
G-Quadruplexes in c-MYC Promoter as Targets for Cancer Therapy

Abstract
:1. Introduction
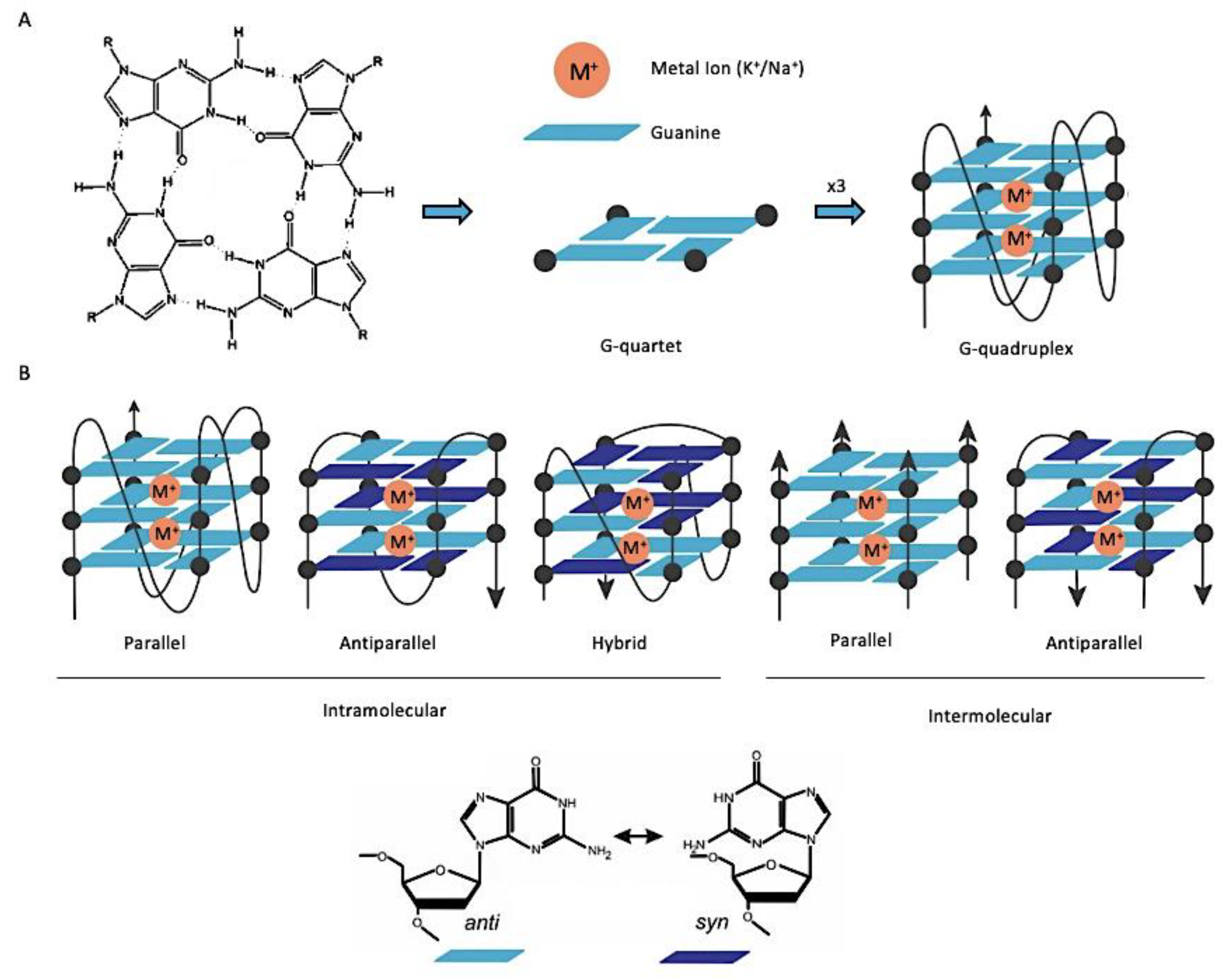
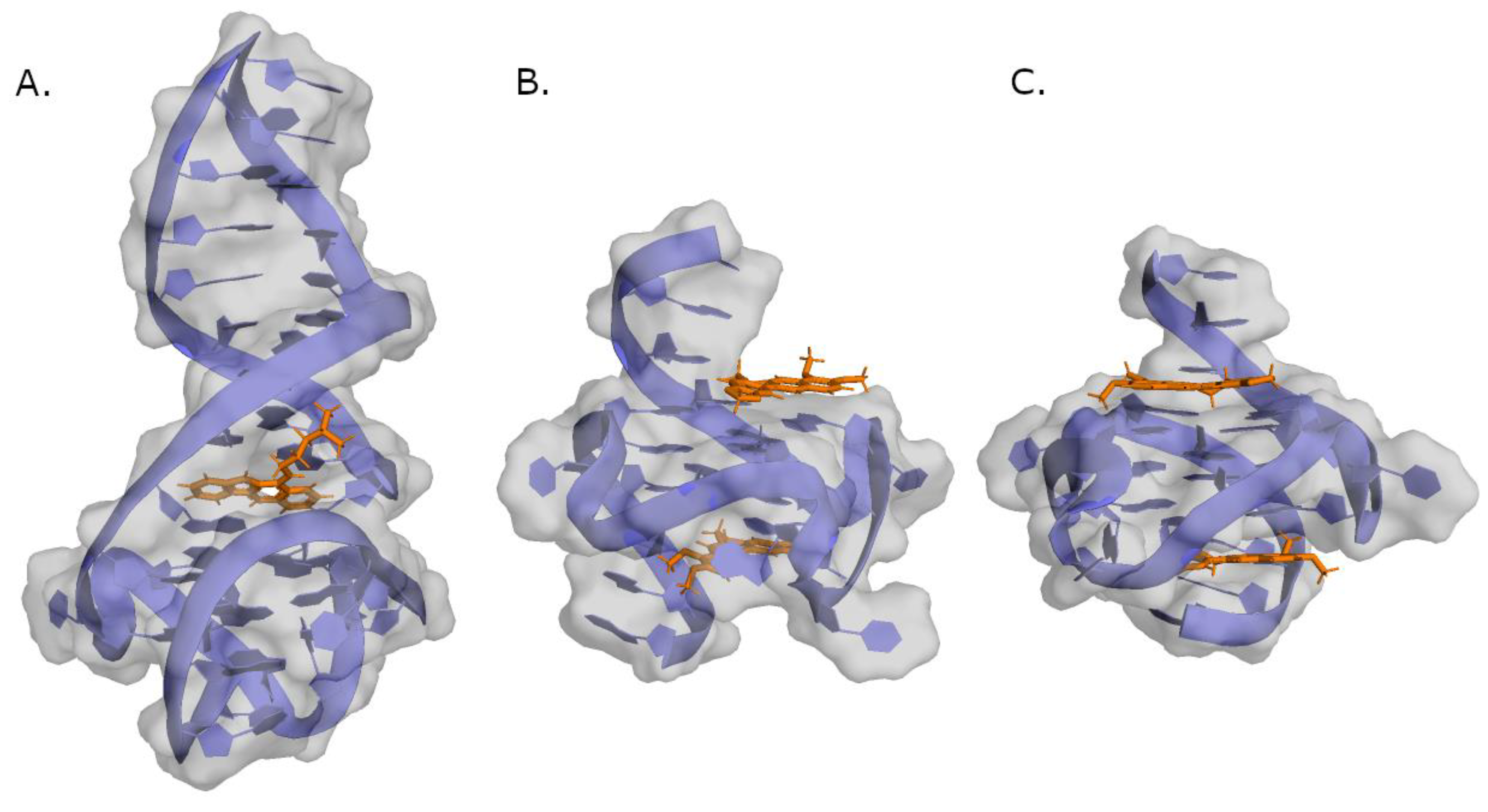
2. G-Quadruplex Nucleic Acids as Drug Targets
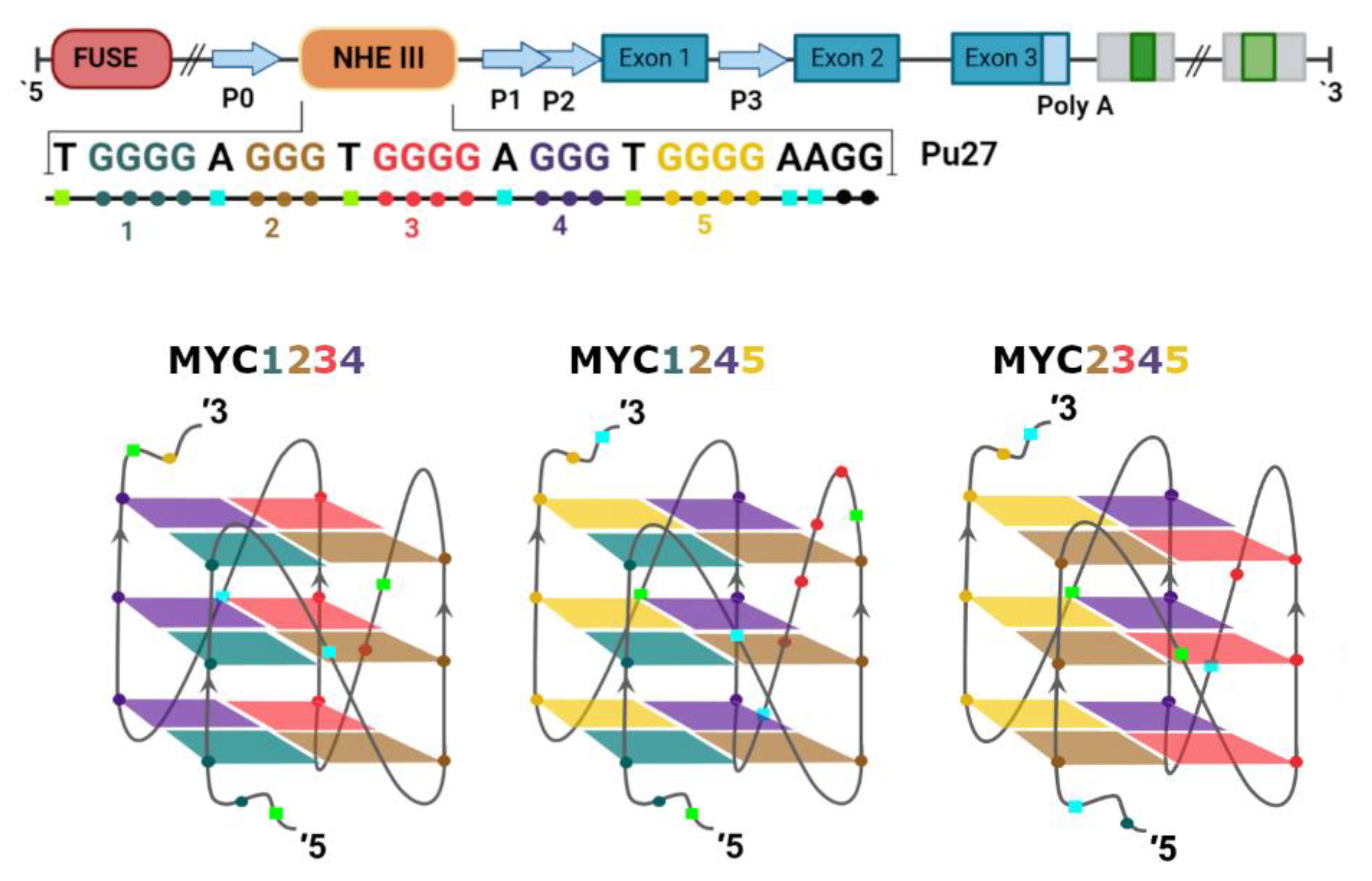
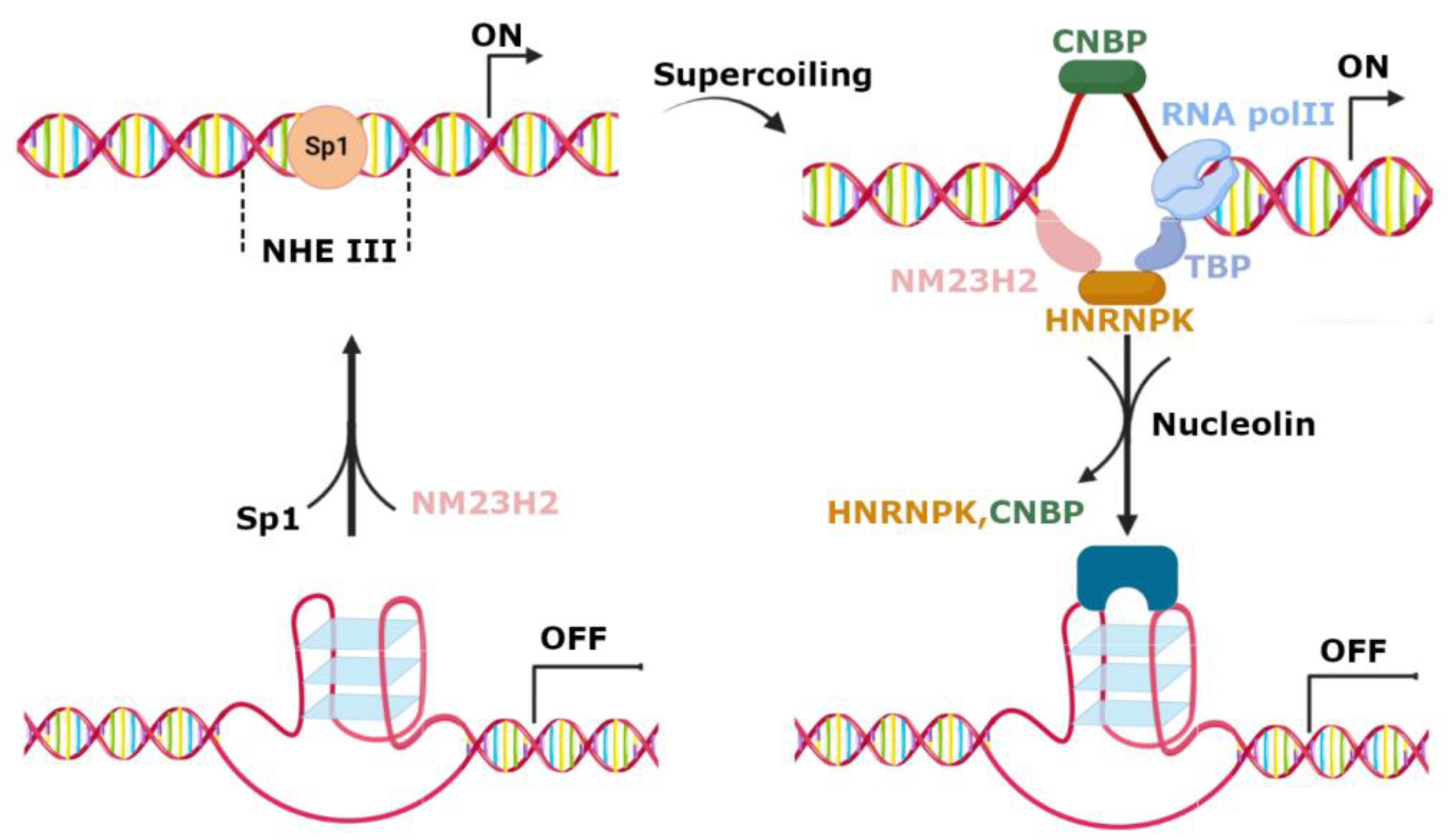
3. Regulation of c-MYC Transcription by G-Quadruplexes in Promoter Region
4. c-MYC G4 Stabilizing Small Molecules with Anticancer Activity
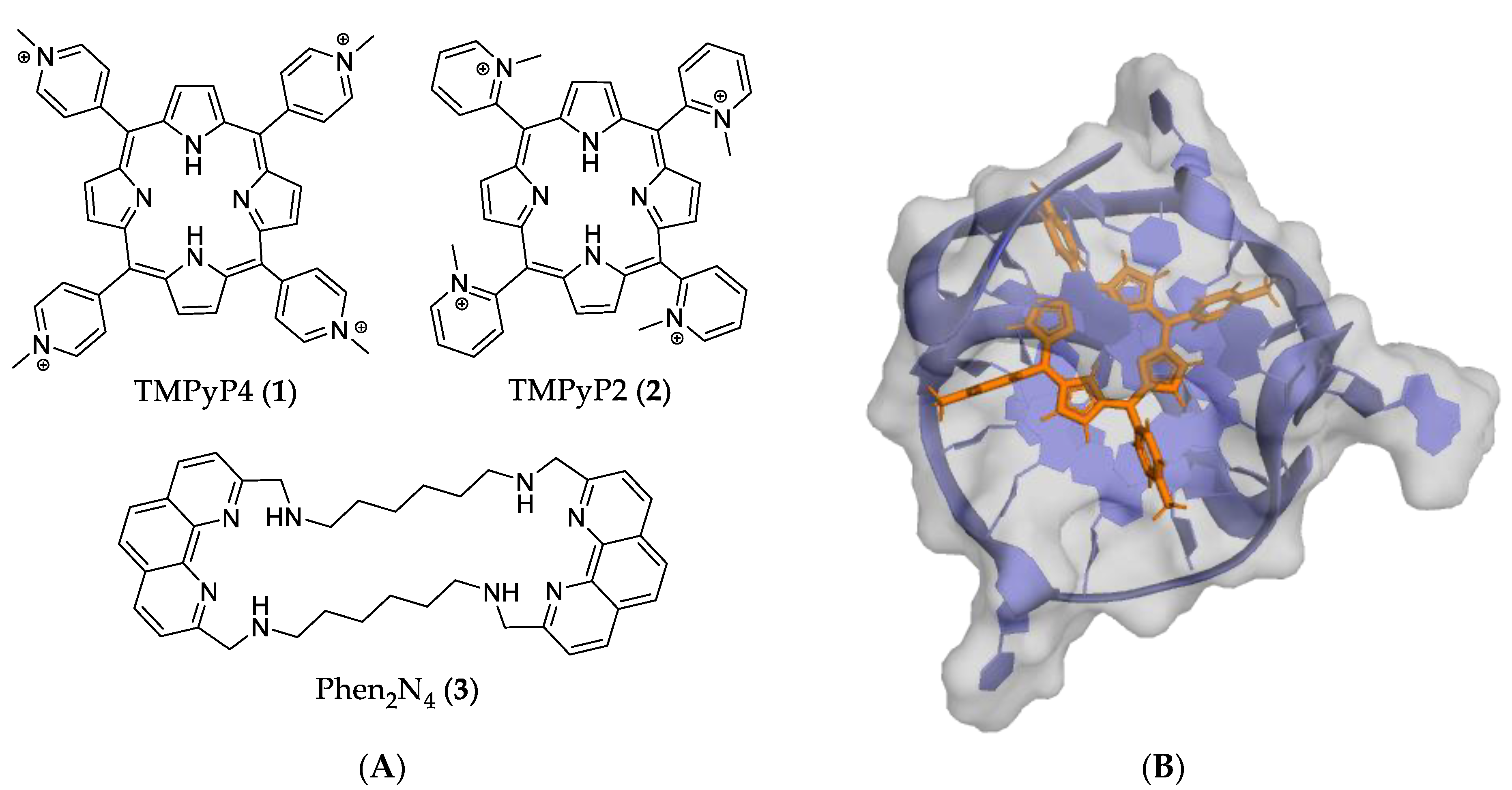
4.1. Macrocycles
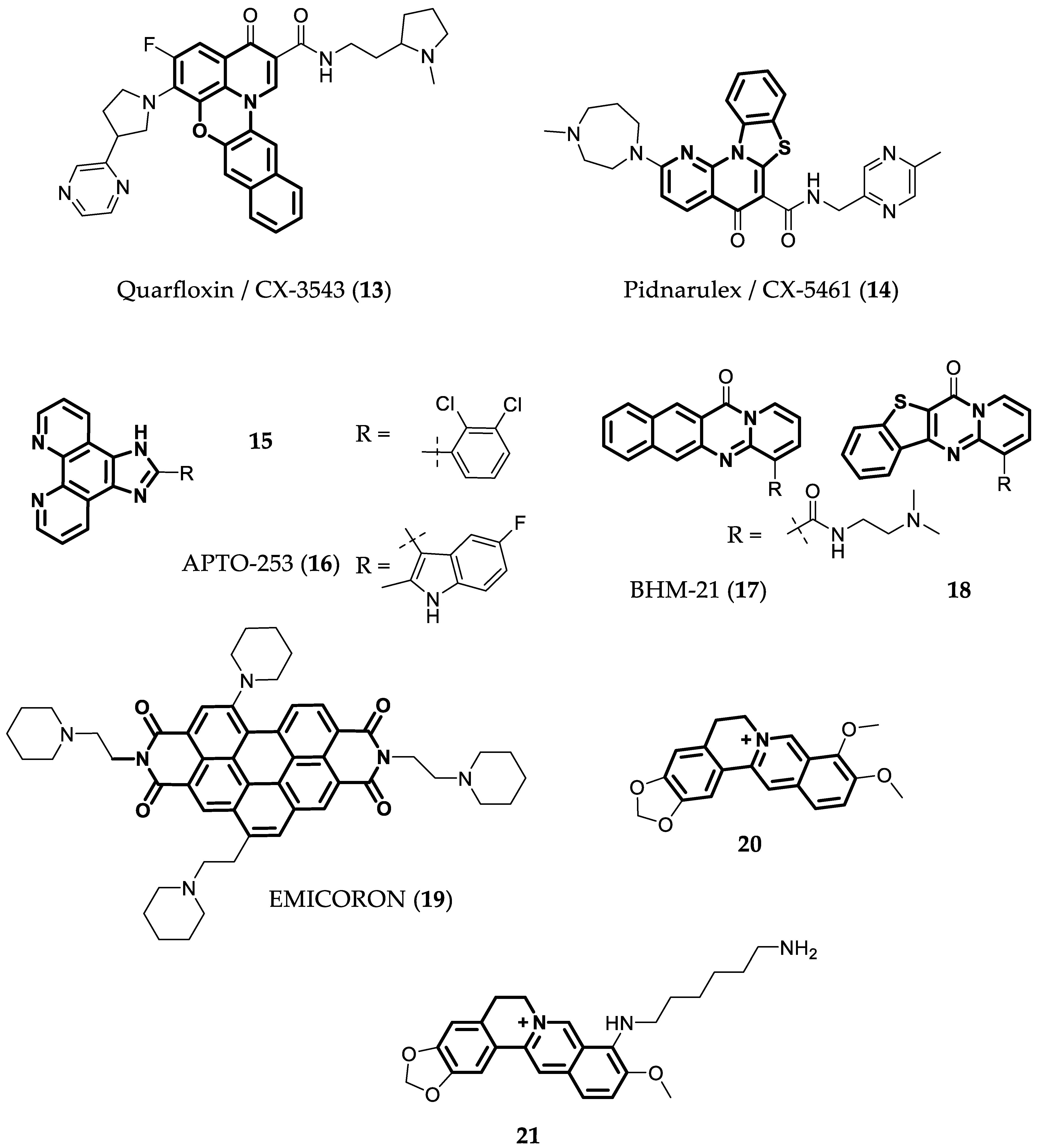
4.2. Ligands with Four or More Fused Aromatic Rings
4.3. Ligands with Three Fused Aromatic Rings
4.4. Ligands with Two Fused Aromatic Rings
4.5. Flexible G4 Ligands
4.6. Metal Complexes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- World Health Organization Estimated Number of New Cases in 2020. Available online: https://gco.iarc.fr/today/online-analysis-table?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&group_cancer=1&include_nmsc=0&include_nmsc_other=1 (accessed on 7 October 2022).
