
Characterization of the Lower Airways and Oral Microbiota in Healthy Young Persons in the Community

 , ,
, ,
Abstract
:1. Introduction
2. Materials & Methods
2.1. Subjects
2.2. Procedures
2.3. Microbiome Profiling
2.4. Microbiome Analyses
2.5. Predicted Functional Metagenomics Analyses
2.6. Statistical Analysis
3. Results
3.1. Study Population
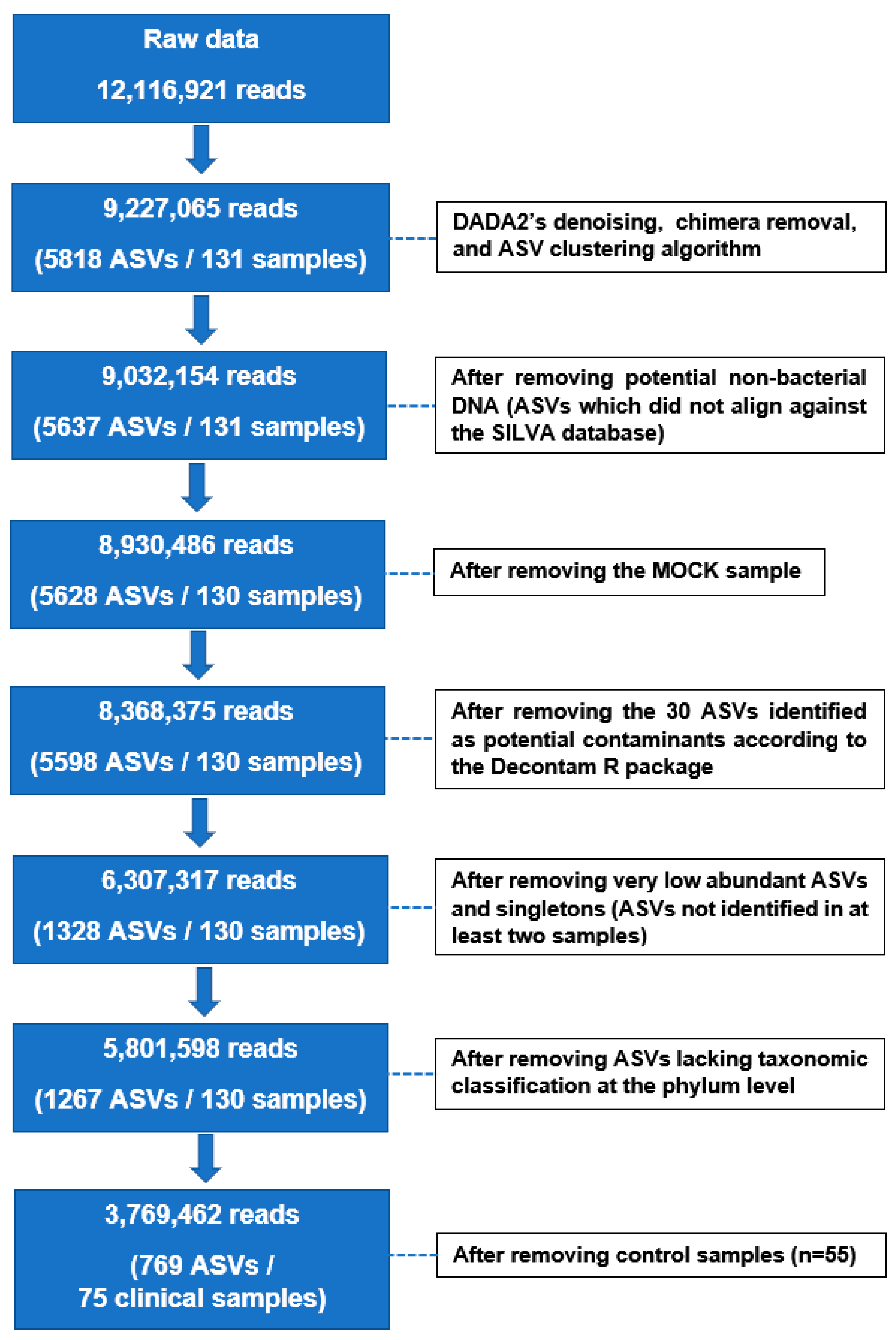
3.2. 16S rRNA Gene Sequencing Data
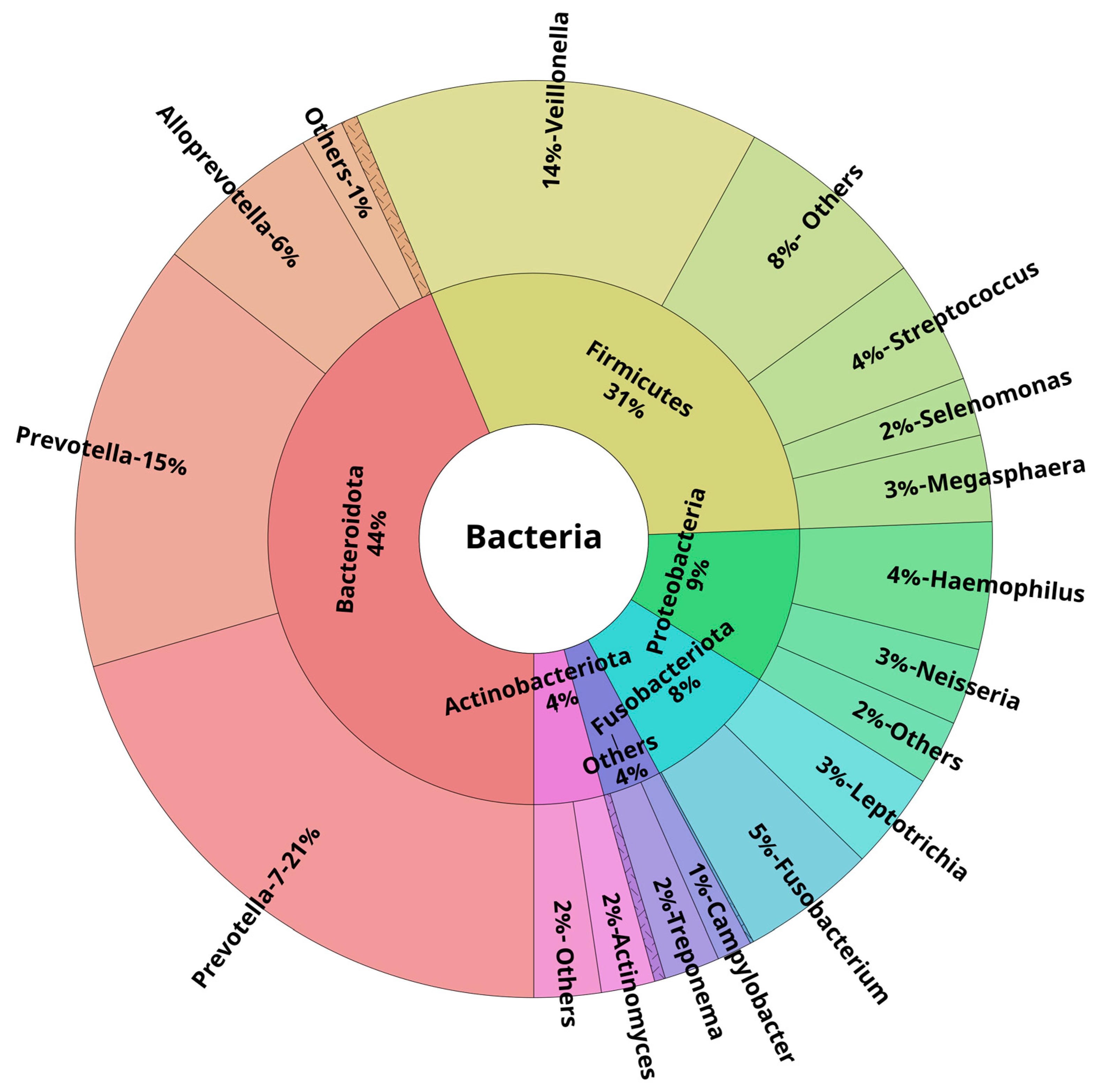
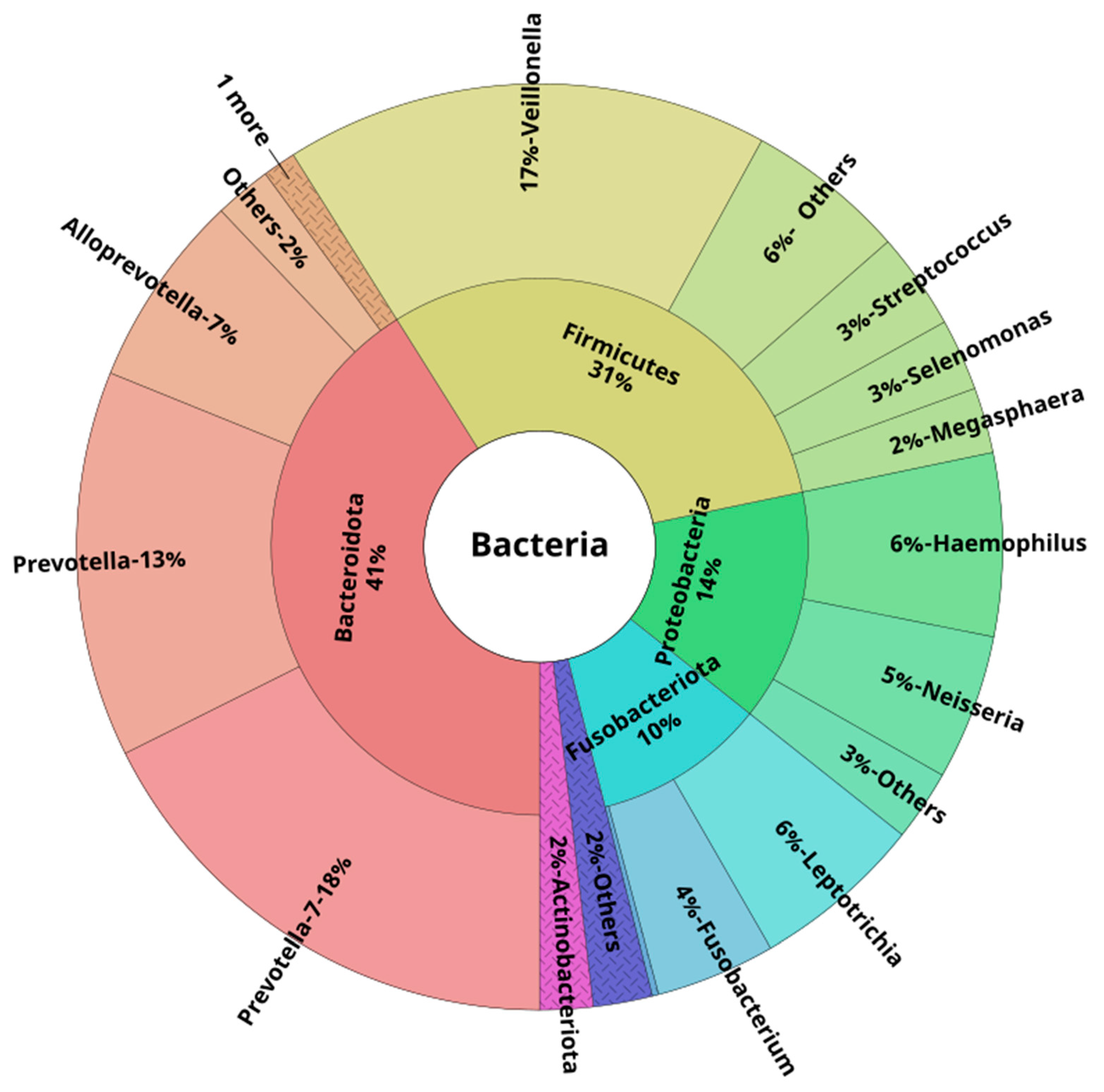
3.3. Oral and Lower Airway Microbiome Composition in Health
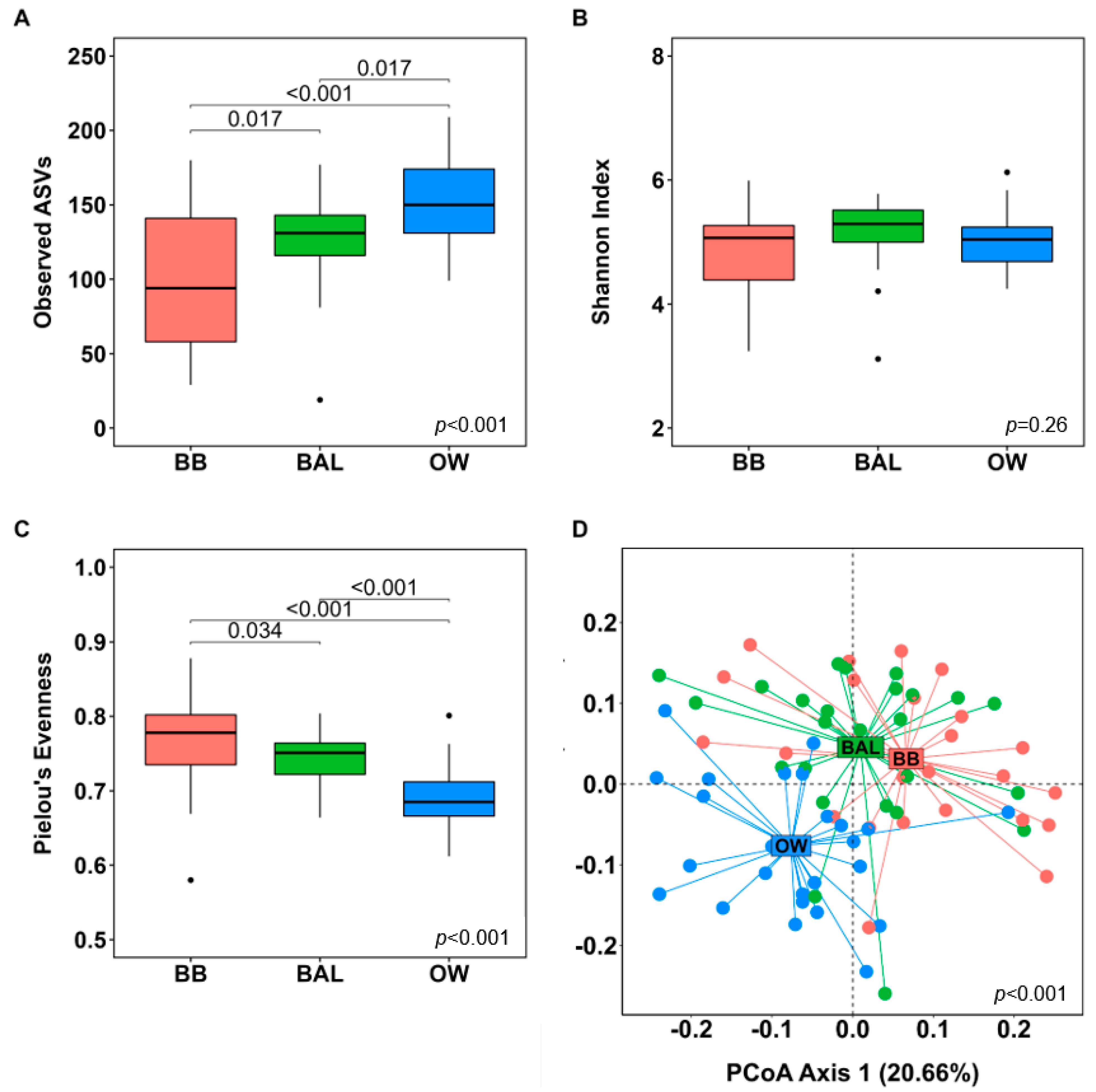
3.4. Microbial Diversity Analyses
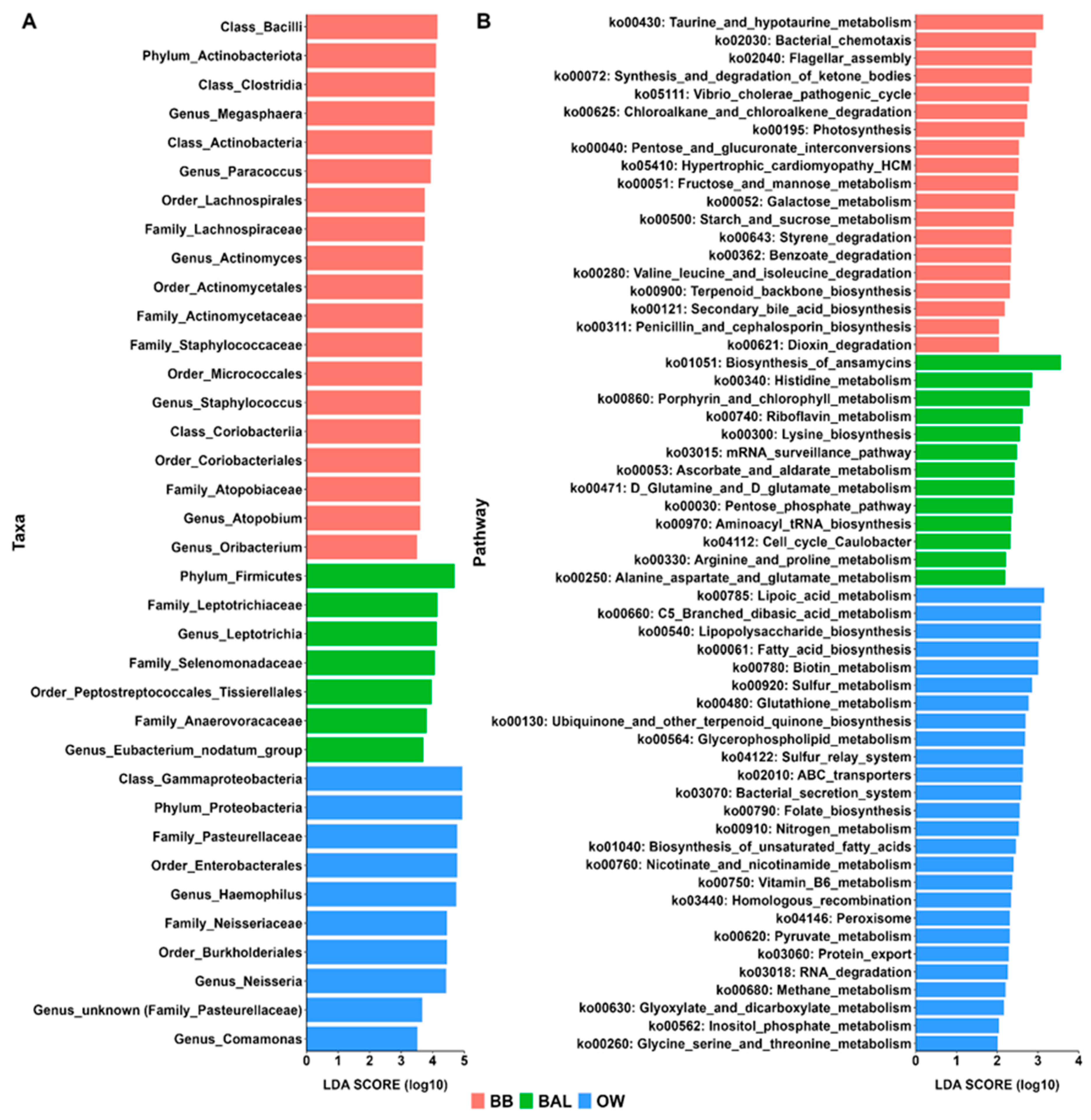
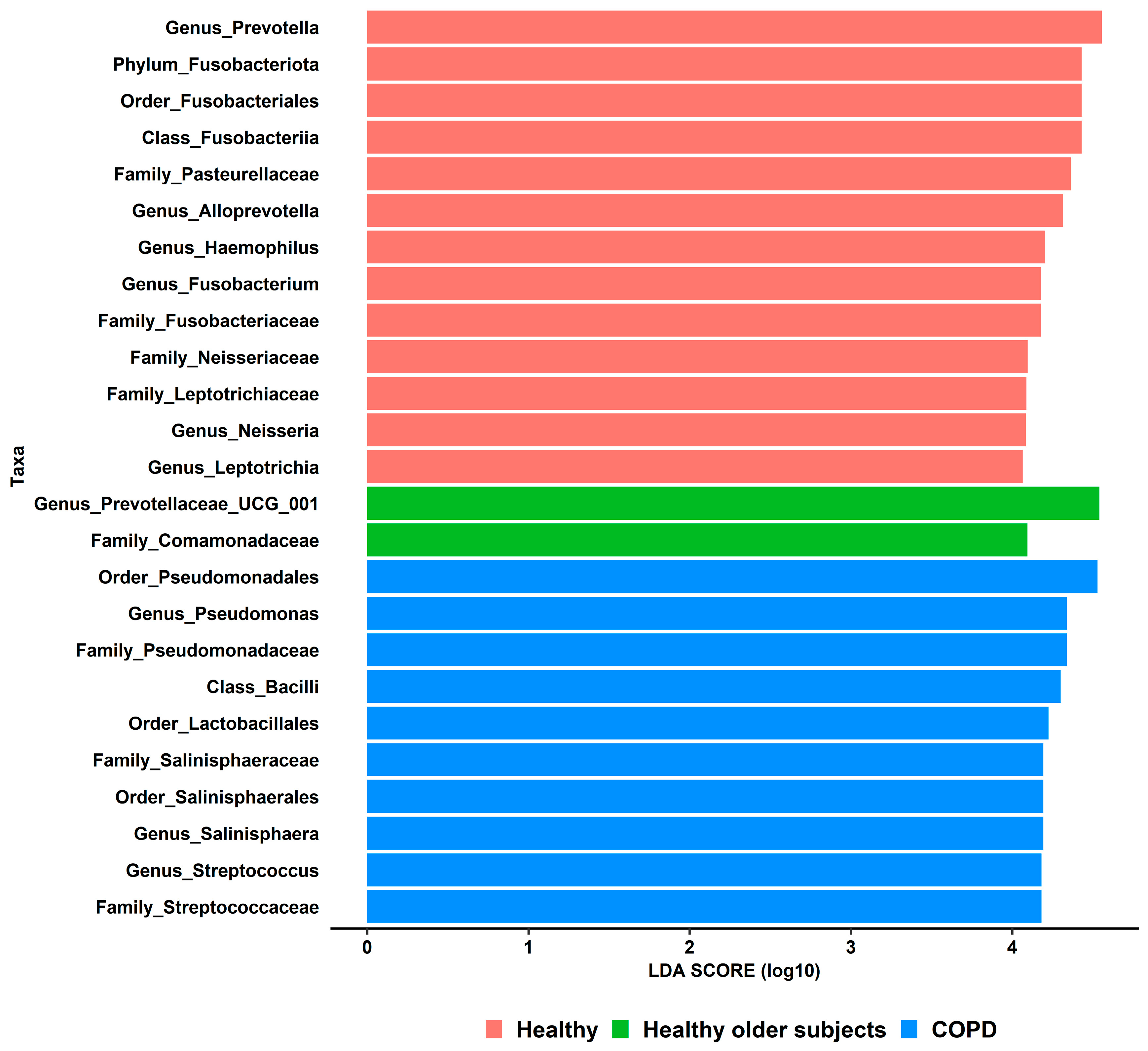
3.5. Differential Taxonomic Analyses
3.6. Metagenome Prediction Analyses
3.7. Lower Airway Microbiota Changes Associated with Local Dysbiosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Segal, L.N.; Rom, W.N.; Weiden, M.D. Lung Microbiome for Clinicians: New Discoveries about Bugs in Healthy and Diseased Lungs. Ann. Am. Thorac. Soc. 2014, 11, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarridge, J.E., 3rd. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef] [Green Version]
- Moffatt, M.F.; Cookson, W.O. The lung microbiome in health and disease. Clin. Med. 2017, 17, 525–529. [Google Scholar] [CrossRef]
