
Endometriosis: Update of Pathophysiology, (Epi) Genetic and Environmental Involvement

Abstract
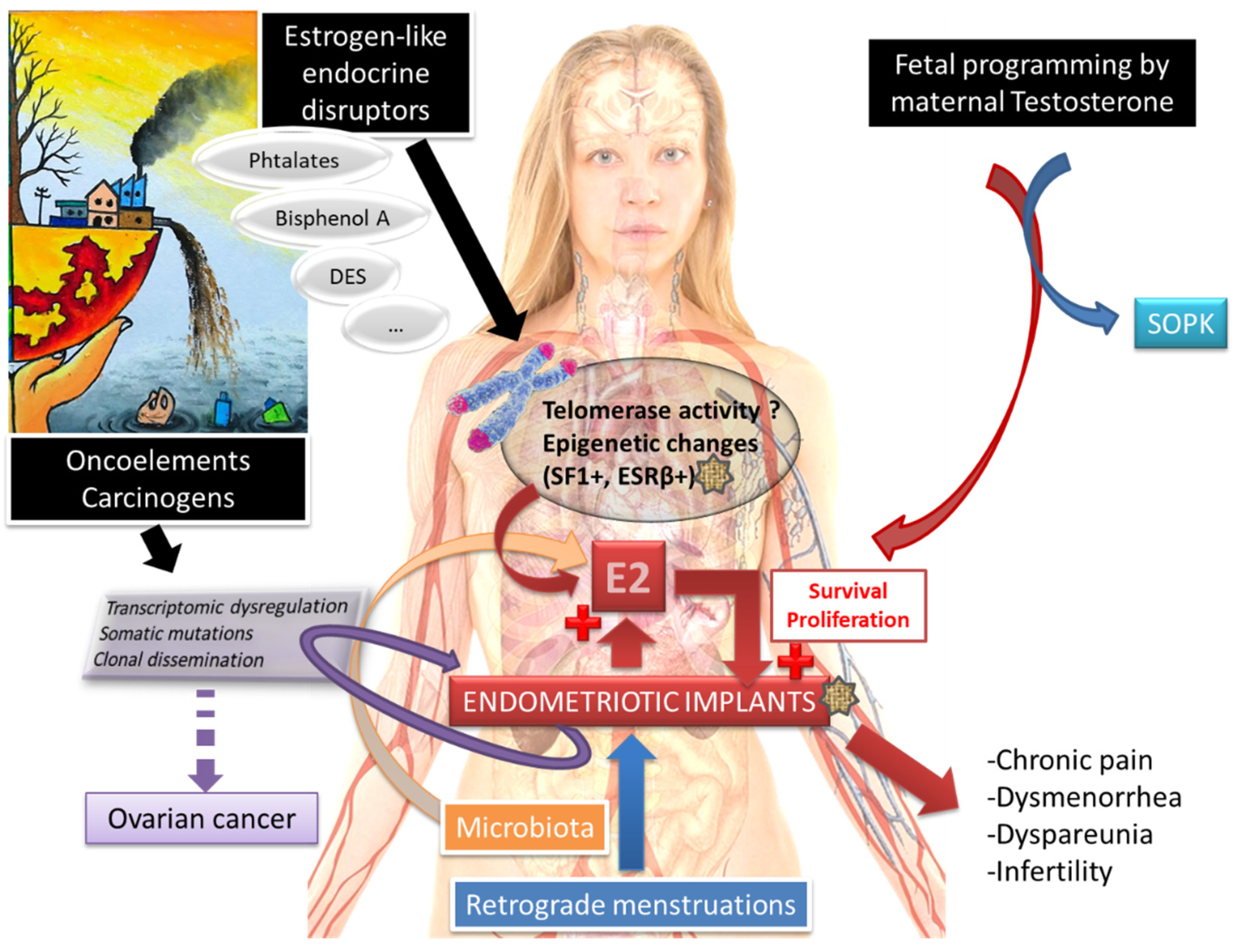
:1. Introduction
2. Theories on Endometriosis
2.1. An Anatomical Predisposition to Endometriosis?
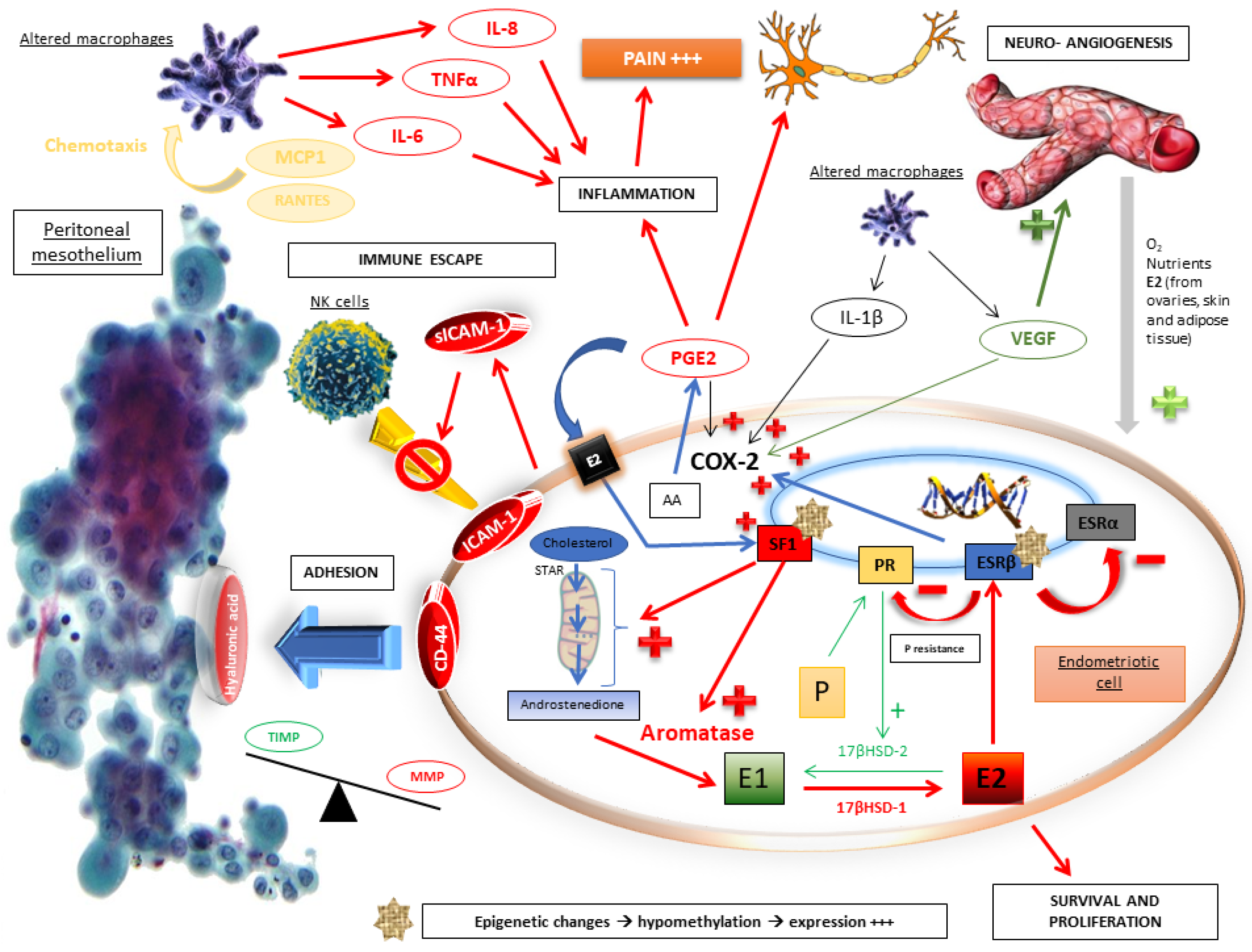
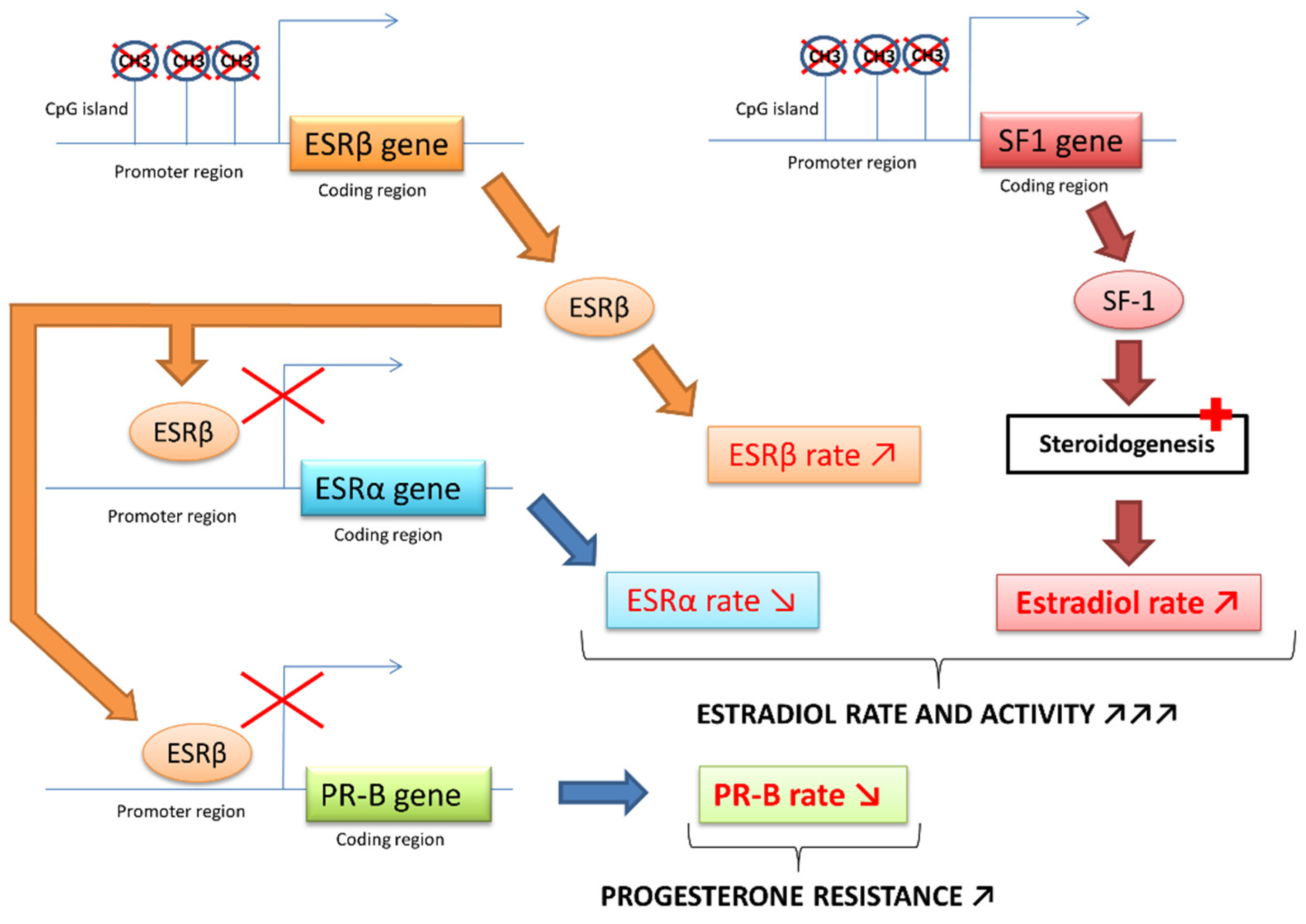
2.2. Molecular Pathophysiological Mechanisms
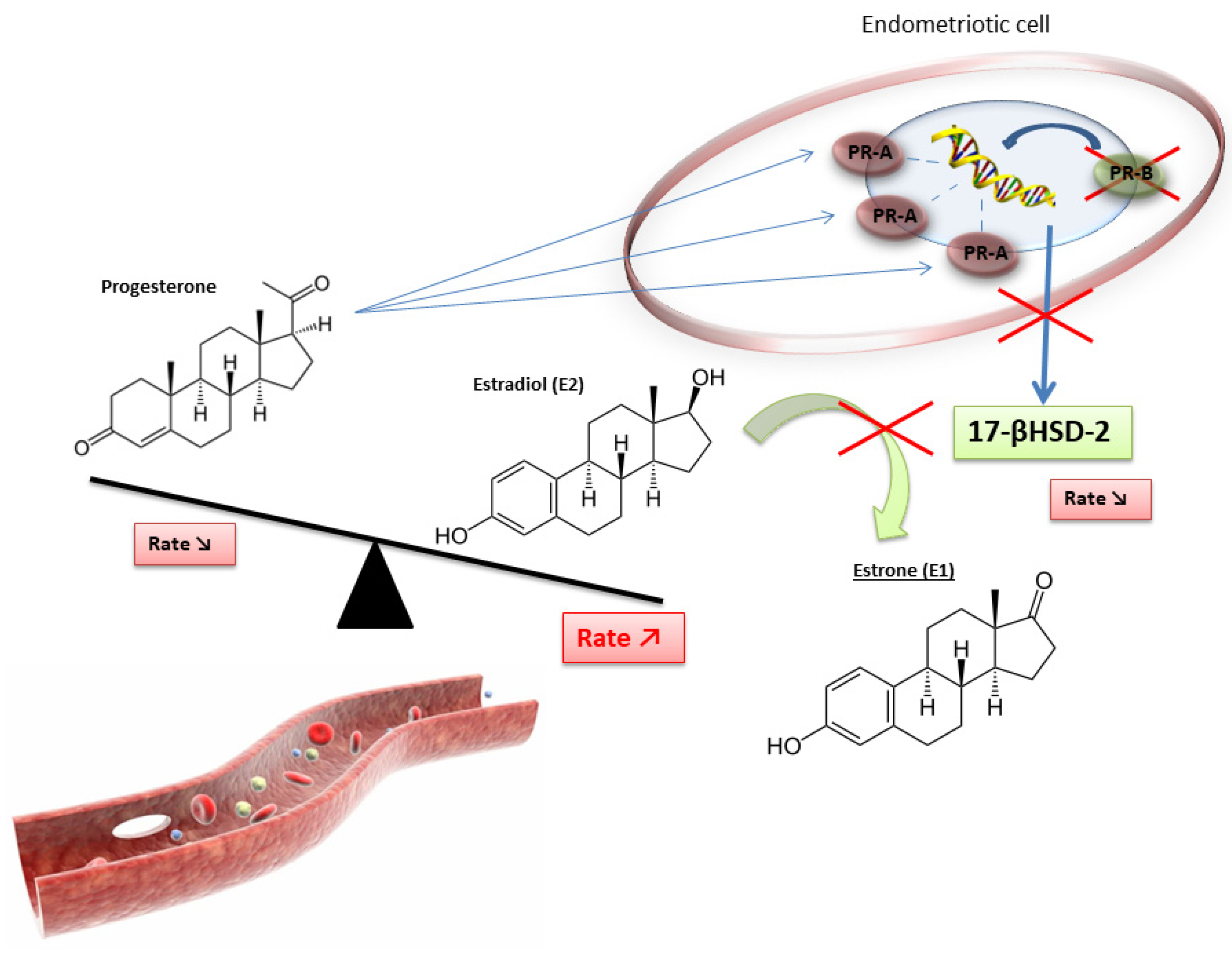
2.3. The Crucial Role of an Altered Hormonal Environment

2.4. Telomeres, Telomerase and Endometriosis
2.5. Immune Escape
2.6. Adhesion, Implanting and Invasion
2.7. Growth and Lesional Neuroangiogenesis
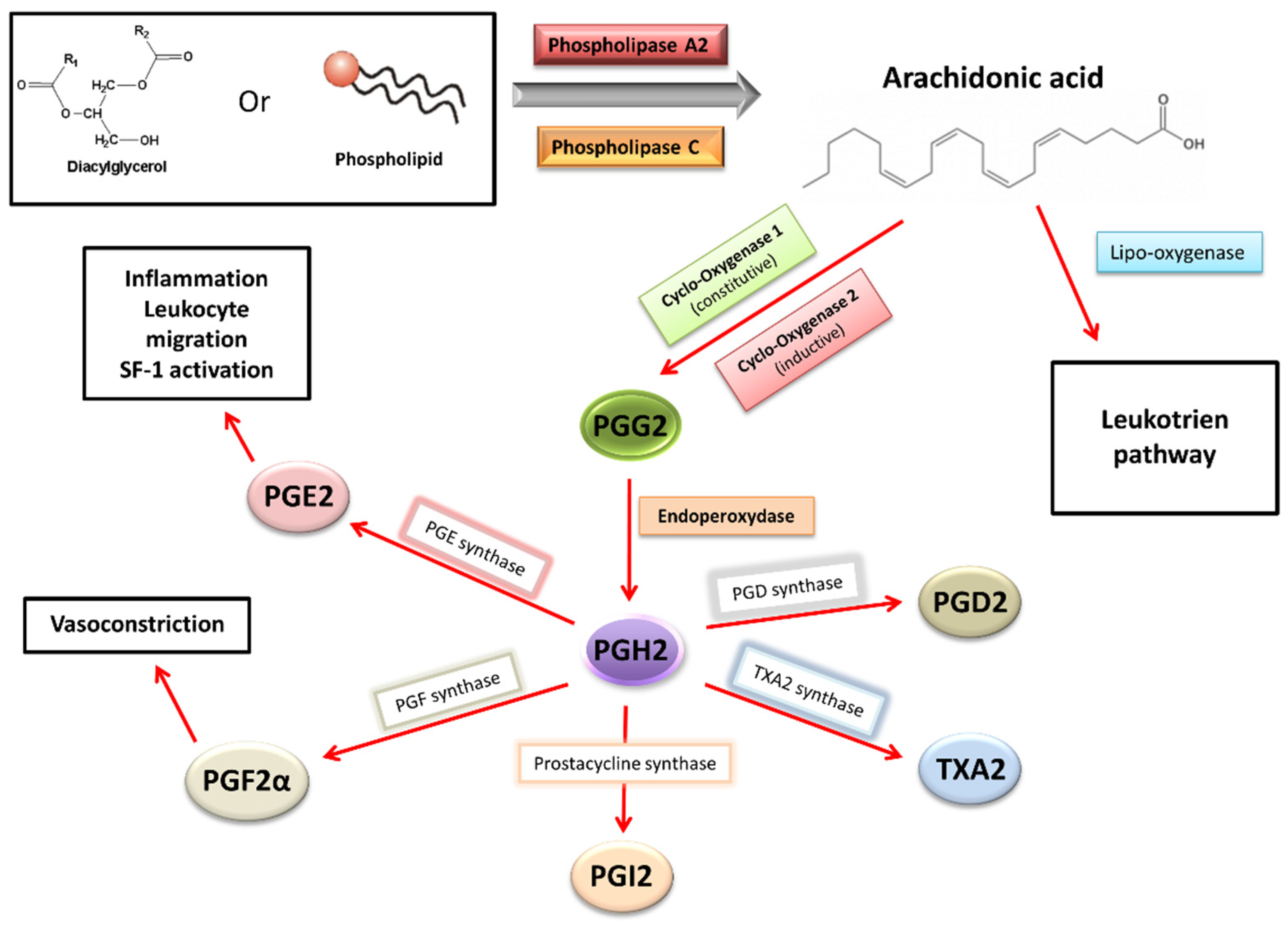
