
Thyroid Hormone and Heart Failure: Charting Known Pathways for Cardiac Repair/Regeneration
 , ,
, ,
Abstract
: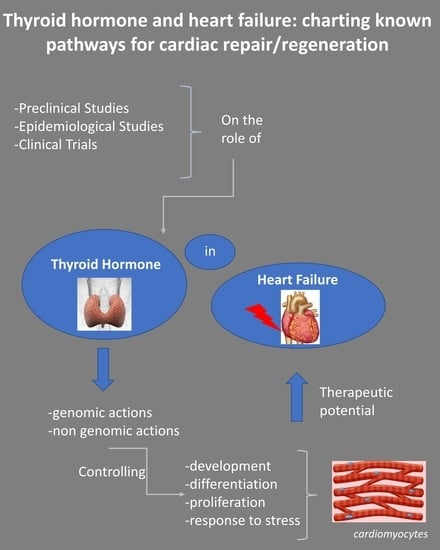
1. Introduction
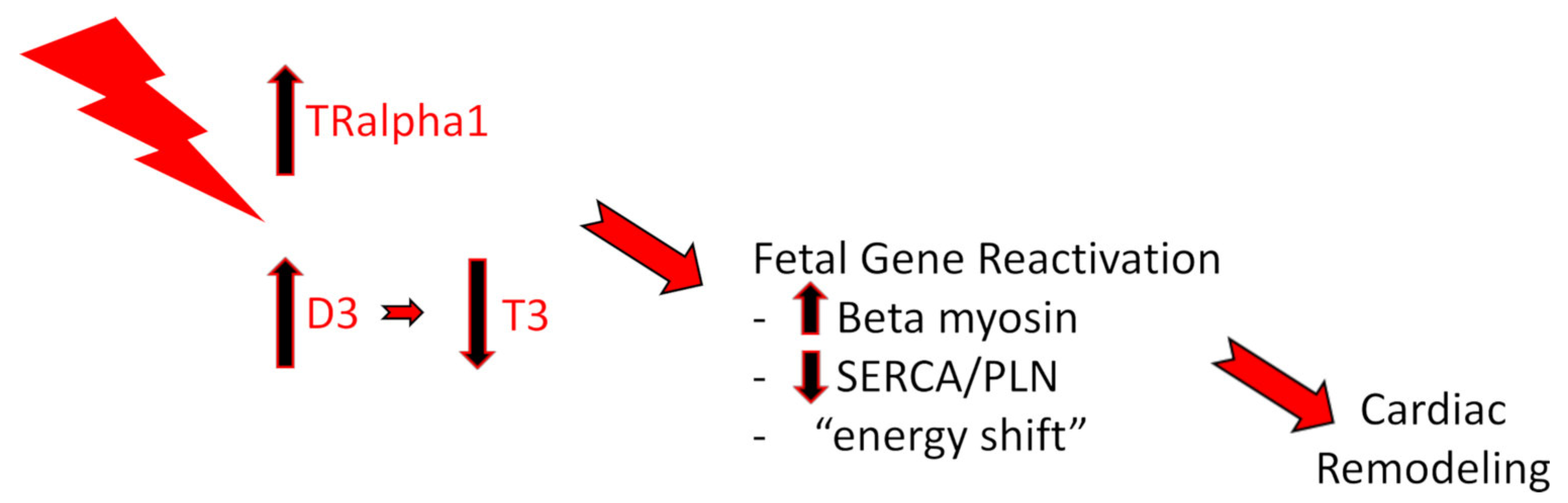
2. TH and HFrEF
2.1. Preclinical Studies
2.2. Epidemiological Studies
2.3. Clinical Trials
2.4. TH and HFpEF
3. The Challenge of Clinical Translation
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2022, 118, 3272–3287. [Google Scholar] [CrossRef] [PubMed]
- Mantzouratou, P.; Lavecchia, A.M.; Xinaris, C. Thyroid Hormone Signalling in Human Evolution and Disease: A Novel Hypothesis. J. Clin. Med. 2021, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoykov, I.; Zandieh-Doulabi, B.; Moorman, A.F.M.; Christoffels, V.; Wiersinga, W.M.; Bakker, O. Expression pattern and ontogenesis of thyroid hormone receptor isoforms in the mouse heart. J. Endocrinol. 2006, 189, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Anyetei-Anum, C.S.; Roggero, V.R.; Allison, L.A. Thyroid hormone receptor localization in target tissues. J. Endocrinol. 2018, 237, R19–R34. [Google Scholar] [CrossRef] [Green Version]
- Pantos, C.; Mourouzis, I. Thyroid hormone receptor α1 as a novel therapeutic target for tissue repair. Ann. Transl. Med. 2018, 6, 254. [Google Scholar] [CrossRef]
- Mourouzis, I.; Lavecchia, A.M.; Xinaris, C. Thyroid Hormone Signalling: From the Dawn of Life to the Bedside. J. Mol. Evol. 2020, 88, 88–103. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I. The emerging role of TRα1 in cardiac repair: Potential therapeutic implications. Oxid. Med. Cell Longev. 2014, 2014, 481482. [Google Scholar] [CrossRef] [Green Version]
- Hirose, K.; Payumo, A.Y.; Cutie, S.; Hoang, A.; Zhang, H.; Guyot, R.; Lunn, D.; Bigley, R.B.; Yu, H.; Wang, J.; et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019, 364, 184–188. [Google Scholar] [CrossRef]
- Lavecchia, A.M.; Pelekanos, K.; Mavelli, F.; Xinaris, C. Cell Hypertrophy: A ‘Biophysical Roadblock’ to Reversing Kidney Injury. Front. Cell Dev. Biol. 2022, 10, 854998. [Google Scholar] [CrossRef]
- Luongo, C.; Dentice, M.; Salvatore, D. Deiodinases and their intricate role in thyroid hormone homeostasis. Nat. Rev. Endocrinol. 2019, 15, 479–488. [Google Scholar] [CrossRef]
- Dentice, M.; Marsili, A.; Zavacki, A.; Larsen, P.R.; Salvatore, D. The deiodinases and the control of intracellular thyroid hormone signaling during cellular differentiation. Biochim. Biophys. Acta 2013, 1830, 3937–3945. [Google Scholar] [CrossRef] [Green Version]
- Barca-Mayo, O.; Liao, X.H.; Alonso, M.; Di Cosmo, C.; Hernandez, A.; Refetoff, S.; Weisse, R.E. Thyroid hormone receptor α and regulation of type 3 deiodinase. Mol. Endocrinol. 2011, 25, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Simonides, W.S.; Mulcahey, M.A.; Redout, E.M.; Muller, A.; Zuidwijk, M.J.; Visser, T.J.; Wassen, F.W.J.S.; Crescenzi, A.; da-Silva, W.S.; Harney, J.; et al. Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J. Clin. Investig. 2008, 118, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Pantos, C.; Xinaris, C.; Mourouzis, I.; Perimenis, P.; Politi, E.; Spanou, D.; Cokkinos, D.V. Thyroid hormone receptor alpha 1: A switch to cardiac cell ‘metamorphosis’? J. Physiol. Pharmacol. 2008, 59, 253–269. [Google Scholar]
