, 174645. [Google Scholar] [CrossRef] [PubMed]
- Ongay, K.K.; Granato, D.; Barreto, G.E. Comparison of Antioxidant Capacity and Network Pharmacology of Phloretin and Phlorizin against Neuroinflammation in Traumatic Brain Injury. Molecules 2023, 28, 919. [Google Scholar] [CrossRef]
- Pereira, M.J.; Eriksson, J.W. Emerging Role of SGLT-2 Inhibitors for the Treatment of Obesity. Drugs 2019, 79, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, G.; Hach, T.; Crowe, S.; Sanghvi, A.; Hall, K.D.; Ferrannini, E. Energy Balance after Sodium-Glucose Cotransporter 2 Inhibition. Diabetes Care 2015, 38, 1730–1735. [Google Scholar] [CrossRef] [Green Version]
- Londzin, P.; Siudak, S.; Cegieła, U.; Pytlik, M.; Janas, A.; Waligóra, A.; Folwarczna, J. Phloridzin, an Apple Polyphenol, Exerted Unfavorable Effects on Bone and Muscle in an Experimental Model of Type 2 Diabetes in Rats. Nutrients 2018, 10, 1701. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to Fatty Substrate Utilization in Response to Sodium-Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients with Type 2 Diabetes. Diabetes 2016, 65, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, R. The Effect of Fasting on Human Metabolism and Psychological Health. Dis. Markers 2022, 2022, 5653739. [Google Scholar] [CrossRef]
- Takii, H.; Matsumoto, K.; Kometani, T.; Okada, S.; Fushiki, T. NII-Electronic Library Service Lowering Effect of Phenolic Glycosides on the Rise in Postprandial Glucose in Mice. Biosci. Biotech. Biochem. 1997, 61, 1531–1535. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like Peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lee, E.; Yoshikawa, H.; Noda, T.; Miyamoto, J.; Kimura, I.; Hatano, R.; Miki, T. Phloretin Suppresses Carbohydrate-Induced GLP-1 Secretion via Inhibiting Short Chain Fatty Acid Release from Gut Microbiome. Biochem. Biophys. Res. Commun. 2022, 621, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Hjørne, A.P.; Modvig, I.M.; Holst, J.J. The Sensory Mechanisms of Nutrient-Induced GLP-1 Secretion. Metabolites 2022, 12, 420. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.W.; De Fontgalland, D.; Rabbitt, P.; Hollington, P.; Sposato, L.; Due, S.L.; Wattchow, D.A.; Rayner, C.K.; Deane, A.M.; Young, R.L.; et al. Mechanisms Controlling Glucose-Induced GLP-1 Secretion in Human Small Intestine. Diabetes 2017, 66, 2144–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutniak, M.; Ørkov, C.; Holst, J.J.; Ahrén, B.; Efendić, S. Antidiabetogenic Effect of Glucagon-like Peptide-1 (7–36)Amide in Normal Subjects and Patients with Diabetes Mellitus. N. Engl. J. Med. 2010, 326, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved Incretin Activity of Glucagon-like Peptide 1 [7-36 Amide] but Not of Synthetic Human Gastric Inhibitory Polypeptide in Patients with Type-2 Diabetes Mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candeias, E.M.; Sebastião, I.C.; Cardoso, S.M.; Correia, S.C.; Carvalho, C.I.; Plácido, A.I.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I.; Duarte, A.I. Gut-Brain Connection: The Neuroprotective Effects of the Anti-Diabetic Drug Liraglutide. World J. Diabetes 2015, 6, 807–827. [Google Scholar] [CrossRef]
- Duarte, A.I.; Candeias, E.; Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Plácido, A.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Crosstalk between Diabetes and Brain: Glucagon-like Peptide-1 Mimetics as a Promising Therapy against Neurodegeneration. Biochim. Biophys. Acta-Mol. Basis Dis. 2013, 1832, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Burcelin, R. The Gut-Brain Axis: A Major Glucoregulatory Player. Diabetes Metab. 2010, 36, S54–S58. [Google Scholar] [CrossRef]
- Baraboi, E.D.; St-Pierre, D.H.; Shooner, J.; Timofeeva, E.; Richard, D. Brain Activation Following Peripheral Administration of the GLP-1 Receptor Agonist Exendin-4. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R1011–R1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastin, A.J.; Akerstrom, V.; Pan, W. Interactions of Glucagon-Like Peptide-1 (GLP-1) with the Blood-Brain Barrier. J. Mol. Neurosci. 2002, 18, 7–14. [Google Scholar] [CrossRef]
- Barilar, J.O.; Knezovic, A.; Homolak, J.; Perhoc, A.B.; Salkovic-petrisic, M. Divergent Effect of Central Incretin Receptors Inhibition in a Rat Model of Sporadic Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 548. [Google Scholar] [CrossRef]