- Man, W.H.; De Steenhuijsen Piters, W.A.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef]
- Natalini, J.G.; Singh, S.; Segal, L.N. The dynamic lung microbiome in health and disease. Nat. Rev. Microbiol. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pandiyan, P.; Bhaskaran, N.; Zou, M.; Schneider, E.; Jayaraman, S.; Huehn, J. Microbiome Dependent Regulation of T(regs) and Th17 Cells in Mucosa. Front. Immunol. 2019, 10, 426. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, L.J.; Monga, M.; Miller, A.W. Defining Dysbiosis for a Cluster of Chronic Diseases. Sci. Rep. 2019, 9, 12918. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, E.; Escribano-Vazquez, U.; Descamps, D.; Cherbuy, C.; Langella, P.; Riffault, S.; Remot, A.; Thomas, M. Paradigms of Lung Microbiota Functions in Health and Disease, Particularly, in Asthma. Front. Physiol. 2018, 9, 1168. [Google Scholar] [CrossRef]
- Paudel, K.R.; Dharwal, V.; Patel, V.K.; Galvao, I.; Wadhwa, R.; Malyla, V.; Shen, S.S.; Budden, K.F.; Hansbro, N.G.; Vaughan, A.; et al. Role of Lung Microbiome in Innate Immune Response Associated with Chronic Lung Diseases. Front. Med. 2020, 7, 554. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Ashley, S.L.; Gurczynski, S.J.; Xia, M.; Wilke, C.; Falkowski, N.R.; Norman, K.C.; Arnold, K.B.; Huffnagle, G.B.; Salisbury, M.L.; et al. Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1127–1138. [Google Scholar] [CrossRef]