- Pantos, C.; Mourouzis, I.; Xinaris, C.; Papadopoulou-Daifoti, Z.; Cokkinos, D. Thyroid hormone and ‘cardiac metamorphosis’: Potential therapeutic implications. Pharmacol. Ther. 2008, 118, 277–294. [Google Scholar] [CrossRef]
- Ojamaa, K.; Kenessey, A.; Shenoy, R.; Klein, I. Thyroid hormone metabolism and cardiac gene expression after acute myocardial infarction in the rat. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1319–E1324. [Google Scholar] [CrossRef]
- Alpert, N.R.; Brosseau, C.; Federico, A.; Krenz, M.; Robbins, J.; Warshaw, D.M. Molecular mechanics of mouse cardiac myosin isoforms. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1446–H1454. [Google Scholar] [CrossRef] [Green Version]
- Pantos, C.; Mourouzis, I.; Cokkinos, D.V. New insights into the role of thyroid hormone in cardiac remodeling: Time to reconsider? Heart Fail. Rev. 2011, 16, 79–96. [Google Scholar] [CrossRef]
- Rajabi, M.; Kassiotis, C.; Razeghi, P.; Taegtmeyer, H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail. Rev. 2007, 12, 331–343. [Google Scholar] [CrossRef]
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognese, L.; Neskovic, A.N.; Parodi, G.; Cerisano, G.; Buonamici, P.; Santoro, G.M.; Antoniucci, D. Left ventricular remodeling after primary coronary angioplasty: Patterns of left ventricular dilation and long-term prognostic implications. Circulation 2002, 106, 2351–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantos, C.; Mourouzis, I.; Galanopoulos, G.; Gavra, M.; Perimenis, P.; Spanou, D.; Cokkinos, D.V. Thyroid hormone receptor alpha1 downregulation in postischemic heart failure progression: The potential role of tissue hypothyroidism. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 2010, 42, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Mourouzis, I.; Kostakou, E.; Galanopoulos, G.; Mantzouratou, P.; Pantos, C. Inhibition of thyroid hormone receptor α1 impairs post-ischemic cardiac performance after myocardial infarction in mice. Mol. Cell Biochem. 2013, 379, 97–105. [Google Scholar] [CrossRef]
- Mourouzis, I.; Mantzouratou, P.; Galanopoulos, G.; Kostakou, E.; Roukounakis, N.; Kokkinos, A.D.; Cokkinos, D.V.; Pantos, C. Dose-dependent effects of thyroid hormone on post-ischemic cardiac performance: Potential involvement of Akt and ERK signalings. Mol. Cell Biochem. 2012, 363, 235–243. [Google Scholar] [CrossRef]