- Yahya, E.B.; Alqadhi, A.M. Recent Trends in Cancer Therapy: A Review on the Current State of Gene Delivery. Life Sci. 2021, 269, 119087. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Cancer. Available online: https://www.who.int/health-topics/cancer#tab=tab_1 (accessed on 7 October 2022).
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging Landscape of Oncogenic Signatures across Human Cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human C-Myc Onc Gene Is Located on the Region of Chromosome 8 That Is Translocated in Burkitt Lymphoma Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlström, T.; Henriksson, M.A. Impact of MYC in Regulation of Tumor Cell Metabolism. Biochim. Biophys. Acta 2015, 1849, 563–569. [Google Scholar] [CrossRef]
- Rohban, S.; Campaner, S. Myc Induced Replicative Stress Response: How to Cope with It and Exploit It. Biochim. Biophys. Acta 2015, 1849, 517–524. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; León, J. Myc and Cell Cycle Control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Tu, W.B.; Helander, S.; Pilstål, R.; Hickman, K.A.; Lourenco, C.; Jurisica, I.; Raught, B.; Wallner, B.; Sunnerhagen, M.; Penn, L.Z. Myc and Its Interactors Take Shape. Biochim. Biophys. Acta 2015, 1849, 469–483. [Google Scholar] [CrossRef]
- Chen, B.-J.; Wu, Y.-L.; Tanaka, Y.; Zhang, W. Small Molecules Targeting C-Myc Oncogene: Promising Anti-Cancer Therapeutics. Int. J. Biol. Sci. 2014, 10, 1084–1096. [Google Scholar] [CrossRef]
- Wolf, E.; Eilers, M. Targeting MYC Proteins for Tumor Therapy. Annu. Rev. Cancer Biol. 2020, 4, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct. Target. Ther. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a Target for Cancer Treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Nakanishi, C.; Seimiya, H. G-Quadruplex in Cancer Biology and Drug Discovery. Biochem. Biophys. Res. Commun. 2020, 531, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Raguseo, F.; Nuccio, S.P.; Liano, D.; Di Antonio, M. DNA G-Quadruplex Structures: More than Simple Roadblocks to Transcription? Nucleic Acids Res. 2021, 49, 8419–8431. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, Topology and Structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-Quadruplexes: A Promising Target for Cancer Therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef]
- Tan, J.; Lan, L. The DNA Secondary Structures at Telomeres and Genome Instability. Cell Biosci. 2020, 10, 47. [Google Scholar] [CrossRef]
- Ma, Y.; Iida, K.; Nagasawa, K. Topologies of G-Quadruplex: Biological Functions and Regulation by Ligands. Biochem. Biophys. Res. Commun. 2020, 531, 3–17. [Google Scholar] [CrossRef]
- Mendes, E.; Aljnadi, I.M.; Bahls, B.; Victor, B.L.; Paulo, A. Major Achievements in the Design of Quadruplex-Interactive Small Molecules. Pharmaceuticals 2022, 15, 300. [Google Scholar] [CrossRef]
- Mendes, E.; Bahls, B.; Aljnadi, I.M.; Paulo, A. Indoloquinolines as Scaffolds for the Design of Potent G-Quadruplex Ligands. Bioorg. Med. Chem. Lett. 2022, 72, 128862. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.P.; Paulo, A. Oncogene Expression Modulation in Cancer Cell Lines by DNA G-Quadruplex-Interactive Small Molecules. Curr. Med. Chem. 2017, 24, 4873–4904. [Google Scholar] [CrossRef] [PubMed]
- Awadasseid, A.; Ma, X.; Wu, Y.; Zhang, W. G-Quadruplex Stabilization via Small-Molecules as a Potential Anti-Cancer Strategy. Biomed. Pharmacother. 2021, 139, 111550. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, E.; Zanin, I.; Terreri, M.; Richter, S.N. G-Quadruplex Targeting in the Fight against Viruses: An Update. Int. J. Mol. Sci. 2021, 22, 10984. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative Approaches to Target Myc for Cancer Treatment. Signal Transduct. Target. Ther. 2021, 6, 117. [Google Scholar] [CrossRef]
- Burton, R.A.; Mattila, S.; Taparowsky, E.J.; Post, C.B. B-Myc: N-Terminal Recognition of Myc Binding Proteins. Biochemistry 2006, 45, 9857–9865. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC Oncogene - the Grand Orchestrator of Cancer Growth and Immune Evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Ryan, K.M.; Birnie, G.D. Myc Oncogenes: The Enigmatic Family. Biochem. J. 1996, 314, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, K.; Yamana, H.; Kitanaka, C.; Mochizuki, T.; Kokubu, A.; Kuchino, Y. Differential Role of the JNK and P38 MAPK Pathway in C-Myc- and s-Myc-Mediated Apoptosis. Biochem. Biophys. Res. Commun. 2000, 267, 221–227. [Google Scholar] [CrossRef]
- González, V.; Hurley, L.H. The C-MYC NHE III(1): Function and Regulation. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 111–129. [Google Scholar] [CrossRef]
- Wang, W.; Hu, S.; Gu, Y.; Yan, Y.; Stovall, D.B.; Li, D.; Sui, G. Human MYC G-Quadruplex: From Discovery to a Cancer Therapeutic Target. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188410. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Thomas, S.D.; Murty, V.V.; Sedoris, K.J.; Miller, D.M. C-Myc Quadruplex-Forming Sequence Pu-27 Induces Extensive Damage in Both Telomeric and Nontelomeric Regions of DNA. J. Biol. Chem. 2014, 289, 8521–8531. [Google Scholar] [CrossRef] [Green Version]
- Simonsson, T.; Pecinka, P.; Kubista, M. DNA Tetraplex Formation in the Control Region of C-Myc. Nucleic Acids Res. 1998, 26, 1167–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, H.; Wu, J.; Shao, F.; Yan, J. Stability and Kinetics of C-MYC Promoter G-Quadruplexes Studied by Single-Molecule Manipulation. J. Am. Chem. Soc. 2015, 137, 2424–2427. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct Evidence for a G-Quadruplex in a Promoter Region and Its Targeting with a Small Molecule to Repress c-MYC Transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [Green Version]
- Phan, A.T.; Modi, Y.S.; Patel, D.J. Propeller-Type Parallel-Stranded G-Quadruplexes in the Human c-Myc Promoter. J. Am. Chem. Soc. 2004, 126, 8710–8716. [Google Scholar] [CrossRef] [Green Version]
- Brázda, V.; Hároníková, L.; Liao, J.C.C.; Fojta, M. DNA and RNA Quadruplex-Binding Proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [Green Version]
- González, V.; Guo, K.; Hurley, L.; Sun, D. Identification and Characterization of Nucleolin as a C-Myc G-Quadruplex-Binding Protein. J. Biol. Chem. 2009, 284, 23622–23635. [Google Scholar] [CrossRef] [Green Version]
- Postel, E.H.; Berberich, S.J.; Flint, S.J.; Ferrone, C.A. Human C-Myc Transcription Factor PuF Identified as Nm23-H2 Nucleoside Diphosphate Kinase, a Candidate Suppressor of Tumor Metastasis. Science 1993, 261, 478–480. [Google Scholar] [CrossRef]
- Fekete, A.; Kenesi, E.; Hunyadi-Gulyas, E.; Durgo, H.; Berko, B.; Dunai, Z.A.; Bauer, P.I. The Guanine-Quadruplex Structure in the Human c-Myc Gene’s Promoter Is Converted into B-DNA Form by the Human Poly(ADP-Ribose)Polymerase-1. PLoS ONE 2012, 7, e42690. [Google Scholar] [CrossRef]
- Cashman, D.J.; Buscaglia, R.; Freyer, M.W.; Dettler, J.; Hurley, L.H.; Lewis, E.A. Molecular Modeling and Biophysical Analysis of the C-MYC NHE-III1 Silencer Element. J. Mol. Model. 2008, 14, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Ohnmacht, S.A.; Neidle, S. Small-Molecule Quadruplex-Targeted Drug Discovery. Bioorg. Med. Chem. Lett. 2014, 24, 2602–2612. [Google Scholar] [CrossRef]
- Neidle, S. Structured Waters Mediate Small Molecule Binding to G-Quadruplex Nucleic Acids. Pharmaceuticals 2021, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Paulo, A.; Castillo, C.C.; Neidle, S. Targeting Promoter Quadruplex Nucleic Acids for Cancer Therapy. In Comprehensive Medicinal Chemistry III; Elsevier: Amsterdam, The Netherlands, 2017; pp. 308–340. ISBN 9780128032015. [Google Scholar]
- Badran, H.M.; Eid, K.M.; Ammar, H.Y. DFT and TD-DFT Studies of Halogens Adsorption on Cobalt-Doped Porphyrin: Effect of the External Electric Field. Results Phys. 2021, 23, 103964. [Google Scholar] [CrossRef]