- Marshall, E.A.; Filho, F.S.L.; Sin, D.D.; Lam, S.; Leung, J.M.; Lam, W.L. Distinct bronchial microbiome precedes clinical diagnosis of lung cancer. Mol. Cancer 2022, 21, 68. [Google Scholar] [CrossRef]
- Willis, J.R.; Gabaldón, T. The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems. Microorganisms 2020, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Martu, M.A.; Maftei, G.A.; Sufaru, I.G.; Jelihovschi, I.; Luchian, I.; Hurjui, L.; Martu, I.; Pasarin, L. COVID-19 And Periodontal Disease—Ethiopathogenic and Clinical Implications. Rom. J. Oral Rehabil. 2020, 12, 9. [Google Scholar]
- Molina, A.; Huck, O.; Herrera, D.; Montero, E. The association between respiratory diseases and periodontitis: A systematic review and meta-analysis. J. Clin. Periodontol. 2023. [Google Scholar] [CrossRef]
- Leitao Filho, F.S.; Alotaibi, N.M.; Ngan, D.; Tam, S.; Yang, J.; Hollander, Z.; Chen, V.; FitzGerald, J.M.; Nislow, C.; Leung, J.M.; et al. Sputum Microbiome Is Associated with 1-Year Mortality after Chronic Obstructive Pulmonary Disease Hospitalizations. Am. J. Respir. Crit. Care Med. 2019, 199, 1205–1213. [Google Scholar] [CrossRef]