- Iliopoulou, I.; Mourouzis, I.; Lambrou, G.I.; Iliopoulou, D.; Koutsouris, D.D.; Pantos, C. Time-dependent and independent effects of thyroid hormone administration following myocardial infarction in rats. Mol. Med. Rep. 2018, 18, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Kobayashi, S.; Chen, J.; Redetzke, R.A.; Said, S.; Liang, Q.; Gerdes, A.M. Short term triiodo-L-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J. Mol. Cell Cardiol. 2008, 44, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Forini, F.; Lionetti, V.; Ardehali, H.; Pucci, A.; Cecchetti, F.; Ghanefar, M.; Nicolini, G.; Ichikawa, Y.; Nannipieri, M.; Recchia, F.A.; et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J. Cell Mol. Med. 2011, 15, 514–524. [Google Scholar] [CrossRef]
- Mourouzis, I.; Giagourta, I.; Galanopoulos, G.; Mantzouratou, P.; Kostakou, E.; Kokkinos, A.D.; Tentolouris, N.; Pantos, C. Thyroid hormone improves the mechanical performance of the post-infarcted diabetic myocardium: A response associated with up-regulation of Akt/mTOR and AMPK activation. Metabolism 2013, 62, 1387–1393. [Google Scholar]
- Pantos, C.; Malliopoulou, V.; Paizis, I.; Moraitis, P.; Mourouzis, I.; Tzeis, S.; Karamanoli, E.; Cokkinos, D.D.; Carageorgiou, H.; Varonos, D.; et al. Thyroid hormone and cardioprotection: Study of p38 MAPK and JNKs during ischaemia and at reperfusion in isolated rat heart. Mol. Cell Biochem. 2003, 242, 173–180. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I.; Saranteas, T.; Clavé, G.; Ligeret, H.; Noack-Fraissignes, P.; Renard, P.-Y.; Massonneau, M.; Perimenis, P.; Spanou, D.; et al. Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: A new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res. Cardiol. 2009, 104, 69–77. [Google Scholar] [CrossRef]
- Lymvaios, I.; Mourouzis, I.; Cokkinos, D.V.; Dimopoulos, M.A.; Toumanidis, S.T.; Pantos, C. Thyroid hormone and recovery of cardiac function in patients with acute myocardial infarction: A strong association? Eur. J. Endocrinol. 2011, 165, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Lazzeri, C.; Sori, A.; Picariello, C.; Chiostri, M.; Gensini, G.F.; Valente, S. Nonthyroidal illness syndrome in ST-elevation myocardial infarction treated with mechanical revascularization. Int. J. Cardiol. 2012, 158, 103–104. [Google Scholar] [CrossRef]
- Lee, Y.; Lim, Y.H.; Shin, J.H.; Park, J.; Shin, J. Impact of subclinical hypothyroidism on clinical outcomes following percutaneous coronary intervention. Int. J. Cardiol. 2018, 253, 155–160. [Google Scholar] [CrossRef]
- Xue, Y.; Zhu, Y.; Shen, J.; Zhou, W.; Xiang, J.; Xiang, Z.; Wang, L.; Luo, S. The Association of Thyroid Hormones With Cardiogenic Shock and Prognosis in Patients with ST Segment Elevation Myocardial Infarction (STEMI) Treated with Primary PCI. Am. J. Med. Sci. 2022, 363, 251–258. [Google Scholar] [CrossRef]
- Gao, S.; Ma, W.; Huang, S.; Lin, X.; Yu, M. Predictive Value of Free Triiodothyronine to Free Thyroxine Ratio in Euthyroid Patients With Myocardial Infarction With Nonobstructive Coronary Arteries. Front. Endocrinol. 2021, 12, 708216. [Google Scholar] [CrossRef]