- Chou, J.-H.; Kosal, M.E.; Nalwa, H.S.; Rakow, N.A.; Suslick, K.S. Applications of Porphyrins and Metalloporphyrins to Materials Chemistry. ChemInform 2003, 34. [Google Scholar] [CrossRef]
- Auwärter, W.; Écija, D.; Klappenberger, F.; Barth, J.V. Porphyrins at Interfaces. Nat. Chem. 2015, 7, 105–120. [Google Scholar] [CrossRef]
- Stojiljkovic, I.; Evavold, B.D.; Kumar, V. Antimicrobial Properties of Porphyrins. Expert Opin. Investig. Drugs 2001, 10, 309–320. [Google Scholar] [CrossRef]
- Huang, H.; Song, W.; Rieffel, J.; Lovell, J.F. Emerging Applications of Porphyrins in Photomedicine. Front. Phys. 2015, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Phan, A.T.; Kuryavyi, V.; Gaw, H.Y.; Patel, D.J. Small-Molecule Interaction with a Five-Guanine-Tract G-Quadruplex Structure from the Human MYC Promoter. Nat. Chem. Biol. 2005, 1, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.C.; Ulven, T. Macrocyclic G-Quadruplex Ligands. Curr. Med. Chem. 2010, 17, 3438–3448. [Google Scholar] [CrossRef]
- Han, F.X.; Wheelhouse, R.T.; Hurley, L.H. Interactions of TMPyP4 and TMPyP2 with Quadruplex DNA. Structural Basis for the Differential Effects on Telomerase Inhibition. J. Am. Chem. Soc. 1999, 121, 3561–3570. [Google Scholar] [CrossRef]
- Grand, C.L.; Han, H.; Muñoz, R.M.; Weitman, S.; Von Hoff, D.D.; Hurley, L.H.; Bearss, D.J. The Cationic Porphyrin TMPyP4 Down-Regulates c-MYC and Human Telomerase Reverse Transcriptase Expression and Inhibits Tumor Growth in Vivo. Mol. Cancer Ther. 2002, 1, 565–573. [Google Scholar] [PubMed]
- Mikami-Terao, Y.; Akiyama, M.; Yuza, Y.; Yanagisawa, T.; Yamada, O.; Yamada, H. Antitumor Activity of G-Quadruplex-Interactive Agent TMPyP4 in K562 Leukemic Cells. Cancer Lett. 2008, 261, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Ruan, T.L.; Davis, S.J.; Powell, B.M.; Harbeck, C.P.; Habdas, J.; Habdas, P.; Yatsunyk, L.A. Lowering the Overall Charge on TMPyP4 Improves Its Selectivity for G-Quadruplex DNA. Biochimie 2017, 132, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; Quintela, T.; Gueddouda, N.M.; Bourdoncle, A.; Mergny, J.-L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. Phenanthroline Polyazamacrocycles as G-Quadruplex DNA Binders. Org. Biomol. Chem. 2018, 16, 2776–2786. [Google Scholar] [CrossRef]
- Cruz, C.; Cairrao, E.; Silvestre, S.; Breitenfeld, L.; Almeida, P.; Queiroz, J.A. Targeting of Mitochondria-Endoplasmic Reticulum by Fluorescent Macrocyclic Compounds. PLoS ONE 2011, 6, e27078. [Google Scholar] [CrossRef]
- Riechert-Krause, F.; Weisz, K. Indoloquinolines as DNA Binding Ligands. Heterocycl. Comm. 2013, 19, 145–166. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Chen, A.-C.; Yin, Q.-K.; Li, Z.; Huang, S.-M.; Du, G.; He, J.-H.; Zan, L.-P.; Wang, S.-K.; Xu, Y.-H.; et al. New Disubstituted Quindoline Derivatives Inhibiting Burkitt’s Lymphoma Cell Proliferation by Impeding c-MYC Transcription. J. Med. Chem. 2017, 60, 5438–5454. [Google Scholar] [CrossRef]
- Vianney, Y.M.; Weisz, K. Indoloquinoline Ligands Favor Intercalation at Quadruplex-Duplex Interfaces. Chemistry 2022, 28, e202103718. [Google Scholar] [CrossRef]
- Ou, T.-M.; Lin, J.; Lu, Y.-J.; Hou, J.-Q.; Tan, J.-H.; Chen, S.-H.; Li, Z.; Li, Y.-P.; Li, D.; Gu, L.-Q.; et al. Inhibition of Cell Proliferation by Quindoline Derivative (SYUIQ-05) through Its Preferential Interaction with c-Myc Promoter G-Quadruplex. J. Med. Chem. 2011, 54, 5671–5679. [Google Scholar] [CrossRef]
- Boddupally, P.V.L.; Hahn, S.; Beman, C.; De, B.; Brooks, T.A.; Gokhale, V.; Hurley, L.H. Anticancer Activity and Cellular Repression of C-MYC by the G-Quadruplex-Stabilizing 11-Piperazinylquindoline Is Not Dependent on Direct Targeting of the G-Quadruplex in the c-MYC Promoter. J. Med. Chem. 2012, 55, 6076–6086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, D.-Y.; Kuang, G.-T.; Wang, S.-K.; Peng, W.; Lin, S.-L.; Zhang, Q.; Su, X.-X.; Hu, M.-H.; Wang, H.; Tan, J.-H.; et al. Discovery of Novel 11-Triazole Substituted Benzofuro[3,2-b]Quinolone Derivatives as c-Myc G-Quadruplex Specific Stabilizers via Click Chemistry. J. Med. Chem. 2017, 60, 5407–5423. [Google Scholar] [CrossRef] [PubMed]
- Funke, A.; Dickerhoff, J.; Weisz, K. Towards the Development of Structure-Selective G-Quadruplex-Binding Indolo[3,2-b]Quinolines. Chemistry 2016, 22, 3170–3181. [Google Scholar] [CrossRef]
- Funke, A.; Karg, B.; Dickerhoff, J.; Balke, D.; Müller, S.; Weisz, K. Ligand-Induced Dimerization of a Truncated Parallel MYC G-Quadruplex. Chembiochem 2018, 19, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.J.; Turnidge, M.A.; Wai, D.H.; Patel, A.R.; Lee, D.W.; Gokhale, V.; Hurley, L.H.; Arceci, R.J.; Wetmore, C.; Azorsa, D.O. In Vitro Activity of a G-Quadruplex-Stabilizing Small Molecule That Synergizes with Navitoclax to Induce Cytotoxicity in Acute Myeloid Leukemia Cells. BMC Cancer 2019, 19, 1251. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration That Drug-Targeted down-Regulation of MYC in Non-Hodgkins Lymphoma Is Directly Mediated through the Promoter G-Quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef] [Green Version]
- Mohamad Anuar, N.N.; Nor Hisam, N.S.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 564108. [Google Scholar] [CrossRef]
- Kaiser, C.E.; Gokhale, V.; Yang, D.; Hurley, L.H. Gaining Insights into the Small Molecule Targeting of the G-Quadruplex in the c-MYC Promoter Using NMR and an Allele-Specific Transcriptional Assay. Top. Curr. Chem. 2013, 330, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Drlica, K. Mechanism of Fluoroquinolone Action. Curr. Opin. Microbiol. 1999, 2, 504–508. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 Is a DNA G-Quadruplex Stabilizer with Selective Lethality in BRCA1/2 Deficient Tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.-Y.; Jiang, Z.-Z.; Guo, M.; Tan, X.-Z.; Chen, F.; Xi, X.-G.; Xu, Y. G-Quadruplex DNA: A Novel Target for Drug Design. Cell. Mol. Life Sci. 2021, 78, 6557–6583. [Google Scholar] [CrossRef] [PubMed]
- Knox, J.J.; Oza, A.M.; Provencher, D.M.; Soong, J.; McCormick, D.; Chen, J.; Chen, J.; Chang, A. Phase 1b Expansion Study of CX-5461 in Patients with Solid Tumors and BRCA2 and/or PALB2 Mutation. J. Clin. Oncol. 2022, 40, TPS622. [Google Scholar] [CrossRef]
- Pharmaceuticals, C. Quarfloxin in Patients with Low to Intermediate Grade Neuroendocrine Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00780663 (accessed on 10 July 2022).
- Singh, M.; Gupta, R.; Comez, L.; Paciaroni, A.; Rani, R.; Kumar, V. BCL2 G Quadruplex-Binding Small Molecules: Current Status and Prospects for the Development of next-Generation Anticancer Therapeutics. Drug Discov. Today 2022, 27, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer Activity of CX-3543: A Direct Inhibitor of RRNA Biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [Green Version]
- Hilton, J.; Gelmon, K.; Bedard, P.L.; Tu, D.; Xu, H.; Tinker, A.V.; Goodwin, R.; Laurie, S.A.; Jonker, D.; Hansen, A.R.; et al. Results of the Phase I CCTG IND.231 Trial of CX-5461 in Patients with Advanced Solid Tumors Enriched for DNA-Repair Deficiencies. Nat. Commun. 2022, 13, 3607. [Google Scholar] [CrossRef]
- Xu, H.; Hurley, L.H. A First-in-Class Clinical G-Quadruplex-Targeting Drug. The Bench-to-Bedside Translation of the Fluoroquinolone QQ58 to CX-5461 (Pidnarulex). Bioorg. Med. Chem. Lett. 2022, 77, 129016. [Google Scholar] [CrossRef]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The Primary Mechanism of Cytotoxicity of the Chemotherapeutic Agent CX-5461 Is Topoisomerase II Poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef]
- Wu, Q.; Song, Y.; Liu, R.; Wang, R.; Mei, W.; Chen, W.; Yang, H.; Wang, X. Synthesis, Docking Studies and Antitumor Activity of Phenanthroimidazole Derivatives as Promising c-Myc G-Quadruplex DNA Stabilizers. Bioorg. Chem. 2020, 102, 104074. [Google Scholar] [CrossRef]
- Aptose Biosciences Inc. A Study of APTO-253 in Patients with Relapsed or Refractory AML or MDS. Available online: https://clinicaltrials.gov/ct2/show/NCT02267863 (accessed on 10 July 2022).