- Langevin, S.; Pichon, M.; Smith, E.; Morrison, J.; Bent, Z.; Green, R.; Barker, K.; Solberg, O.; Gillet, Y.; Javouhey, E.; et al. Early nasopharyngeal microbial signature associated with severe influenza in children: A retrospective pilot study. J. Gen. Virol. 2017, 98, 2425–2437. [Google Scholar] [CrossRef]
- Aho, V.T.; Pereira, P.A.; Haahtela, T.; Pawankar, R.; Auvinen, P.; Koskinen, K. The microbiome of the human lower airways: A next generation sequencing perspective. World Allergy Organ. J. 2015, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Quanjer, P.H.; Stanojevic, S.; Cole, T.J.; Baur, X.; Hall, G.L.; Culver, B.H.; Enright, P.L.; Hankinson, J.L.; Ip, M.S.M.; Zheng, J.; et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: The global lung function 2012 equations. Eur. Respir. J. 2012, 40, 1324–1343. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; McCloskey, L.; Falkowski, N.R.; Huffnagle, G.B.; Curtis, J.L. Bacterial Topography of the Healthy Human Lower Respiratory Tract. MBio 2017, 8, e02287-16. [Google Scholar] [CrossRef] [PubMed]
- Ramsheh, M.Y.; Haldar, K.; Esteve-Codina, A.; Purser, L.F.; Richardson, M.; Müller-Quernheim, J.; Greulich, T.; Nowinski, A.; Barta, I.; Stendardo, M.; et al. Lung microbiome composition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: A bacterial 16S rRNA gene sequencing and host transcriptomic analysis. Lancet Microbe 2021, 2, e300–e310. [Google Scholar] [CrossRef] [PubMed]