- Jabbar, A.; Ingoe, L.; Thomas, H.; Carey, P.; Junejo, S.; Addison, C.; Vernazza, J.; Austin, D.; Greenwood, J.P.; Zaman, A. Prevalence, predictors and outcomes of thyroid dysfunction in patients with acute myocardial infarction: The ThyrAMI-1 study. J. Endocrinol. Investig. 2021, 44, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, A.; Landi, P.; Taddei, M.C.; Ripoli, A.; L’Abbate, A.; Iervasi, G. Triiodothyronine levels for risk stratification of patients with chronic heart failure. Am. J. Med. 2005, 118, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Xinke, Z.; Rongcheng, Z.; Hugang, J.; Kai, L.; Chengxu, M.; Ming, B.; Tao, A.; Younan, Y.; Xinqiang, W.; Ming, W.; et al. Combined Use of Low T3 Syndrome and NT-proBNP as Predictors for Death in Patients with Acute Decompensated Heart Failure. BMC Endocr. Disord. 2021, 21, 140. [Google Scholar]
- Rothberger, G.D.; Gadhvi, S.; Michelakis, N.; Kumar, A.; Calixte, R.; Shapiro, L.E. Usefulness of Serum Triiodothyronine (T3) to Predict Outcomes in Patients Hospitalized with Acute Heart Failure. Am. J. Cardiol. 2017, 119, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshihisa, A.; Kimishima, Y.; Kiko, T.; Kanno, Y.; Yokokawa, T.; Abe, S.; Misaka, T.; Sato, T.; Oikawa, M.; et al. Low T3 Syndrome Is Associated with High Mortality in Hospitalized Patients With Heart Failure. J. Card. Fail. 2019, 25, 195–203. [Google Scholar] [CrossRef]
- Fontana, M.; Passino, C.; Poletti, R.; Zyw, L.; Prontera, C.; Scarlattini, M.; Clerico, A.; Emdin, M.; Iervasi, G. Low triiodothyronine and exercise capacity in heart failure. Int. J. Cardiol. 2012, 154, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Kannan, L.; Shaw, P.A.; Morley, M.P.; Brandimarto, J.; Fang, J.C.; Sweitzer, N.K.; Cappola, T.P.; Cappola, A.R. Thyroid Dysfunction in Heart Failure and Cardiovascular Outcomes. Circ. Heart Fail. 2018, 11, e005266. [Google Scholar] [CrossRef]
- Wang, W.; Guan, H.; Fang, W.; Zhang, K.; Gerdes, A.M.; Iervasi, G.; Tang, Y.D. Free Triiodothyronine Level Correlates with Myocardial Injury and Prognosis in Idiopathic Dilated Cardiomyopathy: Evidence from Cardiac MRI and SPECT/PET Imaging. Sci. Rep. 2016, 6, 39811. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, M.A.; Stevenson, L.W.; Fonarow, G.C.; Steimle, A.; Goldhaber, J.I.; Child, J.S.; Chopra, I.J.; Moriguchi, J.D.; Hage, A. Safety and hemodynamic effects of intravenous triiodothyronine in advanced congestive heart failure. Am. J. Cardiol. 1998, 81, 443–447. [Google Scholar] [CrossRef]
- Pingitore, A.; Galli, E.; Barison, A.; Iervasi, A.; Scarlattini, M.; Nucci, D.; L’abbate, A.; Mariotti, R.; Iervasi, G. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: A randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 2008, 93, 1351–1358. [Google Scholar] [CrossRef] [Green Version]
- Holmager, P.; Schmidt, U.; Mark, P.; Andersen, U.; Dominguez, H.; Raymond, I.; Zerahn, B.; Nygaard, B.; Kistorp, C.; Faber, J. Long-term L-Triiodothyronine (T3) treatment in stable systolic heart failure patients: A randomised, double-blind, cross-over, placebo-controlled intervention study. Clin. Endocrinol. 2015, 83, 931–937. [Google Scholar] [CrossRef]
