
Development of an Animal Model for Traumatic Brain Injury Augmentation of Heterotopic Ossification in Response to Local Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mild Traumatic Brain Injury (mTBI)
2.2. HO Model
2.3. Dual X-ray Absroptiometry
2.4. Micro Computed Tomography (µ-CT)
2.5. Histology
2.6. HO Induction and IOX2 Treatment
2.7. Pericytes Cultures
2.8. mTBI Serum and Osteogenic Activity
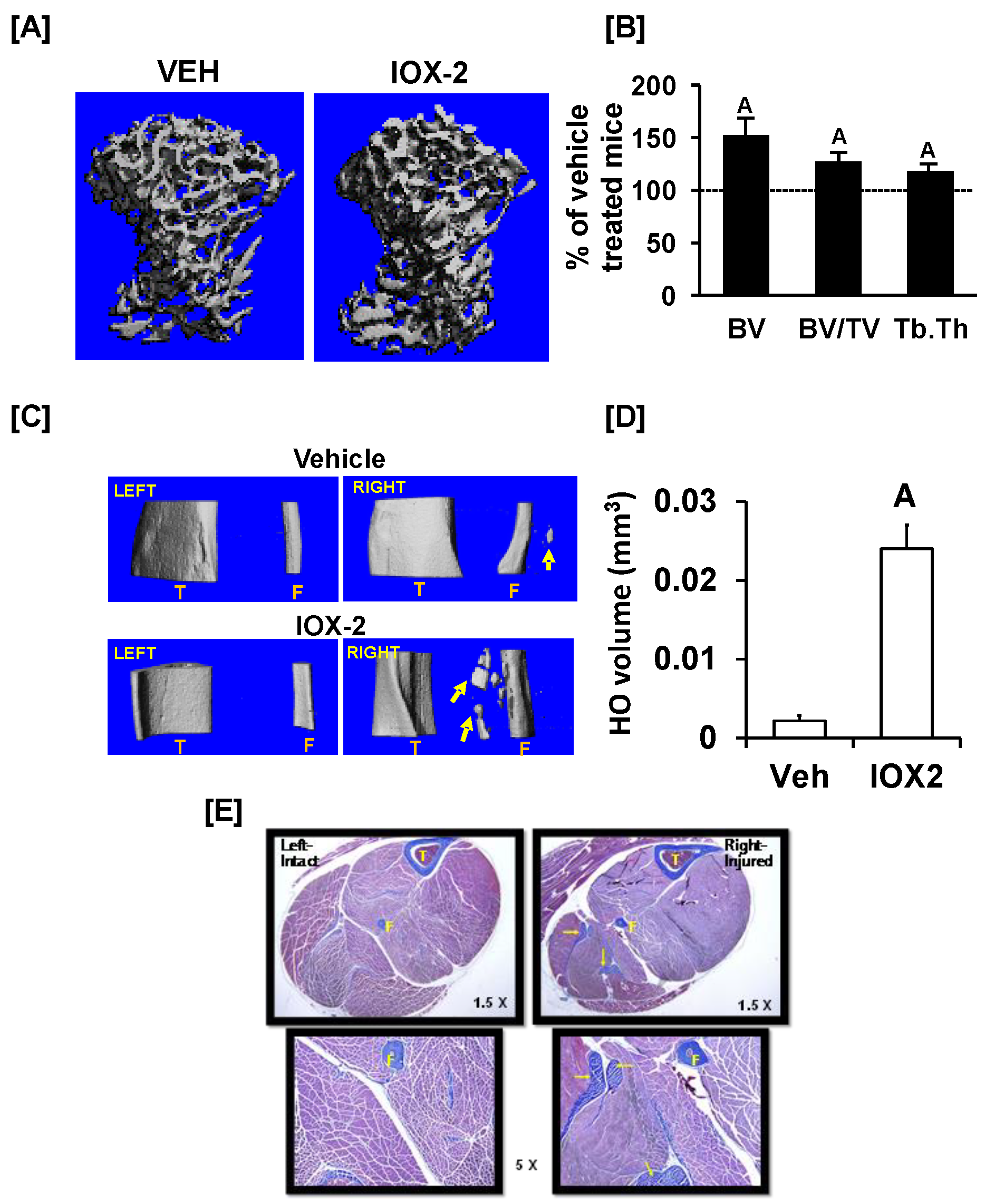
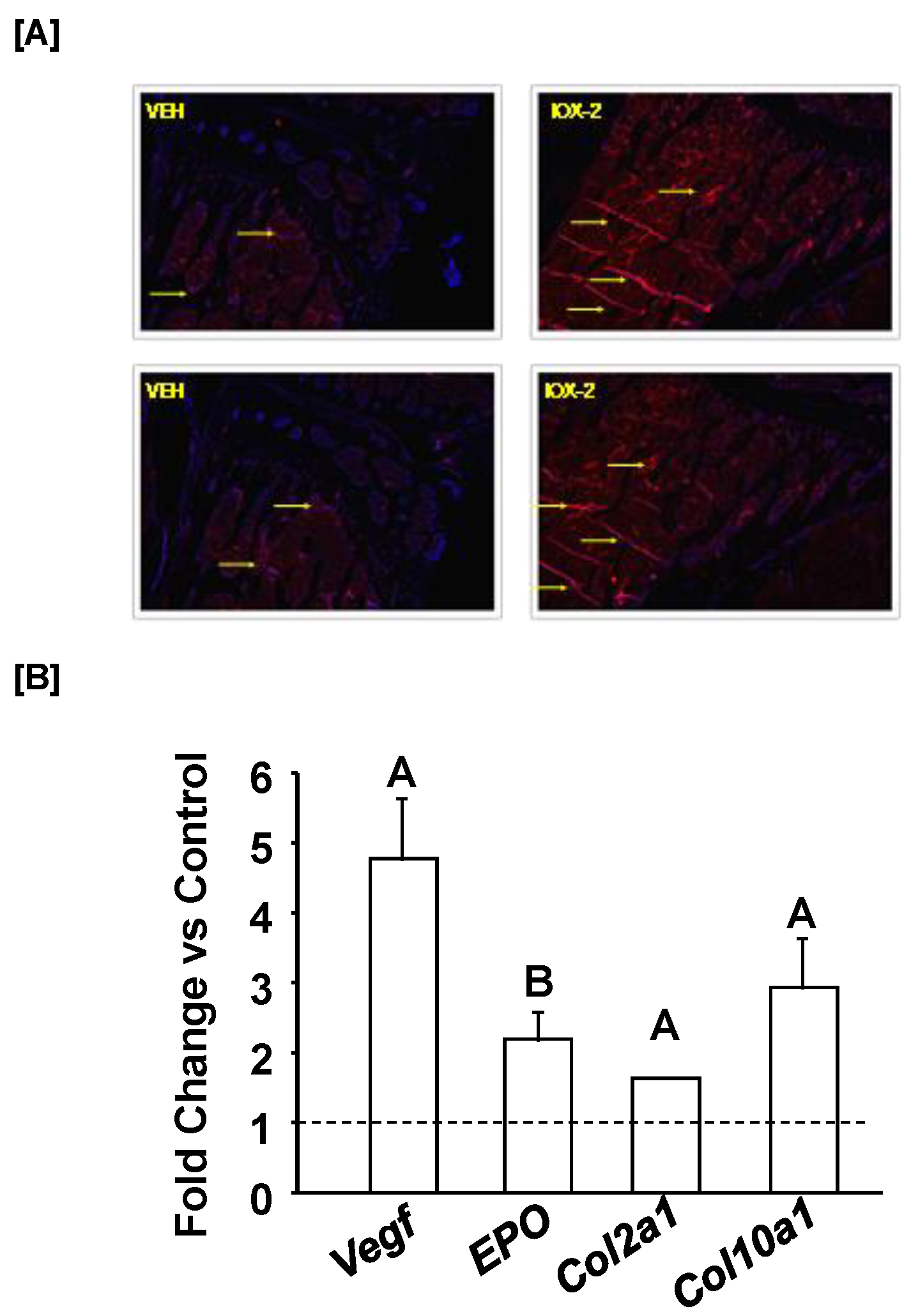
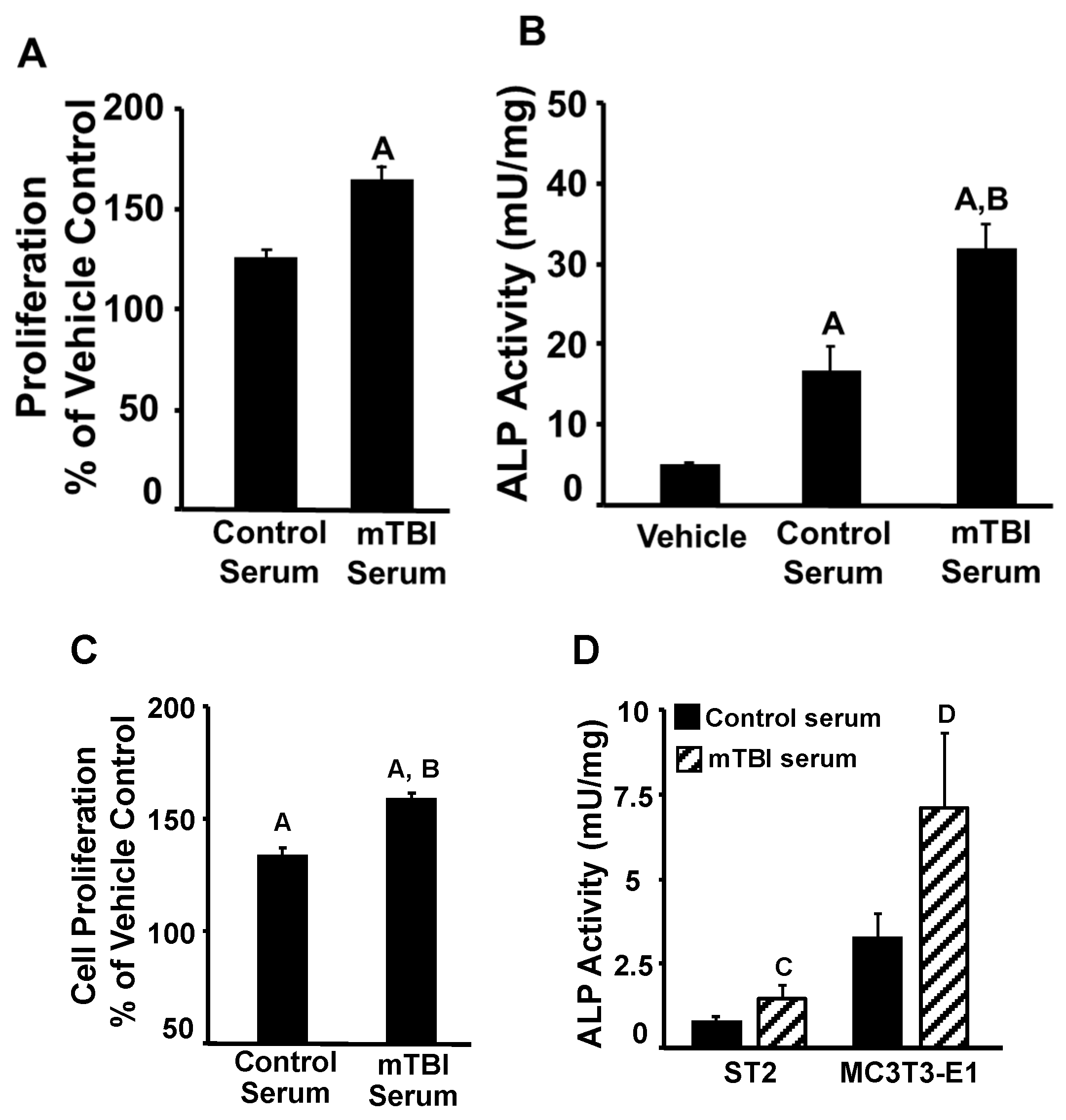
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, J.E.; Lumpkin, R.J.; Grissom, J.R. A Survey of Mild Traumatic Brain Injury Treatment in the Emergency Room and Primary Care Medical Clinics. Mil. Med. 2006, 171, 516–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piper, L.C.; Zogg, C.K.; Schneider, E.B.; Orman, J.A.; Rasmussen, T.E.; Blackbourne, L.H.; Haider, A.H. Guidelines for the Treatment of Severe Traumatic Brain Injury. JAMA Surg. 2015, 150, 1013–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. In Handbook of Clinical Neurology; 2015; Volume 127, pp. 45–66. [Google Scholar] [CrossRef] [Green Version]
- Tverdal, C.; Aarhus, M.; Andelic, N.; Skaansar, O.; Skogen, K.; Helseth, E. Characteristics of traumatic brain injury patients with abnormal neuroimaging in Southeast Norway. Inj. Epidemiol. 2020, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Si, B.; Dumkrieger, G.; Wu, T.; Zafonte, R.; Valadka, A.B.; Okonkwo, D.O.; Manley, G.T.; Wang, L.; Dodick, D.W.; Schwedt, T.J.; et al. Sub-classifying patients with mild traumatic brain injury: A clustering approach based on baseline clinical characteristics and 90-day and 180-day outcomes. PLoS ONE 2018, 13, e0198741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, R.; Schwarzbold, M.; Diaz, A.; Martins, E.T.; Rufino, A.; Amante, L.N.; Thais, M.E.; de Quevedo, J.L.; Hohl, A.; Linhares, M.N. Psychiatric disorders and traumatic brain injury. Neuropsychiatr. Dis. Treat. 