- Mashima, I.; Liao, Y.-C.; Lin, C.-H.; Nakazawa, F.; Haase, E.M.; Kiyoura, Y.; Scannapieco, F.A. Comparative Pan-Genome Analysis of Oral Veillonella Species. Microorganisms 2021, 9, 1775. [Google Scholar] [CrossRef]
- Könönen, E.; Fteita, D.; Gursoy, U.K.; Gursoy, M. Prevotella species as oral residents and infectious agents with potential impact on systemic conditions. J. Oral Microbiol. 2022, 14, 2079814. [Google Scholar] [CrossRef]
- Kosikowska, U.; Biernasiuk, A.; Rybojad, P.; Łoś, R.; Malm, A. Haemophilus parainfluenzae as a marker of the upper respiratory tract microbiota changes under the influence of preoperative prophylaxis with or without postoperative treatment in patients with lung cancer. BMC Microbiol. 2016, 16, 62. [Google Scholar] [CrossRef] [Green Version]
- Erb-Downward, J.R.; Thompson, D.L.; Han, M.K.; Freeman, C.M.; McCloskey, L.; Schmidt, L.A.; Young, V.B.; Toews, G.B.; Curtis, J.L.; Sundaram, B.; et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE 2011, 6, e16384. [Google Scholar] [CrossRef] [Green Version]
- Gambin, D.J.; Vitali, F.C.; De Carli, J.P.; Mazzon, R.R.; Gomes, B.P.F.A.; Duque, T.M.; Trentin, M.S. Prevalence of red and orange microbial complexes in endodontic-periodontal lesions: A systematic review and meta-analysis. Clin. Oral Investig. 2021, 25, 6533–6546. [Google Scholar] [CrossRef]