2.8. Inflammation
2.9. Role of miRNAs in Endometriosis
3. Endometriosis and Cancer
4. Endometriosis and PolyCystic Ovaries Syndrome (PCOS)
5. Infertility and Endometriosis
6. Environmental Impact
6.1. EE and DES
6.2. Dioxins
6.3. Bisphenol A
6.4. Phthalates
6.5. Tobacco
7. Role of the Microbiota in Endometriosis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soave, I.; Caserta, D.; Wenger, J.M.; Dessole, S.; Perino, A.; Marci, R. Environment and Endometriosis: A toxic relationship. Eur. Rev. Med. Pharm. Sci. 2015, 19, 1964–1972. [Google Scholar]
- Vinatier, D.; Orazi, G.; Cosson, M.; Dufour, P. Theories of endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2001, 96, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Huntington, A.; Gilmour, J.A. A life shaped by pain: Women and endometriosis. J. Clin. Nurs. 2005, 14, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E. Endometriosis. N. Engl. J. Med. 2009, 360, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Burney, R.O.; Giudice, L.C. Pathogenesis and pathophysiology of endometriosis. Fertil. Steril. 2012, 98, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- rpcrhésus.qxd—RPC_endometriose.pdf. Available online: http://www.cngof.asso.fr/D_TELE/RPC_endometriose.pdf (accessed on 14 June 2016).