2008, 4, 797–816. [Google Scholar] [CrossRef] [Green Version]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.-R. Traumatic Brain Injury. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.C.; Gupta, R.K.; Kaushal, S.; Suri, V.; Sarkar, C.; Singh, M.; Kale, S.; Sahoo, R.K.; Kumar, L.; Raina, V. A clinicopathological study of primary central nervous system lymphomas & their association with Epstein-Barr virus. Indian J. Med. Res. 2016, 143, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Mallya, S.; Sutherland, J.; Pongracic, S.; Mainland, B.; Ornstein, T.J. The Manifestation of Anxiety Disorders after Traumatic Brain Injury: A Review. J. Neurotrauma 2015, 32, 411–421. [Google Scholar] [CrossRef]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Bajwa, N.M.; Kesavan, C.; Mohan, S. Long-term Consequences of Traumatic Brain Injury in Bone Metabolism. Front. Neurol. 2018, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Dong, B.; Mao, Y.; Guan, W.; Cao, J.; Zhu, R.; Wang, S. Review: Traumatic brain injury and hyperglycemia, a potentially modifiable risk factor. Oncotarget 2016, 7, 71052–71061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheutzow, D.R.W.M.H. Subject Review Panic disorder in a patient with traumatic brain injury: A case report and discussion. Brain Inj. 1999, 13, 705–714. [Google Scholar] [CrossRef]
- Viola-Saltzman, M.; Watson, N.F. Traumatic Brain Injury and Sleep Disorders. Neurol. Clin. 2012, 30, 1299–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, J.; Chan, L.; Flynn, S. A Systematic Review of the Incidence, Prevalence, Costs, and Activity and Work Limitations of Amputation, Osteoarthritis, Rheumatoid Arthritis, Back Pain, Multiple Sclerosis, Spinal Cord Injury, Stroke, and Traumatic Brain Injury in the United States: A 2019 Update. Arch. Phys. Med. Rehabilit. 2021, 102, 115–131. [Google Scholar] [CrossRef]