- Butera, A.; Gallo, S.; Maiorani, C.; Molino, D.; Chiesa, A.; Preda, C.; Esposito, F.; Scribante, A. Probiotic Alternative to Chlorhexidine in Periodontal Therapy: Evaluation of Clinical and Microbiological Parameters. Microorganisms 2021, 9, 69. [Google Scholar] [CrossRef]
- Yan, Z.; Chen, B.; Yang, Y.; Yi, X.; Wei, M.; Ecklu-Mensah, G.; Buschmann, M.M.; Liu, H.; Gao, J.; Liang, W.; et al. Multi-omics analyses of airway host–microbe interactions in chronic obstructive pulmonary disease identify potential therapeutic interventions. Nat. Microbiol. 2022, 7, 1361–1375. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Participants (n = 25) |
|---|---|
| Age, years | 24.3 ± 3.3 |
| Sex, female | 13 (52) |
| Current/previous smoker | 1 (4) |
| Swimmers | 7 (28) |
| BMI, kg/m2 | 22.57 ± 1.96 |
| FVC, liters | 5.4 ± 0.94 |
| FVC, % of predicted | 111.43 ± 11.86 |
| FEV1, liters | 4.39 ± 0.77 |
| FEV1, % of predicted | 106.09 ± 11.38 |
| FEV1/FVC, % | 81.47 ± 6.53 |
| Taxon | MICROBIOME COMPARTMENT | p-Value * | ||||||
|---|---|---|---|---|---|---|---|---|
| Bronchial Brushings | BAL Samples | Oral Wash Samples | ||||||
| Phylum | Median | Q1; Q3 | Median | Q1; Q3 | Median | Q1; Q3 | p | adj. p |
| Bacteroidota | 42.3 | 36.8; 46.2 | 40.2 | 37.3; 45.5 | 41.7 | 35.2; 44.5 | 0.59 | 0.59 |
| Firmicutes | 32.7 | 22.8; 37.6 | 31.4 | 26.9; 35.9 | 20.9 | 17.5; 25.5 | <0.001 | <0.001 |