- Homolak, J.; Perhoc, A.B.; Knezovic, A.; Barilar, J.O.; Salkovic-Petrisic, M. Failure of the Brain Glucagon-like Peptide-1-Mediated Control of Intestinal Redox Homeostasis in a Rat Model of Sporadic Alzheimer’s Disease. Antioxidants 2021, 10, 1118. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Xu, Z.; Zhang, L.; Zhang, C.; Zhao, X.; Mao, Y.; Zhang, H.; Liang, X.; Wu, J.; Yang, Y.; et al. Gut-Derived β-Amyloid: Likely a Centerpiece of the Gut-Brain Axis Contributing to Alzheimer’s Pathogenesis. Gut Microbes 2023, 15, 2167172. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sommerville, N.R.; Liu, J.Y.H.; Ngan, M.P.; Poon, D.; Ponomarev, E.D.; Lu, Z.; Kung, J.S.C.; Rudd, J.A. Intra-Gastrointestinal Amyloid-Β1-42 Oligomers Perturb Enteric Function and Induce Alzheimer’s Disease Pathology. J. Physiol. 2020, 598, 4209–4223. [Google Scholar] [CrossRef]
- Puig, K.L.; Manocha, G.D.; Combs, C.K. Amyloid Precursor Protein Mediated Changes in Intestinal Epithelial Phenotype in Vitro. PLoS ONE 2015, 10, e0119534. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.D.; Ou, Y.; Davis, J.E.; Odle, A.K.; Groves, T.R.; Allen, A.R.; Childs, G.V.; Barger, S.W. Alzheimer Amyloid-β-Peptide Disrupts Membrane Localization of Glucose Transporter 1 in Astrocytes: Implications for Glucose Levels in Brain and Blood. Neurobiol. Aging 2021, 97, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Tharp, W.G.; Gupta, D.; Smith, J.; Jones, K.P.; Jones, A.M.; Pratley, R.E. Effects of Glucose and Insulin on Secretion of Amyloid-β by Human Adipose Tissue Cells. Obesity 2016, 24, 1471–1479. [Google Scholar] [CrossRef] [Green Version]
- Salkovic-Petrisic, M.; Tribl, F.; Schmidt, M.; Hoyer, S.; Riederer, P. Alzheimer-like Changes in Protein Kinase B and Glycogen Synthase Kinase-3 in Rat Frontal Cortex and Hippocampus after Damage to the Insulin Signalling Pathway. J. Neurochem. 2006, 96, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Chilumuri, A.; Ashioti, M.; Nercessian, A.N.; Milton, N.G.N. Immunolocalization of Kisspeptin Associated with Amyloid-β Deposits in the Pons of an Alzheimer’s Disease Patient. J. Neurodegener. Dis. 2013, 2013, 879710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, L.K.; Lee, M.T.C.; Yang, J. Inhibitors of Catalase-Amyloid Interactions Protect Cells from Beta-Amyloid-Induced Oxidative Stress and Toxicity. J. Biol. Chem. 2010, 285, 38933–38943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [Green Version]
- Milton, N.G.N. Amyloid-β Binds Catalase with High Affinity and Inhibits Hydrogen Peroxide Breakdown. Biochem. J. 1999, 344, 293–296. [Google Scholar] [CrossRef]
- Homolak, J.; Babic Perhoc, A.; Knezovic, A.; Osmanovic Barilar, J.; Koc, F.; Stanton, C.; Ross, R.P.; Salkovic-Petrisic, M. Disbalance of the Duodenal Epithelial Cell Turnover and Apoptosis Accompanies Insensitivity of Intestinal Redox Homeostasis to Inhibition of the Brain Glucose-Dependent Insulinotropic Polypeptide Receptors in a Rat Model of Sporadic Alzheimer’s Disease. Neuroendocrinology 2021, 112, 744–762. [Google Scholar] [CrossRef] [PubMed]
- Homolak, J.; De Busscher, J.; Lucio, M.; Joja, M.; Virag, D.; Babic Perhoc, A.; Knezovic, A.; Osmanovic Barilar, J.; Salkovic-Petrisic, M. Altered Secretion, Constitution, and Functional Properties of the Gastrointestinal Mucus in a Rat Model of Sporadic Alzheimer’s Disease. bioRxiv 2022. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osmanović Barilar, J.; Babić Perhoč, A.; Knezović, A.; Homolak, J.; Virag, D.; Šalković-Petrišić, M. The Effect of the Sodium—Glucose Cotransporter Inhibitor on Cognition and Metabolic Parameters in a Rat Model of Sporadic Alzheimer’s Disease. Biomedicines 2023, 11, 1025. https://doi.org/10.3390/biomedicines11041025
Osmanović Barilar J, Babić Perhoč A, Knezović A, Homolak J, Virag D, Šalković-Petrišić M. The Effect of the Sodium—Glucose Cotransporter Inhibitor on Cognition and Metabolic Parameters in a Rat Model of Sporadic Alzheimer’s Disease. Biomedicines. 2023; 11(4):1025. https://doi.org/10.3390/biomedicines11041025
Chicago/Turabian StyleOsmanović Barilar, Jelena, Ana Babić Perhoč, Ana Knezović, Jan Homolak, Davor Virag, and Melita Šalković-Petrišić. 2023. "The Effect of the Sodium—Glucose Cotransporter Inhibitor on Cognition and Metabolic Parameters in a Rat Model of Sporadic Alzheimer’s Disease" Biomedicines 11, no. 4: 1025. https://doi.org/10.3390/biomedicines11041025