- Huang, H.; Cheng, W.-X.; Hu, Y.-P.; Chen, J.-H.; Zheng, Z.-T.; Zhang, P. Relationship between heterotopic ossification and traumatic brain injury. J. Orthop. Transl. 2017, 12, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.P.; Torres, S.J.; Mehta, S.; Ahn, J. Heterotopic ossification after central nervous system trauma. Bone Jt. Res. 2013, 2, 51–57. [Google Scholar] [CrossRef]
- Kluger, G.; Kochs, A.; Holthausen, H. Heterotopic Ossification in Childhood and Adolescence. J. Child Neurol. 2000, 15, 406–413. [Google Scholar] [CrossRef]
- Alfieri, K.A.; Forsberg, J.A.; Potter, B.K. Blast injuries and heterotopic ossification. Bone Jt. Res. 2012, 1, 174–179. [Google Scholar] [CrossRef]
- Towler, O.W.; Shore, E.M. BMP signaling and skeletal development in fibrodysplasia ossificans progressiva (FOP). Dev. Dyn. 2022, 251, 144–157. [Google Scholar] [CrossRef]
- Kaliya-Perumal, A.-K.; Carney, T.J.; Ingham, P.W. Fibrodysplasia ossificans progressiva: Current concepts from bench to bedside. Dis. Model. Mech. 2020, 13, dmm046441. [Google Scholar] [CrossRef]
- Hwang, C.D.; Pagani, C.A.; Nunez, J.H.; Cherief, M.; Qin, Q.; Gomez-Salazar, M.; Kadaikal, B.; Kang, H.; Chowdary, A.R.; Patel, N.; et al. Contemporary perspectives on heterotopic ossification. J. Clin. Investig. 2022, 7, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.A. One of the Primary Functions of Tissue-Resident Pluripotent Pericytes Cells May Be to Regulate Normal Organ Growth and Maturation: Implications for Attempts to Repair Tissues Later in Life. Int. J. Mol. Sci. 2022, 23, 5496. [Google Scholar] [CrossRef]