- Viganò, P.; Parazzini, F.; Somigliana, E.; Vercellini, P. Endometriosis: Epidemiology and aetiological factors. Best Pract. Res. Clin. Obstet. Gynaecol. 2004, 18, 177–200. [Google Scholar] [CrossRef]
- Lindsay, S.F.; Luciano, D.E.; Luciano, A.A. Emerging therapy for endometriosis. Expert Opin. Emerg. Drugs 2015, 20, 449–461. [Google Scholar] [CrossRef]
- Practice Committee of the American Society for Reproductive Medicine. Endometriosis and infertility: A committee opinion. Fertil. Steril. 2012, 98, 591–598. [Google Scholar] [CrossRef]
- Missmer, S.A.; Hankinson, S.E.; Spiegelman, D.; Barbieri, R.L.; Michels, K.B.; Hunter, D.J. In utero exposures and the incidence of endometriosis. Fertil. Steril. 2004, 82, 1501–1508. [Google Scholar] [CrossRef]
- Bouquet de Jolinière, J.; Ayoubi, J.M.; Lesec, G.; Validire, P.; Goguin, A.; Gianaroli, L.; Dubuisson, J.B.; Feki, A.; Gogusev, J. Identification of displaced endometrial glands and embryonic duct remnants in female fetal reproductive tract: Possible pathogenetic role in endometriotic and pelvic neoplastic processes. Front. Physiol. 2012, 3, 444. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.A. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am. J. Pathol. 1927, 3, 93–110.43. [Google Scholar] [PubMed]
- Halme, J.; Hammond, M.G.; Hulka, J.F.; Raj, S.G.; Talbert, L.M. Retrograde menstruation in healthy women and in patients with endometriosis. Obstet. Gynecol. 1984, 64, 151–154. [Google Scholar]
- Greene, A.D.; Lang, S.A.; Kendziorski, J.A.; Sroga-Rios, J.M.; Herzog, T.J.; Burns, K. Endometriosis: Where are We and Where are We Going? Reproduction 2016, 152, R63–R78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafrir, A.L.; Farland, L.V.; Shah, D.K.; Harris, H.R.; Kvaskoff, M.; Zondervan, K.; Missmer, S.A. Risk for and consequences of endometriosis: A critical epidemiologic review. Best Pr. Res. Clin. Obstet. Gynaecol. 2018, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.A.S.; Haro, M.; Wright, K.N.; Lin, X.; Abbasi, F.; Sun, J.; Hernandez, L.; Orr, N.L.; Hong, J.; Choi-Kuaea, Y.; et al. Single-cell transcriptomic analysis of endometriosis. Nat. Genet. 2023, 55, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Wan, Y.; Matei, D. Epithelial Mutations in Endometriosis: Link to Ovarian Cancer. Endocrinology 2019, 160, 626–638. [Google Scholar] [CrossRef] [Green Version]
- Fusco, N.; Malapelle, U.; Fassan, M.; Marchiò, C.; Buglioni, S.; Zupo, S.; Criscitiello, C.; Vigneri, P.; Dei Tos, A.P.; Maiorano, E.; et al. PIK3CA Mutations as a Molecular Target for Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Front. Oncol. 2021, 11, 644737. [Google Scholar] [CrossRef]
- Praetorius, T.H.; Leonova, A.; Lac, V.; Senz, J.; Tessier-Cloutier, B.; Nazeran, T.M.; Köbel, M.; Grube, M.; Kraemer, B.; Yong, P.J.; et al. Molecular analysis suggests oligoclonality and metastasis of endometriosis lesions across anatomically defined subtypes. Fertil. Steril. 2022, 118, 524–534. [Google Scholar] [CrossRef]
- Adashek, J.J.; Kato, S.; Lippman, S.M.; Kurzrock, R. The paradox of cancer genes in non-malignant conditions: Implications for precision medicine. Genome Med. 2020, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Hapangama, D.K.; Kamal, A.M.; Bulmer, J.N. Estrogen receptor β: The guardian of the endometrium. Hum. Reprod. Update 2015, 21, 174–193. [Google Scholar] [CrossRef] [Green Version]
- Bulun, S.E.; Monsavais, D.; Pavone, M.E.; Dyson, M.; Xue, Q.; Attar, E.; Tokunaga, H.; Su, E.J. Role of estrogen receptor-β in endometriosis. Semin. Reprod. Med. 2012, 30, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulun, S.E.; Utsunomiya, H.; Lin, Z.; Yin, P.; Cheng, Y.-H.; Pavone, M.E.; Tokunaga, H.; Trukhacheva, E.; Attar, E.; Gurates, B.; et al. Steroidogenic factor-1 and endometriosis. Mol. Cell. Endocrinol. 2009, 300, 104–108. [Google Scholar] [CrossRef]
- Bulun, S.E.; Monsivais, D.; Kakinuma, T.; Furukawa, Y.; Bernardi, L.; Pavone, M.E.; Dyson, M. Molecular biology of endometriosis: From aromatase to genomic abnormalities. Semin. Reprod. Med. 2015, 33, 220–224. [Google Scholar] [CrossRef]
- Xue, Q.; Lin, Z.; Yin, P.; Milad, M.P.; Cheng, Y.-H.; Confino, E.; Reierstad, S.; Bulun, S.E. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5′ CpG island in endometriosis. J. Clin. Endocrinol. Metab. 2007, 92, 3261–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, P.; Sharma, I.; Saha, S.C.; Srinivasan, R.; Sharma, A. Promoter methylation status of key genes and its implications in the pathogenesis of endometriosis, endometrioid carcinoma of ovary and endometrioid endometrial cancer. J. Cancer Res. Ther. 2022, 18, S328–S334. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, A.; Pereira, R.M.A.; da Rocha, A.M.; Cogliati, B.; Baracat, E.C.; Taylor, H.S.; da Motta, E.L.A.; Serafini, P.C. The Relationship Among HOXA10, Estrogen Receptor α, Progesterone Receptor, and Progesterone Receptor B Proteins in Rectosigmoid Endometriosis: A Tissue Microarray Study. Reprod. Sci. 2014, 22, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thilagavathi, J.; Venkatesh, S.; Dada, R. Telomere length in reproduction. Andrologia 2013, 45, 289–304. [Google Scholar] [CrossRef]
- de Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W. Telomeres and aging. Curr. Opin. Cell Biol. 2018, 52, 1–7. [Google Scholar] [CrossRef]
- Aviv, A. Telomeres and human aging: Facts and fibs. Sci. Aging Knowl. Environ. 2004, 2004, pe43. [Google Scholar] [CrossRef]
- von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, A.; Pantell, M.S.; Puterman, E.; Dhabhar, F.S.; Blackburn, E.H.; Yaffe, K.; Cawthon, R.M.; Opresko, P.L.; Hsueh, W.-C.; Satterfield, S.; et al. Cumulative inflammatory load is associated with short leukocyte telomere length in the Health, Aging and Body Composition Study. PLoS ONE 2011, 6, e19687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dracxler, R.C.; Oh, C.; Kalmbach, K.; Wang, F.; Liu, L.; Kallas, E.G.; Giret, M.T.M.; Seth-Smith, M.L.; Antunes, D.; Keefe, D.L.; et al. Peripheral blood telomere content is greater in patients with endometriosis than in controls. Reprod. Sci. 2014, 21, 1465–1471. [Google Scholar] [CrossRef]