- Amin, A.; Chitsazan, M.; Taghavi, S.; Ardeshiri, M. Effects of triiodothyronine replacement therapy in patients with chronic stable heart failure and low-triiodothyronine syndrome: A randomized, double-blind, placebo-controlled study. ESC Heart Fail. 2015, 2, 5–11. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Zhang, K.; Tian, J.; Zheng, J.L.; Chen, J.; An, S.M.; Wang, S.Y.; Liu, Y.P.; Zhao, Y.; et al. Efficacy and safety of levothyroxine (L-T4) replacement on the exercise capability in chronic systolic heart failure patients with subclinical hypothyroidism: Study protocol for a multi-center, open label, randomized, parallel group trial (ThyroHeart-CHF). Trials 2019, 20, 143. [Google Scholar] [CrossRef] [Green Version]
- Pingitore, A.; Mastorci, F.; Piaggi, P.; Aquaro, G.D.; Molinaro, S.; Ravani, M.; De Caterina, A.; Trianni, G.; Ndreu, R.; Berti, S.; et al. Usefulness of Triiodothyronine Replacement Therapy in Patients with ST Elevation Myocardial Infarction and Borderline/Reduced Triiodothyronine Levels (from the THIRST Study). Am. J. Cardiol. 2019, 123, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Jabbar, A.; Ingoe, L.; Junejo, S.; Carey, P.; Addison, C.; Thomas, H.; Parikh, J.D.; Austin, D.; Hollingsworth, K.G.; Stocken, D.D.; et al. Effect of Levothyroxine on Left Ventricular Ejection Fraction in Patients With Subclinical Hypothyroidism and Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA 2020, 324, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Sinn, M.R.; Lund, G.K.; Muellerleile, K.; Freiwald, E.; Saeed, M.; Avanesov, M.; Lenz, A.; Starekova, J.; von Kodolitsch, Y.; Blankenberg, S.; et al. Prognosis of early pre-discharge and late left ventricular dilatation by cardiac magnetic resonance imaging after acute myocardial infarction. Int. J. Cardiovasc. Imaging 2021, 37, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Breathett, K.; Allen, L.A.; Udelson, J.; Davis, G.; Bristow, M. Changes in Left Ventricular Ejection Fraction Predict Survival and Hospitalization in Heart Failure With Reduced Ejection Fraction. Circ. Heart Fail. 2016, 9, e002962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerra, L.; Anavekar, N.; Skali, H.; Zelenkofske, S.; Velazquez, E.; McMurray, J.; Pfeffer, M.; Solomon, S.D. Association of QRS duration and outcomes after myocardial infarction: The VALIANT trial. Heart Rhythm 2006, 3, 313–316. [Google Scholar] [CrossRef]
- Pantos, C.I.; Trikas, A.G.; Pissimisis, E.G.; Grigoriou, K.P.; Stougiannos, P.N.; Dimopoulos, A.K.; Linardakis, S.I.; Alexopoulos, N.A.; Evdoridis, C.G.; Gavrielatos, G.D.; et al. Effects of Acute Triiodothyronine Treatment in Patients with Anterior Myocardial Infarction Undergoing Primary Angioplasty: Evidence from a Pilot Randomized Clinical Trial (ThyRepair Study). Thyroid 2022, 32, 714–724. [Google Scholar] [CrossRef]