- Wong, K.R.; Mychasiuk, R.; O’Brien, T.J.; Shultz, S.R.; McDonald, S.J.; Brady, R.D. Neurological heterotopic ossification: Novel mechanisms, prognostic biomarkers and prophylactic therapies. Bone Res. 2020, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Watt, H.; Mohan, S. The negative impact of traumatic brain injury (TBI) on bone in a mouse model. Brain Inj. 2014, 28, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Kessler, J.A. Animal Models of Typical Heterotopic Ossification. J. Biomed. Biotechnol. 2011, 2011, 309287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, W.; Larkin, D.; Pourteymoor, S.; Tambunan, W.; Gomez, G.A.; Liu, E.K.; Mohan, S. Lack of Skeletal Effects in Mice with Targeted Disruptionof Prolyl Hydroxylase Domain 1 (Phd1) Gene Expressed in Chondrocytes. Life 2022, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, C.; Bajwa, N.M.; Watt, H.; Mohan, S. Growth Hormone Effects on Bone Loss-Induced by Mild Traumatic Brain Injury and/or Hind Limb Unloading. Sci. Rep. 2019, 9, 18995. [Google Scholar] [CrossRef] [Green Version]
- Tigges, U.; Welser-Alves, J.V.; Boroujerdi, A.; Milner, R. A novel and simple method for culturing pericytes from mouse brain. Microvasc. Res. 2012, 84, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, T.; Tsukamoto, S.; Kuratani, M. Heterotopic bone induction via BMP signaling: Potential therapeutic targets for fibrodysplasia ossificans progressiva. Bone 2018, 109, 241–250. [Google Scholar] [CrossRef]
- Grenier, G.; Leblanc, É.; Faucheux, N.; Lauzier, D.; Kloen, P.; Hamdy, R.C. BMP-9 expression in human traumatic heterotopic ossification: A case report. Skelet. Muscle 2013, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Meyers, C.; Lisiecki, J.; Miller, S.; Levin, A.; Fayad, L.; Ding, C.; Sono, T.; McCarthy, E.; Levi, B.; James, A.W. Heterotopic Ossification: A Comprehensive Review. JBMR Plus 2019, 3, e10172. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Ren, B.; Shi, F.; Hua, P.; Lin, H. BMP and mTOR signaling in heterotopic ossification: Does their crosstalk provide therapeutic opportunities? J. Cell. Biochem. 