- Sasamoto, N.; Yland, J.; Vitonis, A.F.; Cramer, D.W.; Titus, L.J.; De Vivo, I.; Missmer, S.A.; Terry, K.L. Peripheral Blood Leukocyte Telomere Length and Endometriosis. Reprod. Sci. 2020, 27, 1951–1959. [Google Scholar] [CrossRef]
- Hapangama, D.K.; Turner, M.A.; Drury, J.A.; Quenby, S.; Saretzki, G.; Martin-Ruiz, C.; Von Zglinicki, T. Endometriosis is associated with aberrant endometrial expression of telomerase and increased telomere length. Hum. Reprod. 2008, 23, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Valentijn, A.J.; Saretzki, G.; Tempest, N.; Critchley, H.O.D.; Hapangama, D.K. Human endometrial epithelial telomerase is important for epithelial proliferation and glandular formation with potential implications in endometriosis. Hum. Reprod. 2015, 30, 2816–2828. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.M.; Oh, Y.J.; Cho, S.H.; Chung, D.J.; Hwang, J.Y.; Park, K.H.; Cho, D.J.; Choi, Y.M.; Lee, B.S. Increased telomerase activity and human telomerase reverse transcriptase mRNA expression in the endometrium of patients with endometriosis. Hum. Reprod. 2007, 22, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Mafra, F.A.; Christofolini, D.M.; Cavalcanti, V.; Vilarino, F.L.; André, G.M.; Kato, P.; Bianco, B.; Barbosa, C.P. Aberrant telomerase expression in the endometrium of infertile women with deep endometriosis. Arch. Med. Res. 2014, 45, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Hapangama, D.K.; Turner, M.A.; Drury, J.; Heathcote, L.; Afshar, Y.; Mavrogianis, P.A.; Fazleabas, A.T. Aberrant expression of regulators of cell-fate found in eutopic endometrium is found in matched ectopic endometrium among women and in a baboon model of endometriosis. Hum. Reprod. 2010, 25, 2840–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgård, C.; Benetos, A.; Verhulst, S.; Labat, C.; Kark, J.D.; Christensen, K.; Kimura, M.; Kyvik, K.O.; Aviv, A. Leukocyte telomere length dynamics in women and men: Menopause vs age effects. Int. J. Epidemiol. 2015, 44, 1688–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toupance, S.; Fattet, A.-J.; Thornton, S.N.; Benetos, A.; Guéant, J.-L.; Koscinski, I. Ovarian Telomerase and Female Fertility. Biomedicines 2021, 9, 842. [Google Scholar] [CrossRef] [PubMed]
- Berkley, K.J.; Rapkin, A.J.; Papka, R.E. The pains of endometriosis. Science 2005, 308, 1587–1589. [Google Scholar] [CrossRef]
- Olkowska-Truchanowicz, J.; Białoszewska, A.; Zwierzchowska, A.; Sztokfisz-Ignasiak, A.; Janiuk, I.; Dąbrowski, F.; Korczak-Kowalska, G.; Barcz, E.; Bocian, K.; Malejczyk, J. Peritoneal Fluid from Patients with Ovarian Endometriosis Displays Immunosuppressive Potential and Stimulates Th2 Response. Int. J. Mol. Sci. 2021, 22, 8134. [Google Scholar] [CrossRef]
- Xiao, F.; Liu, X.; Guo, S.-W. Interleukin-33 Derived from Endometriotic Lesions Promotes Fibrogenesis through Inducing the Production of Profibrotic Cytokines by Regulatory T Cells. Biomedicines 2022, 10, 2893. [Google Scholar] [CrossRef]
- Shi, J.-L.; Zheng, Z.-M.; Chen, M.; Shen, H.-H.; Li, M.-Q.; Shao, J. IL-17: An important pathogenic factor in endometriosis. Int. J. Med. Sci. 2022, 19, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Raja, M.H.R.; Farooqui, N.; Zuberi, N.; Ashraf, M.; Azhar, A.; Baig, R.; Badar, B.; Rehman, R. Endometriosis, infertility and MicroRNA’s: A review. J. Gynecol. Obstet. Hum. Reprod. 2021, 50, 102157. [Google Scholar] [CrossRef]
- Ghasemi, F.; Alemzadeh, E.; Allahqoli, L.; Alemzadeh, E.; Mazidimoradi, A.; Salehiniya, H.; Alkatout, I. MicroRNAs Dysregulation as Potential Biomarkers for Early Diagnosis of Endometriosis. Biomedicines 2022, 10, 2558. [Google Scholar] [CrossRef]
- Vanhie, A.; O, D.; Peterse, D.; Beckers, A.; Cuéllar, A.; Fassbender, A.; Meuleman, C.; Mestdagh, P.; D’Hooghe, T. Plasma miRNAs as biomarkers for endometriosis. Hum. Reprod. 2019, 34, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Kvaskoff, M.; Mahamat-Saleh, Y.; Farland, L.V.; Shigesi, N.; Terry, K.L.; Harris, H.R.; Roman, H.; Becker, C.M.; As-Sanie, S.; Zondervan, K.T.; et al. Endometriosis and cancer: A systematic review and meta-analysis. Hum. Reprod. Update 2021, 27, 393–420. [Google Scholar] [CrossRef]
- Ye, J.; Peng, H.; Huang, X.; Qi, X. The association between endometriosis and risk of endometrial cancer and breast cancer: A meta-analysis. BMC Women’s Health 2022, 22, 455. [Google Scholar] [CrossRef]
- Hazelwood, E.; Sanderson, E.; Tan, V.Y.; Ruth, K.S.; Frayling, T.M.; Dimou, N.; Gunter, M.J.; Dossus, L.; Newton, C.; Ryan, N.; et al. Identifying molecular mediators of the relationship between body mass index and endometrial cancer risk: A Mendelian randomization analysis. BMC Med. 2022, 20, 125. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B. Variation among human populations in endometriosis and PCOS A test of the inverse comorbidity model. Evol. Med. Public Health 2021, 9, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Throwba, H.; Unnikrishnan, L.; Pangath, M.; Vasudevan, K.; Jayaraman, S.; Li, M.; Iyaswamy, A.; Palaniyandi, K.; Gnanasampanthapandian, D. The epigenetic correlation among ovarian cancer, endometriosis and PCOS: A review. Crit. Rev. Oncol. Hematol. 2022, 180, 103852. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Lobo, R.A. Cancer risk and PCOS. Steroids 2013, 78, 782–785. [Google Scholar] [CrossRef]
- De Ziegler, D.; Borghese, B.; Chapron, C. Endometriosis and infertility: Pathophysiology and management. Lancet 2010, 376, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Osborn, B.H.; Haney, A.F.; Misukonis, M.A.; Weinberg, J.B. Inducible nitric oxide synthase expression by peritoneal macrophages in endometriosis-associated infertility. Fertil. Steril. 2002, 77, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Lebovic, D.I.; Mueller, M.D.; Taylor, R.N. Immunobiology of endometriosis. Fertil. Steril. 2001, 75, 1–10. [Google Scholar] [CrossRef]
- Gruber, T.M.; Mechsner, S. Pathogenesis of Endometriosis: The Origin of Pain and Subfertility. Cells 2021, 10, 1381. [Google Scholar] [CrossRef]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef] [Green Version]
- Sirohi, D.; Al Ramadhani, R.; Knibbs, L.D. Environmental exposures to endocrine disrupting chemicals (EDCs) and their role in endometriosis: A systematic literature review. Rev. Environ. Health 2021, 36, 101–115. [Google Scholar] [CrossRef]