- Ohanian, M.; Arellano, M.L.; Levy, M.Y.; O’Dwyer, K.; Babiker, H.; Mahadevan, D.; Zhang, H.; Rastgoo, N.; Jin, Y.; Marango, J.; et al. A Phase 1a/b Dose Escalation Study of the MYC Repressor Apto-253 in Patients with Relapsed or Refractory AML or High-Risk MDS. Blood 2021, 138, 3411. [Google Scholar] [CrossRef]
- Cercek, A.; Wheler, J.; Murray, P.E.; Zhou, S.; Saltz, L. Phase 1 Study of APTO-253 HCl, an Inducer of KLF4, in Patients with Advanced or Metastatic Solid Tumors. Investig. New Drugs 2015, 33, 1086–1092. [Google Scholar] [CrossRef]
- Local, A.; Zhang, H.; Benbatoul, K.D.; Folger, P.; Sheng, X.; Tsai, C.-Y.; Howell, S.B.; Rice, W.G. APTO-253 Stabilizes G-Quadruplex DNA, Inhibits MYC Expression, and Induces DNA Damage in Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2018, 17, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, L.; Mazzini, S.; Rossini, A.; Castagnoli, L.; Scaglioni, L.; Artali, R.; Di Nicola, M.; Zunino, F.; Dallavalle, S. C-MYC G-Quadruplex Binding by the RNA Polymerase I Inhibitor BMH-21 and Analogues Revealed by a Combined NMR and Biochemical Approach. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Micheli, E.; Altieri, A.; Cianni, L.; Cingolani, C.; Iachettini, S.; Bianco, A.; Leonetti, C.; Cacchione, S.; Biroccio, A.; Franceschin, M.; et al. Perylene and Coronene Derivatives Binding to G-Rich Promoter Oncogene Sequences Efficiently Reduce Their Expression in Cancer Cells. Biochimie 2016, 125, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Porru, M.; Zizza, P.; Franceschin, M.; Leonetti, C.; Biroccio, A. EMICORON: A Multi-Targeting G4 Ligand with a Promising Preclinical Profile. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1362–1370. [Google Scholar] [CrossRef]
- Dickerhoff, J.; Brundridge, N.; McLuckey, S.A.; Yang, D. Berberine Molecular Recognition of the Parallel MYC G-Quadruplex in Solution. J. Med. Chem. 2021, 64, 16205–16212. [Google Scholar] [CrossRef]
- Wen, L.; Han, Z.; Li, J.; Du, Y. C-MYC and HIF1α Promoter G-Quadruplexes Dependent Metabolic Regulation Mechanism of Berberine in Colon Cancer. J. Gastrointest. Oncol. 2022, 13, 1152–1168. [Google Scholar] [CrossRef]
- Ma, Y.; Ou, T.-M.; Hou, J.-Q.; Lu, Y.-J.; Tan, J.-H.; Gu, L.-Q.; Huang, Z.-S. 9-N-Substituted Berberine Derivatives: Stabilization of G-Quadruplex DNA and down-Regulation of Oncogene c-Myc. Bioorg. Med. Chem. 2008, 16, 7582–7591. [Google Scholar] [CrossRef]
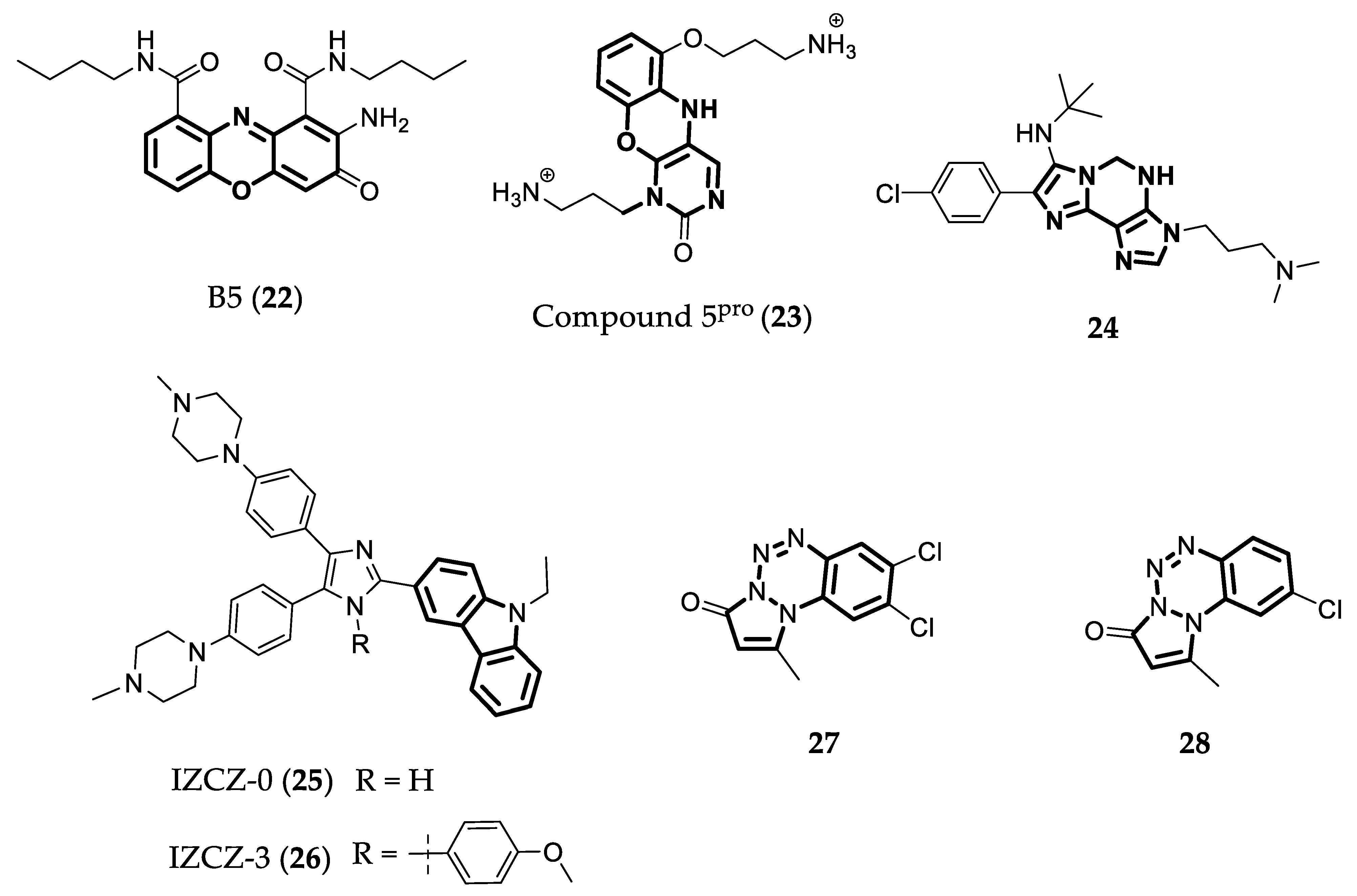
- Tsvetkov, V.B.; Varizhuk, A.M.; Lizunova, S.A.; Nikolenko, T.A.; Ivanov, I.A.; Severov, V.V.; Belyaev, E.S.; Shitikov, E.A.; Pozmogova, G.E.; Aralov, A.V. Phenoxazine-Based Scaffold for Designing G4-Interacting Agents. Org. Biomol. Chem. 2020, 18, 6147–6154. [Google Scholar] [CrossRef]
- Tsvetkov, V.B.; Turaev, A.V.; Petrunina, N.A.; Melnik, D.M.; Khodarovich, Y.M.; Pozmogova, G.E.; Zatsepin, T.S.; Varizhuk, A.M.; Aralov, A.V. Phenoxazine Pseudonucleotides in DNA I-Motifs Allow Precise Profiling of Small Molecule Binders by Fluorescence Monitoring. Analyst 2021, 146, 4436–4440. [Google Scholar] [CrossRef]
- Kang, H.-J.; Park, H.-J. In Silico Identification of Novel Ligands for G-Quadruplex in the c-MYC Promoter. J. Comput. Aided Mol. Des. 2015, 29, 339–348. [Google Scholar] [CrossRef]
- Pelliccia, S.; Amato, J.; Capasso, D.; Di Gaetano, S.; Massarotti, A.; Piccolo, M.; Irace, C.; Tron, G.C.; Pagano, B.; Randazzo, A.; et al. Bio-Inspired Dual-Selective BCL-2/c-MYC G-Quadruplex Binders: Design, Synthesis, and Anticancer Activity of Drug-like Imidazo[2,1-i]Purine Derivatives. J. Med. Chem. 2020, 63, 2035–2050. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-H.; Wang, Y.-Q.; Yu, Z.-Y.; Hu, L.-N.; Ou, T.-M.; Chen, S.-B.; Huang, Z.-S.; Tan, J.-H. Discovery of a New Four-Leaf Clover-like Ligand as a Potent c-MYC Transcription Inhibitor Specifically Targeting the Promoter G-Quadruplex. J. Med. Chem. 2018, 61, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Mulliri, S.; Laaksonen, A.; Spanu, P.; Farris, R.; Farci, M.; Mingoia, F.; Roviello, G.N.; Mocci, F. Spectroscopic and in Silico Studies on the Interaction of Substituted Pyrazolo[1,2-a]Benzo[1,2,3,4]Tetrazine-3-One Derivatives with c-Myc G4-DNA. Int. J. Mol. Sci. 2021, 22, 6028. [Google Scholar] [CrossRef] [PubMed]