| Proteobacteria | 7.0 | 4.6; 14.7 | 12.5 | 7.7; 18.5 | 27.9 | 23.0; 32.8 | <0.001 | <0.001 |
| Fusobacteriota | 6.2 | 3.0; 10.3 | 9.1 | 6.4; 14.0 | 4.6 | 3.1; 7.1 | 0.011 | 0.014 |
| Actinobacteriota | 3.7 | 2.9; 5.1 | 2.1 | 1.4; 2.4 | 1.7 | 1.0; 2.5 | <0.001 | <0.001 |
| Taxon | MICROBIOME COMPARTMENT | p-Value * | ||||||
|---|---|---|---|---|---|---|---|---|
| Bronchial Brushings | BAL Samples | Oral Wash Samples | ||||||
| Genus | Median | Q1; Q3 | Median | Q1; Q3 | Median | Q1; Q3 | p | adj. p |
| Prevotella-7 | 21.0 | 16.2;25.0 | 18.0 | 15.5; 20.7 | 19.7 | 15.7; 21.8 | 0.29 | 0.40 |
| Veillonella | 15.5 | 9.4; 17.9 | 16.8 | 13.9; 21.2 | 15.3 | 11.4; 16.8 | 0.28 | 0.40 |
| Prevotella | 14.0 | 9.4; 16.0 | 12.3 | 9.8; 14.1 | 11.0 | 8.8; 13.9 | 0.39 | 0.48 |
| Haemophilus | 3.1 | 1.8; 7.2 | 5.5 | 4.7; 7.2 | 16.3 | 12.2; 18.7 | <0.001 | <0.001 |
| Alloprevotella | 4.8 | 2.3; 10.0 | 7.2 | 4.4; 8.6 | 5.2 | 3.8; 10.1 | 0.47 | 0.51 |
| Neisseria | 1.7 | 0.5; 5.0 | 4.2 | 1.5; 7.1 | 7.1 | 3.7; 11.5 | <0.001 | 0.001 |
| Leptotrichia | 1.8 | 1.0; 4.5 | 5.7 | 2.7; 7.7 | 2.6 | 1.4; 4.0 | 0.006 | 0.014 |
| Fusobacterium | 3.1 | 1.8; 6.2 | 2.5 | 1.9; 3.9 | 1.5 | 1.0; 2.9 | 0.07 | 0.12 |
| Streptococcus | 4.3 | 2.6; 5.7 | 3.2 | 1.9; 4.1 | 2.2 | 1.7; 4.1 | 0.06 | 0.11 |
| Megasphaera | 2.7 | 0.3; 4.2 | 2.4 | 1.5; 3.2 | 0.5 | 0.2; 0.8 | <0.001 | 0.002 |