- McLachlan, J.A.; Simpson, E.; Martin, M. Endocrine disrupters and female reproductive health. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Koike, E.; Yasuda, Y.; Shiota, M.; Shimaoka, M.; Tsuritani, M.; Konishi, H.; Yamasaki, H.; Okumoto, K.; Hoshiai, H. Exposure to ethinyl estradiol prenatally and/or after sexual maturity induces endometriotic and precancerous lesions in uteri and ovaries of mice. Congenit. Anom. 2013, 53, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Haney, A.F.; Hammond, M.G. Infertility in women exposed to diethylstilbestrol in utero. J. Reprod. Med. 1983, 28, 851–856. [Google Scholar] [PubMed]
- Benagiano, G.; Brosens, I. In utero exposure and endometriosis. J. Matern. Fetal Neonatal. Med. 2014, 27, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Wolff, E.F.; Sun, L.; Hediger, M.L.; Sundaram, R.; Peterson, C.M.; Chen, Z.; Buck Louis, G.M. In utero exposures and endometriosis: The Endometriosis, Natural History, Disease, Outcome (ENDO) Study. Fertil. Steril. 2013, 99, 790–795. [Google Scholar] [CrossRef] [Green Version]
- Stillman, R.J.; Miller, L.C. Diethylstilbestrol exposure in utero and endometriosis in infertile females. Fertil. Steril. 1984, 41, 369–372. [Google Scholar] [CrossRef]
- Ostrander, P.L.; Mills, K.T.; Bern, H.A. Long-term responses of the mouse uterus to neonatal diethylstilbestrol treatment and to later sex hormone exposure. J. Natl. Cancer Inst. 1985, 74, 121–135. [Google Scholar]
- Berger, M.J.; Alper, M.M. Intractable primary infertility in women exposed to diethylstilbestrol in utero. J. Reprod. Med. 1986, 31, 231–235. [Google Scholar]
- Newbold, R. Cellular and molecular effects of developmental exposure to diethylstilbestrol: Implications for other environmental estrogens. Environ. Health Perspect. 1995, 103, 83–87. [Google Scholar]
- Golden, R.J.; Noller, K.L.; Titus-Ernstoff, L.; Kaufman, R.H.; Mittendorf, R.; Stillman, R.; Reese, E.A. Environmental endocrine modulators and human health: An assessment of the biological evidence. Crit. Rev. Toxicol. 1998, 28, 109–227. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, L.; Guo, R.; Sheng, W. Role of epidermal growth factor signaling system in the pathogenesis of endometriosis under estrogen deprivation conditions. Zhonghua Fu Chan Ke Za Zhi 2013, 48, 447–452. [Google Scholar] [PubMed]
- Polak, G.; Banaszewska, B.; Filip, M.; Radwan, M.; Wdowiak, A. Environmental Factors and Endometriosis. Int. J. Environ. Res. Public Health 2021, 18, 11025. [Google Scholar] [CrossRef] [PubMed]
- Verga, J.U.; Huff, M.; Owens, D.; Wolf, B.J.; Hardiman, G. Integrated Genomic and Bioinformatics Approaches to Identify Molecular Links between Endocrine Disruptors and Adverse Outcomes. Int. J. Environ. Res. Public Health 2022, 19, 574. [Google Scholar] [CrossRef]
- Wei, M.; Chen, X.; Zhao, Y.; Cao, B.; Zhao, W. Effects of Prenatal Environmental Exposures on the Development of Endometriosis in Female Offspring. Reprod. Sci. 2016, 23, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- White, S.S.; Birnbaum, L.S. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Rideout, K.; Teschke, K. Potential for increased human foodborne exposure to PCDD/F when recycling sewage sludge on agricultural land. Environ. Health Perspect. 2004, 112, 959–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, H.; Arisawa, K.; Hiyoshi, M.; Satoh, H.; Sumiyoshi, Y.; Morinaga, K.; Kodama, K.; Suzuki, T.-I.; Nagai, M.; Suzuki, T. PCDDs/PCDFs and dioxin-like PCBs: Recent body burden levels and their determinants among general inhabitants in Japan. Chemosphere 2008, 73, 30–37. [Google Scholar] [CrossRef]
- Van den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Sofo, V.; Götte, M.; Laganà, A.S.; Salmeri, F.M.; Triolo, O.; Sturlese, E.; Retto, G.; Alfa, M.; Granese, R.; Abrão, M.S. Correlation between dioxin and endometriosis: An epigenetic route to unravel the pathogenesis of the disease. Arch. Gynecol. Obstet. 2015, 292, 973–986. [Google Scholar] [CrossRef]
- Bruner-Tran, K.L.; Ding, T.; Osteen, K.G. Dioxin and endometrial progesterone resistance. Semin. Reprod. Med. 2010, 28, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Cummings, A.M.; Hedge, J.M.; Birnbaum, L.S. Effect of prenatal exposure to TCDD on the promotion of endometriotic lesion growth by TCDD in adult female rats and mice. Toxicol. Sci. 1999, 52, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Rier, S.E.; Martin, D.C.; Bowman, R.E.; Dmowski, W.P.; Becker, J.L. Endometriosis in rhesus monkeys (Macaca mulatta) following chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam. Appl. Toxicol. 1993, 21, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, B.; Mocarelli, P.; Warner, M.; Samuels, S.; Vercellini, P.; Olive, D.; Needham, L.L.; Patterson, D.G.; Brambilla, P.; Gavoni, N.; et al. Serum dioxin concentrations and endometriosis: A cohort study in Seveso, Italy. Environ. Health Perspect. 2002, 110, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Heilier, J.-F.; Ha, A.T.; Lison, D.; Donnez, J.; Tonglet, R.; Nackers, F. Increased serum polychlorobiphenyl levels in Belgian women with adenomyotic nodules of the rectovaginal septum. Fertil. Steril. 2004, 81, 456–458. [Google Scholar] [CrossRef]
- Louis, G.M.B.; Weiner, J.M.; Whitcomb, B.W.; Sperrazza, R.; Schisterman, E.F.; Lobdell, D.T.; Crickard, K.; Greizerstein, H.; Kostyniak, P.J. Environmental PCB exposure and risk of endometriosis. Hum. Reprod. 2005, 20, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porpora, M.G.; Ingelido, A.M.; di Domenico, A.; Ferro, A.; Crobu, M.; Pallante, D.; Cardelli, M.; Cosmi, E.V.; De Felip, E. Increased levels of polychlorobiphenyls in Italian women with endometriosis. Chemosphere 2006, 63, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Rozati, R.; Reddy, S.; Kodampur, S.; Reddy, P.; Reddy, R. High plasma concentrations of polychlorinated biphenyls and phthalate esters in women with endometriosis: A prospective case control study. Fertil. Steril. 2006, 85, 775–779. [Google Scholar] [CrossRef]