- Mingoia, F.; Di Sano, C.; Di Blasi, F.; Fazzari, M.; Martorana, A.; Almerico, A.M.; Lauria, A. Exploring the Anticancer Potential of Pyrazolo[1,2-a]Benzo[1,2,3,4]Tetrazin-3-One Derivatives: The Effect on Apoptosis Induction, Cell Cycle and Proliferation. Eur. J. Med. Chem. 2013, 64, 345–356. [Google Scholar] [CrossRef]
- Das, T.; Panda, D.; Saha, P.; Dash, J. Small Molecule Driven Stabilization of Promoter G-Quadruplexes and Transcriptional Regulation of c-MYC. Bioconjug. Chem. 2018, 29, 2636–2645. [Google Scholar] [CrossRef]
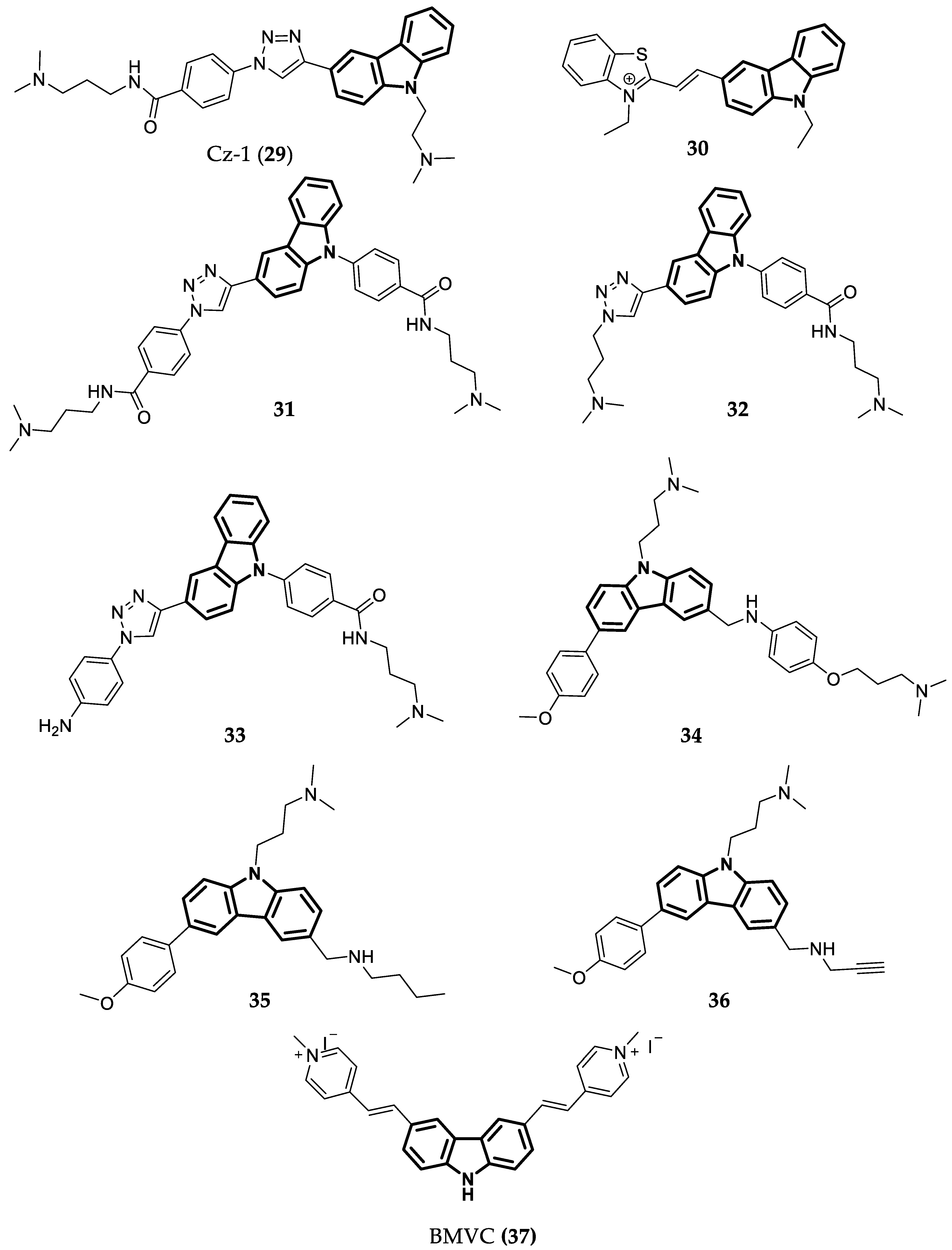
- Głuszyńska, A.; Juskowiak, B.; Kuta-Siejkowska, M.; Hoffmann, M.; Haider, S. Carbazole Ligands as C-Myc G-Quadruplex Binders. Int. J. Biol. Macromol. 2018, 114, 479–490. [Google Scholar] [CrossRef]
- Panda, D.; Saha, P.; Das, T.; Dash, J. Target Guided Synthesis Using DNA Nano-Templates for Selectively Assembling a G-Quadruplex Binding c-MYC Inhibitor. Nat. Commun. 2017, 8, 16103. [Google Scholar] [CrossRef]
- Jana, S.; Panda, D.; Saha, P.; Pantos, G.D.; Dash, J. Dynamic Generation of G-Quadruplex DNA Ligands by Target-Guided Combinatorial Chemistry on a Magnetic Nanoplatform. J. Med. Chem. 2019, 62, 762–773. [Google Scholar] [CrossRef]
- Liu, W.; Lin, C.; Wu, G.; Dai, J.; Chang, T.-C.; Yang, D. Structures of 1:1 and 2:1 Complexes of BMVC and MYC Promoter G-Quadruplex Reveal a Mechanism of Ligand Conformation Adjustment for G4-Recognition. Nucleic Acids Res. 2019, 47, 11931–11942. [Google Scholar] [CrossRef]
- Wang, D.; Gao, F. Quinazoline Derivatives: Synthesis and Bioactivities. Chem. Cent. J. 2013, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Theivendren, P. &.; Kumar, P. Quinazoline Marketed Drugs—A Review. Res. Pharm. 2011, 1, 1–21. [Google Scholar]
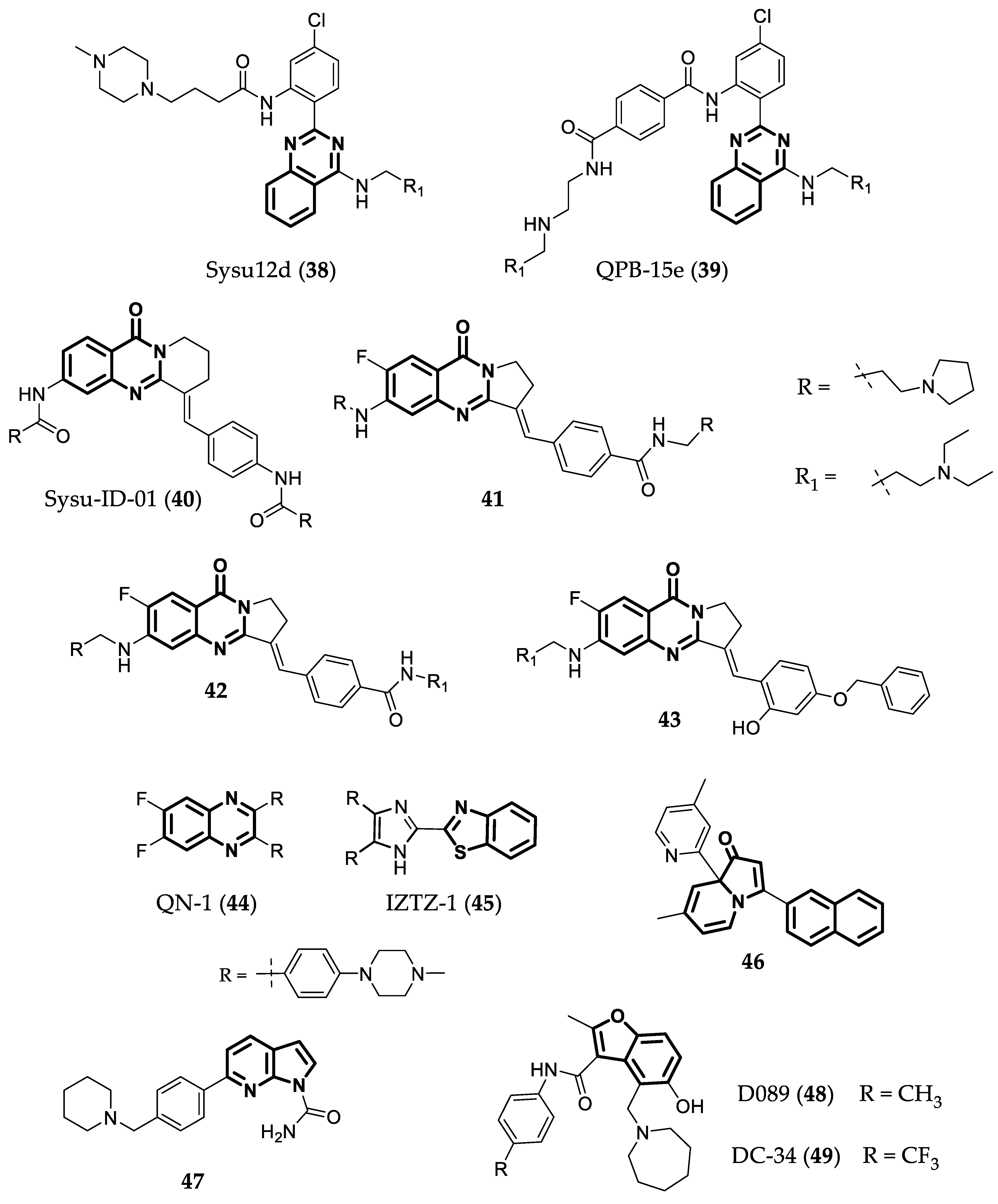
- Su, L.; Zheng, H.; Li, Z.; Qiu, J.; Chen, S.; Liu, J.; Ou, T.-M.; Tan, J.-H.; Gu, L.-Q.; Huang, Z.-S.; et al. Mechanistic Studies on the Anticancer Activity of 2,4-Disubstituted Quinazoline Derivative. Biochim. Biophys. Acta 2014, 1840, 3123–3130. [Google Scholar] [CrossRef] [PubMed]
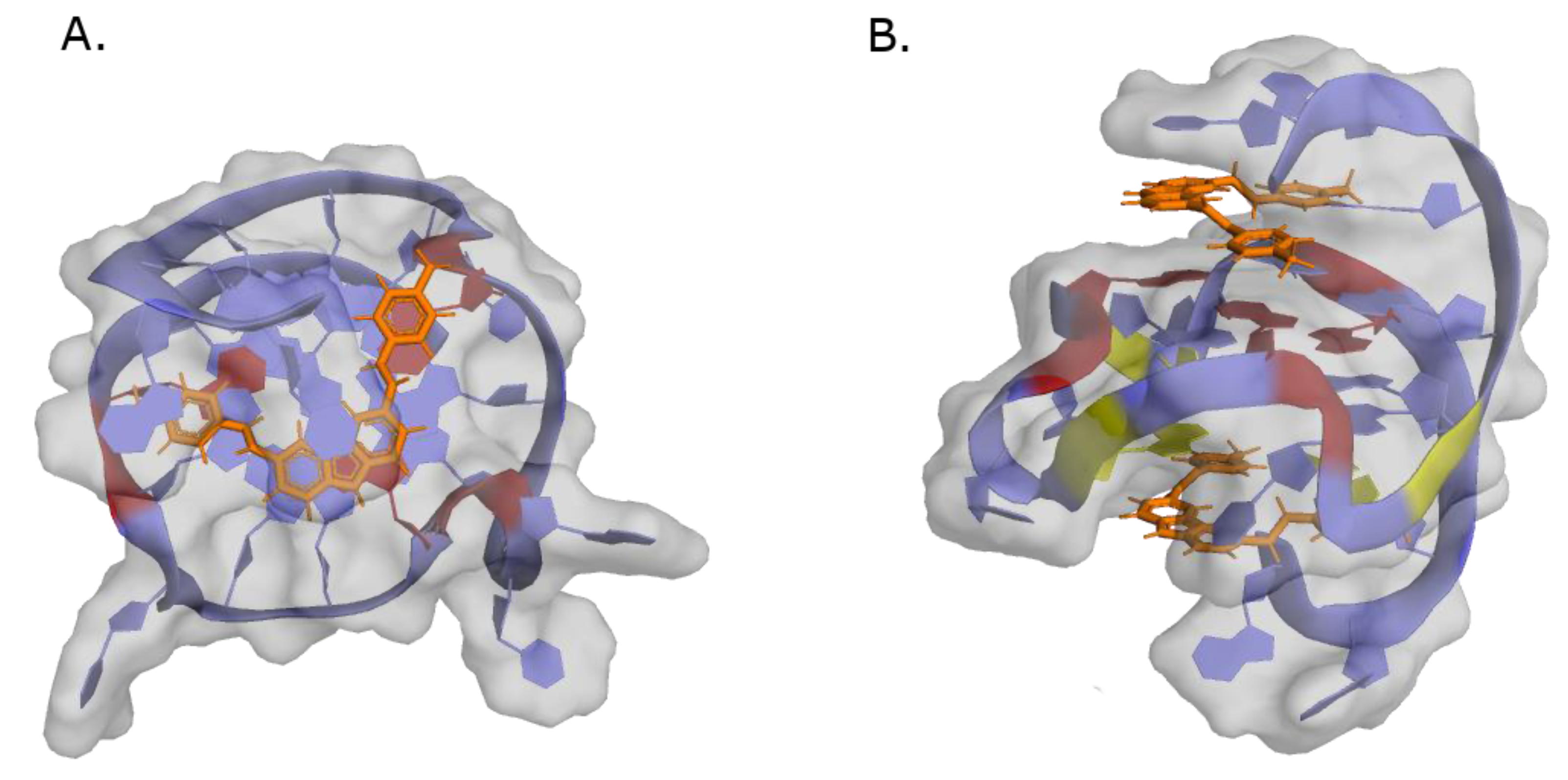
- Li, Z.; Liu, C.; Huang, C.; Meng, X.; Zhang, L.; He, J.; Li, J. Quinazoline Derivative QPB-15e Stabilizes the c-Myc Promoter G-Quadruplex and Inhibits Tumor Growth in Vivo. Oncotarget 2016, 7, 34266–34276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, C.; Lin, J.; Hou, J.-Q.; Liu, H.-Y.; Chen, S.-B.; Chen, A.-C.; Ou, T.-M.; Tan, J.-H.; Li, D.; Gu, L.-Q.; et al. Chemical Intervention of the NM23-H2 Transcriptional Programme on c-MYC via a Novel Small Molecule. Nucleic Acids Res. 2015, 43, 6677–6691. [Google Scholar] [CrossRef] [Green Version]
- Shan, C.; Yan, J.-W.; Wang, Y.-Q.; Che, T.; Huang, Z.-L.; Chen, A.-C.; Yao, P.-F.; Tan, J.-H.; Li, D.; Ou, T.-M.; et al. Design, Synthesis, and Evaluation of Isaindigotone Derivatives to Downregulate c-Myc Transcription via Disrupting the Interaction of NM23-H2 with G-Quadruplex. J. Med. Chem. 2017, 60, 1292–1308. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Huang, Z.-L.; Chen, S.-B.; Wang, C.-X.; Shan, C.; Yin, Q.-K.; Ou, T.-M.; Li, D.; Gu, L.-Q.; Tan, J.-H.; et al. Design, Synthesis, and Evaluation of New Selective NM23-H2 Binders as c-MYC Transcription Inhibitors via Disruption of the NM23-H2/G-Quadruplex Interaction. J. Med. Chem. 2017, 60, 6924–6941. [Google Scholar] [CrossRef]