- Novitzky, D.; Mi, Z.; Sun, Q.; Collins, J.F.; Cooper, D.K. Thyroid hormone therapy in the management of 63,593 brain-dead organ donors: A retrospective review. Transplantation 2014, 98, 1119–1127. [Google Scholar] [CrossRef]
- Novitzky, D.; Cooper, D.K. Thyroid hormones and the stunned myocardium. J. Endocrinol. 2014, 223, R1–R8. [Google Scholar] [CrossRef] [Green Version]
- Omote, K.; Verbrugge, F.H.; Borlaug, B.A. Heart Failure with Preserved Ejection Fraction: Mechanisms and Treatment Strategies. Annu. Rev. Med. 2022, 73, 321–337. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Kataria, R.; Zannad, F.; Sauer, A.J.; Damman, K.; Sharma, K.; Shah, S.J.; Van Spall, H.G.C. Heart failure with preserved ejection fraction: Recent concepts in diagnosis, mechanisms and management. Heart Br. Card Soc. 2022, 108, 1342–1350. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Boen, J.R.A.; Segers, V.F.; Van Craenenbroeck, E.M. Heart Failure with Preserved Ejection Fraction: A Review of Cardiac and Noncardiac Pathophysiology. Front. Physiol. 2019, 10, 638. [Google Scholar] [CrossRef]
- Runte, K.E.; Bell, S.P.; Selby, D.E.; Häußler, T.N.; Ashikaga, T.; LeWinter, M.M.; Palmer, B.M.; Meyer, M. Relaxation and the Role of Calcium in Isolated Contracting Myocardium from Patients with Hypertensive Heart Disease and Heart Failure With Preserved Ejection Fraction. Circ. Heart Fail. 2017, 10, e004311. [Google Scholar] [CrossRef]
- Neves, J.S.; Vale, C.; von Hafe, M.; Borges-Canha, M.; Leite, A.R.; Almeida-Coelho, J.; Lourenço, A.; Falcão-Pires, I.; Carvalho, D.; Leite-Moreira, A. Thyroid hormones and modulation of diastolic function: A promising target for heart failure with preserved ejection fraction. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820958331. [Google Scholar] [CrossRef]
- von Hafe, M.; Neves, J.S.; Vale, C.; Borges-Canha, M.; Leite-Moreira, A. The impact of thyroid hormone dysfunction on ischemic heart disease. Endocr. Connect. 2019, 8, R76–R90. [Google Scholar] [CrossRef] [Green Version]
- Dan, G.-A. Thyroid Hormones and the Heart. Heart Fail. Rev. 2016, 21, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Madathil, A.; Hollingsworth, K.G.; Blamire, A.M.; Razvi, S.; Newton, J.L.; Taylor, R.; Weaver, J.U. Levothyroxine Improves Abnormal Cardiac Bioenergetics in Subclinical Hypothyroidism: A Cardiac Magnetic Resonance Spectroscopic Study. J. Clin. Endocrinol. Metab. 2015, 100, E607–E610. [Google Scholar] [CrossRef] [Green Version]
- Vale, C.; Neves, J.S.; von Hafe, M.; Borges-Canha, M.; Leite-Moreira, A. The Role of Thyroid Hormones in Heart Failure. Cardiovasc. Drugs Ther. 2019, 33, 179–188. [Google Scholar] [CrossRef]
- Biondi, B.; Fazio, S.; Palmieri, E.A.; Carella, C.; Panza, N.; Cittadini, A.; Bonè, F.; Lombardi, G.; Saccà, L. Left ventricular diastolic dysfunction in patients with subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 1999, 84, 2064–2067. [Google Scholar] [CrossRef]