- Gennings, C.; Sabo, R.; Carney, E. Identifying subsets of complex mixtures most associated with complex diseases: Polychlorinated biphenyls and endometriosis as a case study. Epidemiology 2010, 21, S77–S84. [Google Scholar] [CrossRef]
- Simsa, P.; Mihalyi, A.; Schoeters, G.; Koppen, G.; Kyama, C.M.; Den Hond, E.M.; Fülöp, V.; D’Hooghe, T.M. Increased exposure to dioxin-like compounds is associated with endometriosis in a case-control study in women. Reprod. Biomed. Online 2010, 20, 681–688. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Zamora, M.A.; Mattioli, L.; Parera, J.; Abad, E.; Coloma, J.L.; van Babel, B.; Galceran, M.T.; Balasch, J.; Carmona, F. Increased levels of dioxin-like substances in adipose tissue in patients with deep infiltrating endometriosis. Hum. Reprod. 2015, 30, 1059–1068. [Google Scholar] [CrossRef] [Green Version]
- Somigliana, E.; Vigano, P.; Abbiati, A.; Paffoni, A.; Benaglia, L.; Vercellini, P.; Fedele, L. Perinatal environment and endometriosis. Gynecol. Obstet. Investig. 2011, 72, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Mayani, A.; Barel, S.; Soback, S.; Almagor, M. Dioxin concentrations in women with endometriosis. Hum. Reprod. 1997, 12, 373–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebel, G.; Dodin, S.; Ayotte, P.; Marcoux, S.; Ferron, L.A.; Dewailly, E. Organochlorine exposure and the risk of endometriosis. Fertil. Steril. 1998, 69, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, A.; Schepens, P.J.; D’Hooghe, T.; Delbeke, L.; Dhont, M.; Brouwer, A.; Weyler, J. The risk of endometriosis and exposure to dioxins and polychlorinated biphenyls: A case-control study of infertile women. Hum. Reprod. 2001, 16, 2050–2055. [Google Scholar] [CrossRef] [Green Version]
- Fierens, S.; Mairesse, H.; Heilier, J.-F.; De Burbure, C.; Focant, J.-F.; Eppe, G.; De Pauw, E.; Bernard, A. Dioxin/polychlorinated biphenyl body burden, diabetes and endometriosis: Findings in a population-based study in Belgium. Biomarkers 2003, 8, 529–534. [Google Scholar] [CrossRef]
- Tsukino, H.; Hanaoka, T.; Sasaki, H.; Motoyama, H.; Hiroshima, M.; Tanaka, T.; Kabuto, M.; Niskar, A.S.; Rubin, C.; Patterson, D.G.; et al. Associations between serum levels of selected organochlorine compounds and endometriosis in infertile Japanese women. Environ. Res. 2005, 99, 118–125. [Google Scholar] [CrossRef]
- Hoffman, C.S.; Small, C.M.; Blanck, H.M.; Tolbert, P.; Rubin, C.; Marcus, M. Endometriosis among women exposed to polybrominated biphenyls. Ann. Epidemiol. 2007, 17, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Niskar, A.S.; Needham, L.L.; Rubin, C.; Turner, W.E.; Martin, C.A.; Patterson, D.G.; Hasty, L.; Wong, L.-Y.; Marcus, M. Serum dioxins, polychlorinated biphenyls, and endometriosis: A case-control study in Atlanta. Chemosphere 2009, 74, 944–949. [Google Scholar] [CrossRef]
- Trabert, B.; De Roos, A.J.; Schwartz, S.M.; Peters, U.; Scholes, D.; Barr, D.B.; Holt, V.L. Non-dioxin-like polychlorinated biphenyls and risk of endometriosis. Environ. Health Perspect. 2010, 118, 1280–1285. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.Y.; Izumi, S.; Suzuki, T.; Goya, K.; Nakamura, E.; Sugiyama, T.; Kobayashi, H. Dioxins in ascites and serum of women with endometriosis: A pilot study. Hum. Reprod. 2011, 26, 117–126. [Google Scholar] [CrossRef]
- Tsutsumi, O.; Momoeda, M.; Takai, Y.; Ono, M.; Taketani, Y. Breast-fed infants, possibly exposed to dioxins in milk, have unexpectedly lower incidence of endometriosis in adult life. Int. J. Gynecol. Obstet. 2000, 68, 151–153. [Google Scholar] [CrossRef]
- Giampaolino, P.; Della Corte, L.; Foreste, V.; Barra, F.; Ferrero, S.; Bifulco, G. Dioxin and endometriosis: A new possible relation based on epigenetic theory. Gynecol. Endocrinol. 2020, 36, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Banu, S.K.; Arosh, J.A. Endocrine disruptors and endometriosis. Reprod. Toxicol. 2023, 115, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Upson, K.; Sathyanarayana, S.; De Roos, A.J.; Koch, H.M.; Scholes, D.; Holt, V.L. A population-based case-control study of urinary bisphenol A concentrations and risk of endometriosis. Hum. Reprod. 2014, 29, 2457–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorile, P.G.; Spugnini, E.P.; Mita, L.; Mellone, P.; D’Avino, A.; Bianco, M.; Diano, N.; Caputo, L.; Rea, F.; Viceconte, R.; et al. Pre-natal exposure of mice to bisphenol A elicits an endometriosis-like phenotype in female offspring. Gen. Comp. Endocrinol. 2010, 168, 318–325. [Google Scholar] [CrossRef]
- Wen, X.; Xiong, Y.; Jin, L.; Zhang, M.; Huang, L.; Mao, Y.; Zhou, C.; Qiao, Y.; Zhang, Y. Bisphenol A Exposure Enhances Endometrial Stromal Cell Invasion and Has a Positive Association with Peritoneal Endometriosis. Reprod. Sci. 2020, 27, 704–712. [Google Scholar] [CrossRef]
- Xue, W.; Yao, X.; Ting, G.; Ling, J.; Huimin, L.; Yuan, Q.; Chun, Z.; Ming, Z.; Yuanzhen, Z. BPA modulates the WDR5/TET2 complex to regulate ERβ expression in eutopic endometrium and drives the development of endometriosis. Environ. Pollut. 2021, 268, 115748. [Google Scholar] [CrossRef]
- Jones, R.L.; Lang, S.A.; Kendziorski, J.A.; Greene, A.D.; Burns, K.A. Use of a Mouse Model of Experimentally Induced Endometriosis to Evaluate and Compare the Effects of Bisphenol A and Bisphenol AF Exposure. Environ. Health Perspect. 2018, 126, 127004. [Google Scholar] [CrossRef]
- Saillenfait, A.-M. Les phtalates. Point sur la réglementation en vigueur. Arch. Des Mal. Prof. L’Environnement 2015, 76, 32–35. [Google Scholar] [CrossRef]