- Pereira, J.A.; Pessoa, A.M.; Cordeiro, M.N.D.S.; Fernandes, R.; Prudêncio, C.; Noronha, J.P.; Vieira, M. Quinoxaline, Its Derivatives and Applications: A State of the Art Review. Eur. J. Med. Chem. 2015, 97, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Tariq, S.; Somakala, K.; Amir, M. Quinoxaline: An Insight into the Recent Pharmacological Advances. Eur. J. Med. Chem. 2018, 143, 542–557. [Google Scholar] [CrossRef]
- Hu, M.-H.; Wu, T.-Y.; Huang, Q.; Jin, G. New Substituted Quinoxalines Inhibit Triple-Negative Breast Cancer by Specifically Downregulating the c-MYC Transcription. Nucleic Acids Res. 2019, 47, 10529–10542. [Google Scholar] [CrossRef]
- Wu, T.-Y.; Huang, Q.; Huang, Z.-S.; Hu, M.-H.; Tan, J.-H. A Drug-like Imidazole-Benzothiazole Conjugate Inhibits Malignant Melanoma by Stabilizing the c-MYC G-Quadruplex. Bioorg. Chem. 2020, 99, 103866. [Google Scholar] [CrossRef]
- Yang, F.; Sun, X.; Wang, L.; Li, Q.; Guan, A.; Shen, G.; Tang, Y. Selective Recognition of C-Myc Promoter G-Quadruplex and down-Regulation of Oncogene c-Myc Transcription in Human Cancer Cells by 3,8a-Disubstituted Indolizinone. RSC Adv. 2017, 7, 51965–51969. [Google Scholar] [CrossRef] [Green Version]
- Dallavalle, S.; Musso, L.; Artali, R.; Aviñó, A.; Scaglioni, L.; Eritja, R.; Gargallo, R.; Mazzini, S. G-Quadruplex Binding Properties of a Potent PARP-1 Inhibitor Derived from 7-Azaindole-1-Carboxamide. Sci. Rep. 2021, 11, 3869. [Google Scholar] [CrossRef] [PubMed]
- Cincinelli, R.; Musso, L.; Merlini, L.; Giannini, G.; Vesci, L.; Milazzo, F.M.; Carenini, N.; Perego, P.; Penco, S.; Artali, R.; et al. 7-Azaindole-1-Carboxamides as a New Class of PARP-1 Inhibitors. Bioorg. Med. Chem. 2014, 22, 1089–1103. [Google Scholar] [CrossRef]
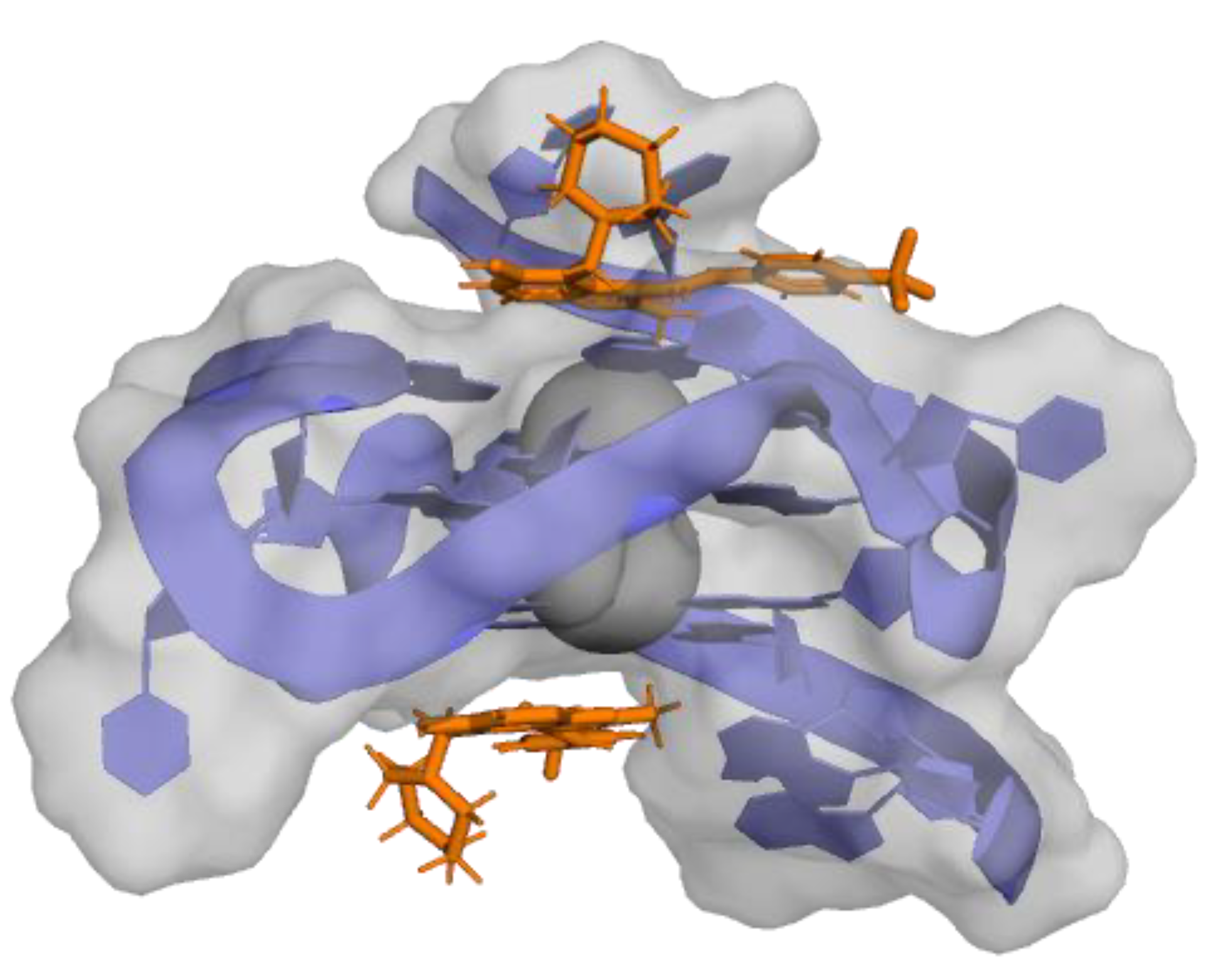
- Gaikwad, S.M.; Phyo, Z.; Arteaga, A.Q.; Gorjifard, S.; Calabrese, D.R.; Connors, D.; Huang, J.; Michalowski, A.M.; Zhang, S.; Liu, Z.-G.; et al. A Small Molecule Stabilizer of the MYC G4-Quadruplex Induces Endoplasmic Reticulum Stress, Senescence and Pyroptosis in Multiple Myeloma. Cancers 2020, 12, 2952. [Google Scholar] [CrossRef]
- Felsenstein, K.M.; Saunders, L.B.; Simmons, J.K.; Leon, E.; Calabrese, D.R.; Zhang, S.; Michalowski, A.; Gareiss, P.; Mock, B.A.; Schneekloth, J.S., Jr. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem. Biol. 2016, 11, 139–148. [Google Scholar] [CrossRef]
- Calabrese, D.R.; Chen, X.; Leon, E.C.; Gaikwad, S.M.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and Structural Studies Provide a Mechanistic Basis for Recognition of the MYC G-Quadruplex. Nat. Commun. 2018, 9, 4229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
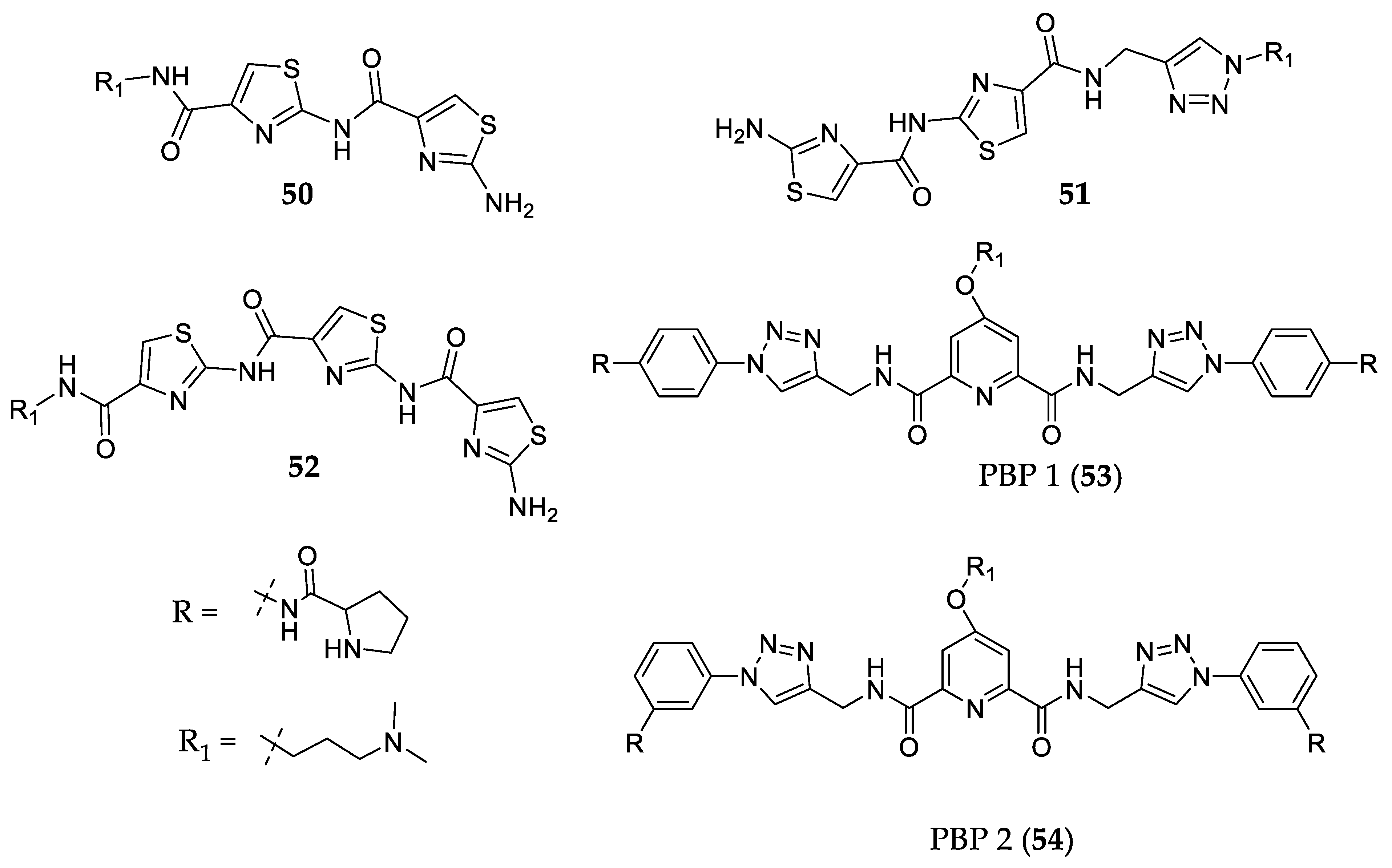
- Dutta, D.; Debnath, M.; Müller, D.; Paul, R.; Das, T.; Bessi, I.; Schwalbe, H.; Dash, J. Cell Penetrating Thiazole Peptides Inhibit C-MYC Expression via Site-Specific Targeting of c-MYC G-Quadruplex. Nucleic Acids Res. 2018, 46, 5355–5365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meunier, B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Debnath, M.; Ghosh, S.; Chauhan, A.; Paul, R.; Bhattacharyya, K.; Dash, J. Preferential Targeting of I-Motifs and G-Quadruplexes by Small Molecules. Chem. Sci. 2017, 8, 7448–7456. [Google Scholar] [CrossRef] [Green Version]
- Paul, R.; Das, T.; Debnath, M.; Chauhan, A.; Dash, J. G-Quadruplex-Binding Small Molecule Induces Synthetic Lethality in Breast Cancer Cells by Inhibiting c-MYC and BCL2 Expression. Chembiochem 2020, 21, 963–970. [Google Scholar] [CrossRef]
- Vilar, R. Interaction of Metal Complexes with G-Quadruplex DNA. In Medicinal Chemistry; Advances in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 2020; pp. 425–445. ISBN 9780128191965. [Google Scholar]