| Selenomonas | 1.2 | 0.0; 2.4 | 2.1 | 1.0; 3.7 | 0.7 | 0.4; 1.1 | 0.004 | 0.012 |
| Treponema | 0.7 | 0.3; 1.0 | 0.6 | 0.2; 0.9 | 0.3 | 0.1; 1.2 | 0.48 | 0.51 |
| Actinomyces | 1.8 | 1.2; 2.5 | 1.0 | 0.7; 1.3 | 0.9 | 0.7; 1.3 | 0.003 | 0.01 |
| Porphyromonas | 0.4 | 0.1; 0.9 | 0.9 | 0.2; 1.6 | 0.9 | 0.4; 1.7 | 0.03 | 0.08 |
| Campylobacter | 0.8 | 0.5; 1.2 | 0.9 | 0.5; 1.3 | 0.7 | 0.6; 1.1 | 0.75 | 0.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leitao Filho, F.S.; Monica Peters, C.; Sheel, A.W.; Yang, J.; Nislow, C.; Lam, S.; Leung, J.M.; Sin, D.D. Characterization of the Lower Airways and Oral Microbiota in Healthy Young Persons in the Community. Biomedicines 2023, 11, 841. https://doi.org/10.3390/biomedicines11030841
Leitao Filho FS, Monica Peters C, Sheel AW, Yang J, Nislow C, Lam S, Leung JM, Sin DD. Characterization of the Lower Airways and Oral Microbiota in Healthy Young Persons in the Community. Biomedicines. 2023; 11(3):841. https://doi.org/10.3390/biomedicines11030841
Chicago/Turabian StyleLeitao Filho, Fernando Sergio, Carli Monica Peters, Andrew William Sheel, Julia Yang, Corey Nislow, Stephen Lam, Janice M. Leung, and Don D. Sin. 2023. "Characterization of the Lower Airways and Oral Microbiota in Healthy Young Persons in the Community" Biomedicines 11, no. 3: 841. https://doi.org/10.3390/biomedicines11030841