- Mankidy, R.; Wiseman, S.; Ma, H.; Giesy, J.P. Biological impact of phthalates. Toxicol. Lett. 2013, 217, 50–58. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.H. Exposure to Phthalate Esters and the Risk of Endometriosis. Dev. Reprod. 2020, 24, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-C.; Tsai, E.-M.; Li, W.-F.; Liao, P.-C.; Chung, M.-C.; Wang, Y.-H.; Wang, S.-L. Association between phthalate exposure and glutathione S-transferase M1 polymorphism in adenomyosis, leiomyoma and endometriosis. Hum. Reprod. 2010, 25, 986–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weuve, J.; Hauser, R.; Calafat, A.M.; Missmer, S.A.; Wise, L.A. Association of exposure to phthalates with endometriosis and uterine leiomyomata: Findings from NHANES, 1999–2004. Environ. Health Perspect. 2010, 118, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Iwasaki, M.; Hanaoka, T.; Sasaki, H.; Tanaka, T.; Tsugane, S. Urinary phthalate monoesters and endometriosis in infertile Japanese women. Sci. Total Environ. 2009, 408, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Upson, K.; Sathyanarayana, S.; De Roos, A.J.; Thompson, M.L.; Scholes, D.; Dills, R.; Holt, V.L. Phthalates and risk of endometriosis. Environ. Res. 2013, 126, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck Louis, G.M.; Peterson, C.M.; Chen, Z.; Croughan, M.; Sundaram, R.; Stanford, J.; Varner, M.W.; Kennedy, A.; Giudice, L.; Fujimoto, V.Y.; et al. Bisphenol A and phthalates and endometriosis: The Endometriosis: Natural History, Diagnosis and Outcomes Study. Fertil. Steril. 2013, 100, 162–169.e2. [Google Scholar] [CrossRef] [Green Version]
- Buck Louis, G.M.; Hediger, M.L.; Peña, J.B. Intrauterine exposures and risk of endometriosis. Hum. Reprod. 2007, 22, 3232–3236. [Google Scholar] [CrossRef] [Green Version]
- Saha, R.; Kuja-Halkola, R.; Tornvall, P.; Marions, L. Reproductive and Lifestyle Factors Associated with Endometriosis in a Large Cross-Sectional Population Sample. J. Women’s Health 2017, 26, 152–158. [Google Scholar] [CrossRef]
- Hemmert, R.; Schliep, K.C.; Willis, S.; Peterson, C.M.; Louis, G.B.; Allen-Brady, K.; Simonsen, S.E.; Stanford, J.B.; Byun, J.; Smith, K.R. Modifiable life style factors and risk for incident endometriosis. Paediatr. Périnat. Epidemiol. 2019, 33, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bravi, F.; Parazzini, F.; Cipriani, S.; Chiaffarino, F.; Ricci, E.; Chiantera, V.; Viganò, P.; La Vecchia, C. Tobacco smoking and risk of endometriosis: A systematic review and meta-analysis. BMJ Open 2014, 4, e006325. [Google Scholar] [CrossRef]
- Sahin Ersoy, G.; Zhou, Y.; İnan, H.; Taner, C.E.; Cosar, E.; Taylor, H.S. Cigarette Smoking Affects Uterine Receptivity Markers. Reprod. Sci. 2017, 24, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Sasamoto, N.; Farland, L.V.; Vitonis, A.F.; Harris, H.R.; DiVasta, A.D.; Laufer, M.R.; Terry, K.L.; Missmer, S.A. In utero and early life exposures in relation to endometriosis in adolescents and young adults. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 252, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Jiang, I.; Yong, P.; Allaire, C.; Bedaiwy, M. Intricate Connections between the Microbiota and Endometriosis. Int. J. Mol. Sci. 2021, 22, 5644. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Shao, J.; Yang, Y.; Niu, X.; Liao, J.; Zhao, Q.; Wang, D.; Li, S.; Hu, J. Gut Microbiota in Patients with Polycystic Ovary Syndrome: A Systematic Review. Reprod. Sci. 2022, 29, 69–83. [Google Scholar] [CrossRef]
- Bedaiwy, M.A. Endometrial macrophages, endometriosis, and microbiota: Time to unravel the complexity of the relationship. Fertil. Steril. 2019, 112, 1049–1050. [Google Scholar] [CrossRef] [Green Version]
- Ata, B.; Yildiz, S.; Turkgeldi, E.; Brocal, V.P.; Dinleyici, E.C.; Moya, A.; Urman, B. The Endobiota Study: Comparison of Vaginal, Cervical and Gut Microbiota between Women with Stage 3/4 Endometriosis and Healthy Controls. Sci. Rep. 2019, 9, 2204. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Zhang, X.; Tang, H.; Zeng, L.; Wu, R. Microbiota composition and distribution along the female reproductive tract of women with endometriosis. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Hopeman, M.M.; Riley, J.K.; Frolova, A.I.; Jiang, H.; Jungheim, E.S. Serum Polyunsaturated Fatty Acids and Endometriosis. Reprod. Sci. 2015, 22, 1083–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadchan, S.B.; Cheng, M.; Parnell, L.A.; Yin, Y.; Schriefer, A.; Mysorekar, I.U.; Kommagani, R. Antibiotic therapy with metronidazole reduces endometriosis disease progression in mice: A potential role for gut microbiota. Hum. Reprod. 2019, 34, 1106–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, N.M.; Sola-Leyva, A.; Saez-Lara, M.J.; Plaza-Diaz, J.; Tubić-Pavlović, A.; Romero, B.; Clavero, A.; Mozas-Moreno, J.; Fontes, J.; Altmäe, S. New Opportunities for Endometrial Health by Modifying Uterine Microbial Composition: Present or Future? Biomolecules 2020, 10, 593. [Google Scholar] [CrossRef] [Green Version]
- Quaranta, G.; Sanguinetti, M.; Masucci, L. Fecal Microbiota Transplantation: A Potential Tool for Treatment of Human Female Reproductive Tract Diseases. Front. Immunol. 2019, 10, 2653. [Google Scholar] [CrossRef] [PubMed]
- Fabozzi, G.; Rebuzzini, P.; Cimadomo, D.; Allori, M.; Franzago, M.; Stuppia, L.; Garagna, S.; Ubaldi, F.M.; Zuccotti, M.; Rienzi, L. Endocrine-Disrupting Chemicals, Gut Microbiota, and Human (In)Fertility—It Is Time to Consider the Triad. Cells 2022, 11, 3335. [Google Scholar] [CrossRef]
- García-Peñarrubia, P.; Ruiz-Alcaraz, A.J.; Martínez-Esparza, M.; Marín, P.; Machado-Linde, F. Hypothetical roadmap towards endometriosis: Prenatal endocrine-disrupting chemical pollutant exposure, anogenital distance, gut-genital microbiota and subclinical infections. Hum. Reprod. Update 2020, 26, 214–246. [Google Scholar] [CrossRef]
- Rossi, M.; Amaretti, A.; Raimondi, S. Folate Production by Probiotic Bacteria. Nutrients 2011, 3, 118–134. [Google Scholar] [CrossRef] [Green Version]
- The Gut Microbiota and Endometriosis: From Pathogenesis to Diagnosis and Treatment–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/36506023/ (accessed on 2 February 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monnin, N.; Fattet, A.J.; Koscinski, I. Endometriosis: Update of Pathophysiology, (Epi) Genetic and Environmental Involvement. Biomedicines 2023, 11, 978. https://doi.org/10.3390/biomedicines11030978
Monnin N, Fattet AJ, Koscinski I. Endometriosis: Update of Pathophysiology, (Epi) Genetic and Environmental Involvement. Biomedicines. 2023; 11(3):978. https://doi.org/10.3390/biomedicines11030978
Chicago/Turabian StyleMonnin, Nicolas, Anne Julie Fattet, and Isabelle Koscinski. 2023. "Endometriosis: Update of Pathophysiology, (Epi) Genetic and Environmental Involvement" Biomedicines 11, no. 3: 978. https://doi.org/10.3390/biomedicines11030978