- Razvi, S. Novel uses of thyroid hormones in cardiovascular conditions. Endocrine 2019, 66, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.; Klein, I.; Danzi, S.; Akhter, N.; Bonow, R.O.; Shah, S.J. Association of serum triiodothyronine with B-type natriuretic peptide and severe left ventricular diastolic dysfunction in heart failure with preserved ejection fraction. Am. J. Cardiol. 2012, 110, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Weltman, N.Y.; Pol, C.J.; Zhang, Y.; Wang, Y.; Koder, A.; Raza, S.; Zucchi, R.; Saba, A.; Colligiani, D.; Gerdes, A.M. Long-term physiological T3 supplementation in hypertensive heart disease in rats. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1059–H1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, A. Developing Oral LT3 Therapy for Heart Failure with Preserved Ejection Fraction. clinicaltrials.gov. 2021 Dec. Report No.: NCT04111536. Available online: https://clinicaltrials.gov/ct2/show/NCT04111536 (accessed on 9 June 2022).
- The Coronary Drug Project. Findings leading to discontinuation of the 2.5-mg day estrogen group. The coronary Drug Project Research Group. JAMA 1973, 226, 652–657. [Google Scholar] [CrossRef]
- Goldman, S.; McCarren, M.; Morkin, E.; Ladenson, P.W.; Edson, R.; Warren, S.; Ohm, J.; Thai, H.; Churby, L.; Barnhill, J.; et al. DITPA (3,5-Diiodothyropropionic Acid), a thyroid hormone analog to treat heart failure: Phase II trial veterans affairs cooperative study. Circulation 2009, 119, 3093–3100. [Google Scholar] [CrossRef] [PubMed]
- Trivieri, M.G.; Oudit, G.Y.; Sah, R.; Kerfant, B.G.; Sun, H.; Gramolini, A.O.; Pan, Y.; Wickenden, A.D.; Croteau, W.; Morreale de Escobar, G.; et al. Cardiac-specific elevations in thyroid hormone enhance contractility and prevent pressure overload-induced cardiac dysfunction. Proc. Natl. Acad. Sci. USA 2006, 103, 6043–6048. [Google Scholar] [CrossRef] [Green Version]
- Pingitore, A.; Mastorci, F.; Berti, S.; Sabatino, L.; Palmieri, C.; Iervasi, G.; Vassalle, C. Hypovitaminosis D and Low T3 Syndrome: A Link for Therapeutic Challenges in Patients with Acute Myocardial Infarction. J. Clin. Med. 2021, 10, 5267. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I. Translating thyroid hormone effects into clinical practice: The relevance of thyroid hormone receptor α1 in cardiac repair. Heart Fail. Rev. 2015, 20, 273–282. [Google Scholar] [CrossRef]
- Karakus, O.O.; Darwish, N.H.E.; Sudha, T.; Salaheldin, T.A.; Fujioka, K.; Dickinson, P.C.T.; Weil, B.; Mousa, S.A. Development of Triiodothyronine Polymeric Nanoparticles for Targeted Delivery in the Cardioprotection against Ischemic Insult. Biomedicines 2021, 9, 1713. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [Green Version]
- Regenerating the Diabetic Heart and Kidney by Using Stress-Specific Thyroid Hormone Nanocarriers—ERA-LEARN. Available online: https://www.era-learn.eu/network-information/networks/euronanomed-iii/joint-transnational-call-2019/regenerating-the-diabetic-heart-and-kidney-by-using-stress-specific-thyroid-hormone-nanocarriersAuthor (accessed on 13 June 2022).