2019, 120, 12108–12122. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocks, M.; Mohan, A.; Meyers, C.; Ding, C.; Levi, B.; McCarthy, E.; James, A.W. Vascular patterning in human heterotopic ossification. Hum. Pathol. 2017, 63, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Yang, Y.; Zhou, W.; Dai, C.; Chen, X.; Xie, Y.; Han, S.; Liu, H.; Hu, Y.; Tang, C.; et al. Single cell analysis reveals inhibition of angiogenesis attenuates the progression of heterotopic ossification in Mkx−/− mice. Bone Res. 2022, 10, 4. [Google Scholar] [CrossRef]
- Heikal, L.; Ghezzi, P.; Mengozzi, M.; Ferns, G. Assessment of HIF-1α expression and release following endothelial injury in-vitro and in-vivo. Mol. Med. 2018, 24, 22. [Google Scholar] [CrossRef] [Green Version]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Agarwal, S.; Loder, S.; Brownley, C.; Cholok, D.; Mangiavini, L.; Li, J.; Breuler, C.; Sung, H.H.; Li, S.; Ranganathan, K.; et al. Inhibition of Hif1α prevents both trauma-induced and genetic heterotopic ossification. Proc. Natl. Acad. Sci. USA 2016, 113, E338–E347. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Aghajanian, P.; Pourteymoor, S.; Alarcon, C.; Mohan, S. Prolyl Hydroxylase Domain-Containing Protein 2 (Phd2) Regulates Chondrocyte Differentiation and Secondary Ossification in Mice. Sci. Rep. 2016, 6, 35748. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Xing, W.; Pourteymoor, S.; Schulte, J.; Mohan, S. Conditional Deletion of Prolyl Hydroxylase Domain-Containing Protein 2 (Phd2) Gene Reveals Its Essential Role in Chondrocyte Function and Endochondral Bone Formation. Endocrinology 2016, 157, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Pourteymoor, S.; Alarcon, C.; Mohan, S. Conditional Deletion of the Phd2 Gene in Articular Chondrocytes Accelerates Differentiation and Reduces Articular Cartilage Thickness. Sci. Rep. 2017, 7, srep45408. [Google Scholar] [CrossRef] [PubMed]