- Palma, E.; Carvalho, J.; Cruz, C.; Paulo, A. Metal-Based G-Quadruplex Binders for Cancer Theranostics. Pharmaceuticals 2021, 14, 605. [Google Scholar] [CrossRef] [PubMed]
- Gama, S.; Rodrigues, I.; Mendes, F.; Santos, I.C.; Gabano, E.; Klejevskaja, B.; Gonzalez-Garcia, J.; Ravera, M.; Vilar, R.; Paulo, A. Anthracene-Terpyridine Metal Complexes as New G-Quadruplex DNA Binders. J. Inorg. Biochem. 2016, 160, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-Z.; Qin, Q.-P.; Meng, T.; Deng, C.-X.; Liang, H.; Chen, Z.-F. 5-Bromo-Oxoisoaporphine Platinum(II) Complexes Exhibit Tumor Cell Cytotoxcicity via Inhibition of Telomerase Activity and Disruption of c-Myc G-Quadruplex DNA and Mitochondrial Functions. Eur. J. Med. Chem. 2018, 145, 360–369. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Meng, Z.; Xu, D.; Shao, F. Dual Functional Dinuclear Platinum Complex with Selective Reactivity towards C-Myc G-Quadruplex. Sci. Rep. 2018, 8, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding to c-MYC G4 | Anticancer Activity | |||||
|---|---|---|---|---|---|---|
| Group | N° | Strength (Kd, Ka, ΔTm) | Preference for c-MYC G4 (Yes/No) | In Vitro | In Vivo | Ref. |
| Macrocycles | 1 | No |
|
| [55,56] | |
| 3 | ΔTm = 17.2 °C | No |
| [58,59] | ||
| Ligands with four or more fused aromatic rings | 4 | Yes |
| [63] | ||
| 5 | ΔTm = 26.6 °C | __ |
|
| [61] | |
| 6 7 | ΔTm = 7 °C ΔTm = 17 °C | No |
| [64] | ||
| 8 9 10 | ΔTm = 22.0 °C ΔTm = 16.6 °C ΔTm = 13.7 °C | Yes |
|
| [65] | |
| 12 | ΔTm = 21 °C | __ |
| [69] | ||
| 13 | ΔTm > 15 °C | __ |
| Reached Phase II clinical trial. | [73,76] | |
| 14 | ΔTm > 15 °C | No |
|
| [81,131] | |
| 15 | ΔTm = 4.4 °C | __ |
|
| [82] | |
| 16 | - | No |
|
| [83,85,86] | |
| 17 18 | ΔTm ~ 10 °C | Yes |
| [87] | ||
| 19 | ΔTm = 16.4 °C | No |
|
| [88,89] | |
| 20 | ΔTm > 6 °C | No |
|
| [91] | |
| 21 | ΔTm = 29 °C | Yes |
| [92] | ||
| Ligands with three fused aromatic rings | 22 | Yes |
| - | [95] | |
| 24 | ΔTm = 12.8 °C | No |
| [96] | ||
| 26 | ΔTm = 20 °C | Yes |
|
| [97] | |
| 27 28 | ΔTm = 4 °C ΔTm = 1.9 °C | Yes |
| [98,99] | ||
| 29 | ΔTm= 15.8 °C | Yes |
| [100] | ||
| 31 | Kd = 0.17 µM | __ |
| [102] | ||
| 34 | ΔTm = 23.4 °C | __ |
| [103] | ||
| 37 | Kd = 36 nM | Yes |
| [104] | ||
| 38 | ΔTm = 15.0 °C | __ |
| [107] | ||
| Ligands with two fused aromatic rings | 39 | ΔTm = 23.7 °C | __ |
|
| [108] |
| 40 | ΔTm = 9 °C | __ |
| [109] | ||
| 41 42 | ΔTm = 12.1 °C ΔTm = 12.9 °C | __ |
|
| [110] | |
| 43 | Binding affinity to NM23-H2 protein (KD = 3.1 μM) | No |
|
| [111] | |
| 44 | KD = 1.3 μM | Yes |
|
| [114] | |
| 45 | ΔTm = 15 °C | Yes |
|
| [115] | |
| 46 | Ka = 9.9 × 105 M−1 | Yes |
| [116] | ||
| 47 | Ka = 106.1 M−1 | No |
|
| [117,118] | |
| 48 | Yes |
| [119,120] | |||
| 49 | ΔTm = 7.5 °C | Yes |
| [121] | ||
| Flexible ligands | 52 | ΔTm = 22 °C | Yes |
| [122] | |
| 53 54 | ΔTm = 15.0 °C ΔTm = 6.0 °C | No |
| - | [124,125] | |
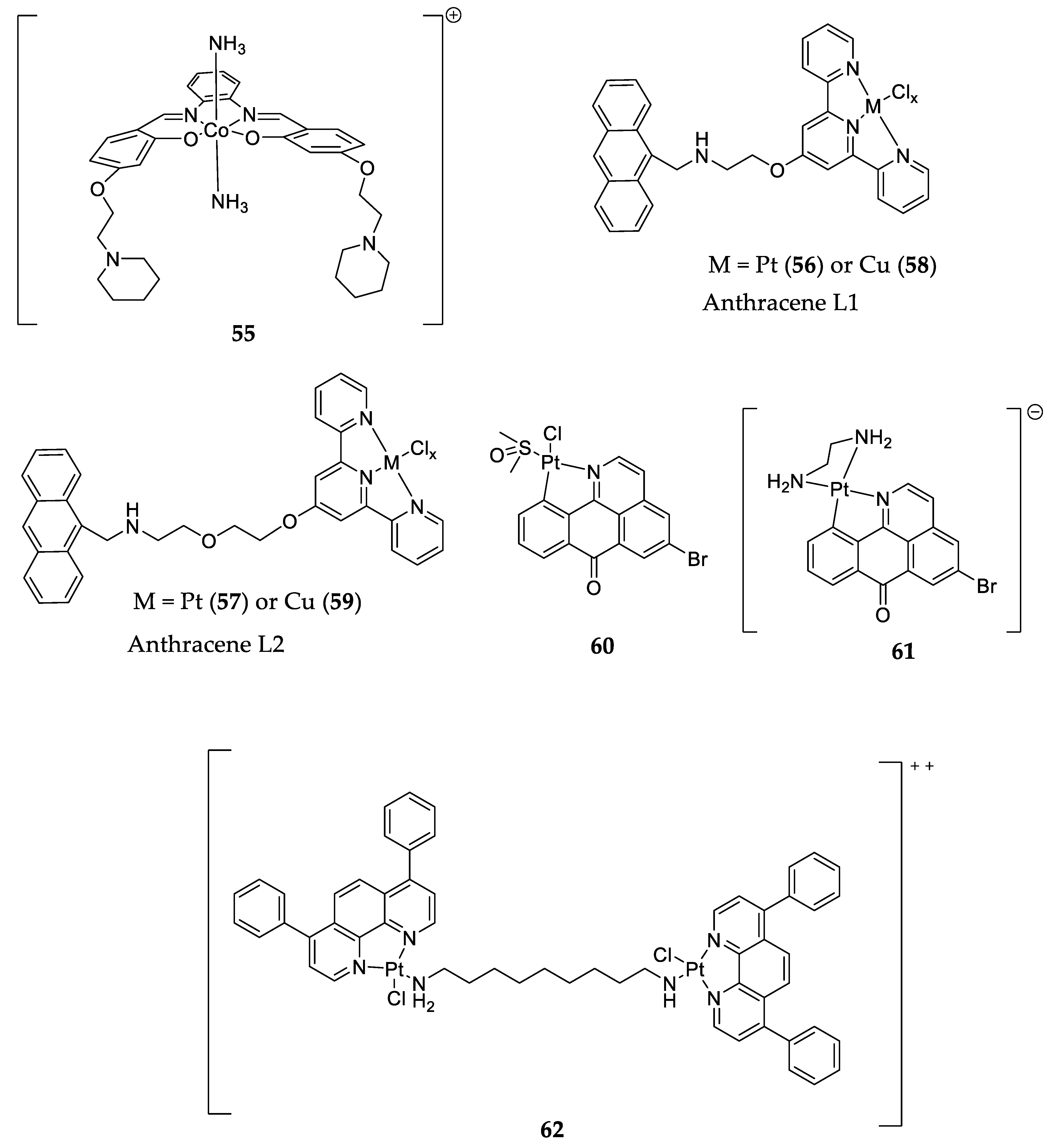
| Metal complexes | 56 57 58 59 | ΔTm = 10.2 °C ΔTm = 15.6 °C ΔTm = 2.0 °C ΔTm = 7.3 °C | No |
| [128] | |
| 60 61 | __ |
| [129] | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahls, B.; Aljnadi, I.M.; Emídio, R.; Mendes, E.; Paulo, A. G-Quadruplexes in c-MYC Promoter as Targets for Cancer Therapy. Biomedicines 2023, 11, 969. https://doi.org/10.3390/biomedicines11030969
Bahls B, Aljnadi IM, Emídio R, Mendes E, Paulo A. G-Quadruplexes in c-MYC Promoter as Targets for Cancer Therapy. Biomedicines. 2023; 11(3):969. https://doi.org/10.3390/biomedicines11030969
Chicago/Turabian StyleBahls, Bárbara, Israa M. Aljnadi, Rita Emídio, Eduarda Mendes, and Alexandra Paulo. 2023. "G-Quadruplexes in c-MYC Promoter as Targets for Cancer Therapy" Biomedicines 11, no. 3: 969. https://doi.org/10.3390/biomedicines11030969