{kind=link}
{kind=link}
| Clinical Study | Patients (N) | Setting | Treatment | Outcome | Safety |
|---|---|---|---|---|---|
| Hamilton et al. [46] | 23 | Advanced HF and low T3 | 0.15–2.7 ìg/kg (iv) T3 for 6–12 h | Increased CO and reduction in SVR | No AEs |
| Pingitore et al. [47] | 20 | HF and low T3 | 35.6 ìg LT3 (iv) in the first 24 h and 15 ìg/day until 72 h | Increased SV and lower HR; decrease in NT-proBNP, noradrenaline, and aldosterone | No AEs |
| Holmager et al. [48] | 13 | Stable systolic HF and low T3 | 20 μg oral T3 per day for 3 months | No changes in cardiac function and neurohormonal profile | No AEs |
| Amin et al. [49] | 50 | Chronic stable HF and low T3 | T3 replacement dose by oral liothyronine for 6 weeks | Increased 6 min walk test, decreased hsCRP, decrease in NTproBNP | No AEs |
| Zhang et al. [50] | 124 (estimated) | Chronic HF and low T3 | Oral levothyroxine with a starting dose of 12.5 μg | Ongoing | Ongoing |
| Pingitore et al. [51] | 37 | AMI and low T3 | Oral liothyronine (T3) (maximum dosage 15 mcg/m2/die for 6 months | Significant reduction in WMSI difference value (discharge/follow-up), increased stroke volume at follow-up | No AEs |
| Jabbar et al. [52] | 95 | AMI and subclinical hypothyroidism | Oral levothyroxine (25 µg titrated to serum thyrotropin levels between 0.4 and 2.5 mU/L | No significant differences | No AEs |
| Pantos et al. [56] | 52 | Anterior STEMI undergoing PCI | (i.v.) bolus injection of 0.8 μg/kg of LT3 followed by a constant infusion of 0.113 μg/kg/h i.v. for 48 h | Significantly lower LV end-diastolic volume index and LV end-systolic volume index at discharge, CMR IV tended to be lower in the LT3-treated group at 6 months | Tendency for an increased incidence of AF during the first 48 h |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantzouratou, P.; Malaxianaki, E.; Cerullo, D.; Lavecchia, A.M.; Pantos, C.; Xinaris, C.; Mourouzis, I. Thyroid Hormone and Heart Failure: Charting Known Pathways for Cardiac Repair/Regeneration. Biomedicines 2023, 11, 975. https://doi.org/10.3390/biomedicines11030975
Mantzouratou P, Malaxianaki E, Cerullo D, Lavecchia AM, Pantos C, Xinaris C, Mourouzis I. Thyroid Hormone and Heart Failure: Charting Known Pathways for Cardiac Repair/Regeneration. Biomedicines. 2023; 11(3):975. https://doi.org/10.3390/biomedicines11030975
Chicago/Turabian StyleMantzouratou, Polyxeni, Eleftheria Malaxianaki, Domenico Cerullo, Angelo Michele Lavecchia, Constantinos Pantos, Christodoulos Xinaris, and Iordanis Mourouzis. 2023. "Thyroid Hormone and Heart Failure: Charting Known Pathways for Cardiac Repair/Regeneration" Biomedicines 11, no. 3: 975. https://doi.org/10.3390/biomedicines11030975