- Postacchini, R.; Carbone, S.; Mastantuono, M.; Della Rocca, C.; Postacchini, F. Ossification of the Interosseous Membrane of the Leg in a Football Player: Case Report and Review of the Literature. Case Rep. Orthop. 2016, 2016, 2930324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-G.; Zhang, J.-N.; Zhou, S.; Yin, D.-P.; Wang, Y.; Tian, Y. Dynamic changes in growth factor levels over a 7-day period predict the functional outcomes of traumatic brain injury. Neural Regen. Res. 2018, 13, 2134–2140. [Google Scholar] [CrossRef]
- Tweedie, D.; Karnati, H.K.; Mullins, R.; Pick, C.G.; Hoffer, B.J.; Goetzl, E.J.; Kapogiannis, D.; Greig, N.H. Time-dependent cytokine and chemokine changes in mouse cerebral cortex following a mild traumatic brain injury. Elife 2020, 9, e55827. [Google Scholar] [CrossRef] [PubMed]
- Cash, A.; Theus, M.H. Mechanisms of Blood–Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kesavan, C.; Gomez, G.A.; Pourteymoor, S.; Mohan, S. Development of an Animal Model for Traumatic Brain Injury Augmentation of Heterotopic Ossification in Response to Local Injury. Biomedicines 2023, 11, 943. https://doi.org/10.3390/biomedicines11030943
Kesavan C, Gomez GA, Pourteymoor S, Mohan S. Development of an Animal Model for Traumatic Brain Injury Augmentation of Heterotopic Ossification in Response to Local Injury. Biomedicines. 2023; 11(3):943. https://doi.org/10.3390/biomedicines11030943
Chicago/Turabian StyleKesavan, Chandrasekhar, Gustavo A. Gomez, Sheila Pourteymoor, and Subburaman Mohan. 2023. "Development of an Animal Model for Traumatic Brain Injury Augmentation of Heterotopic Ossification in Response to Local Injury" Biomedicines 11, no. 3: 943. https://doi.org/10.3390/biomedicines11030943