
Dilated Cardiomyopathy: A Comprehensive Approach to Diagnosis and Risk Stratification
 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
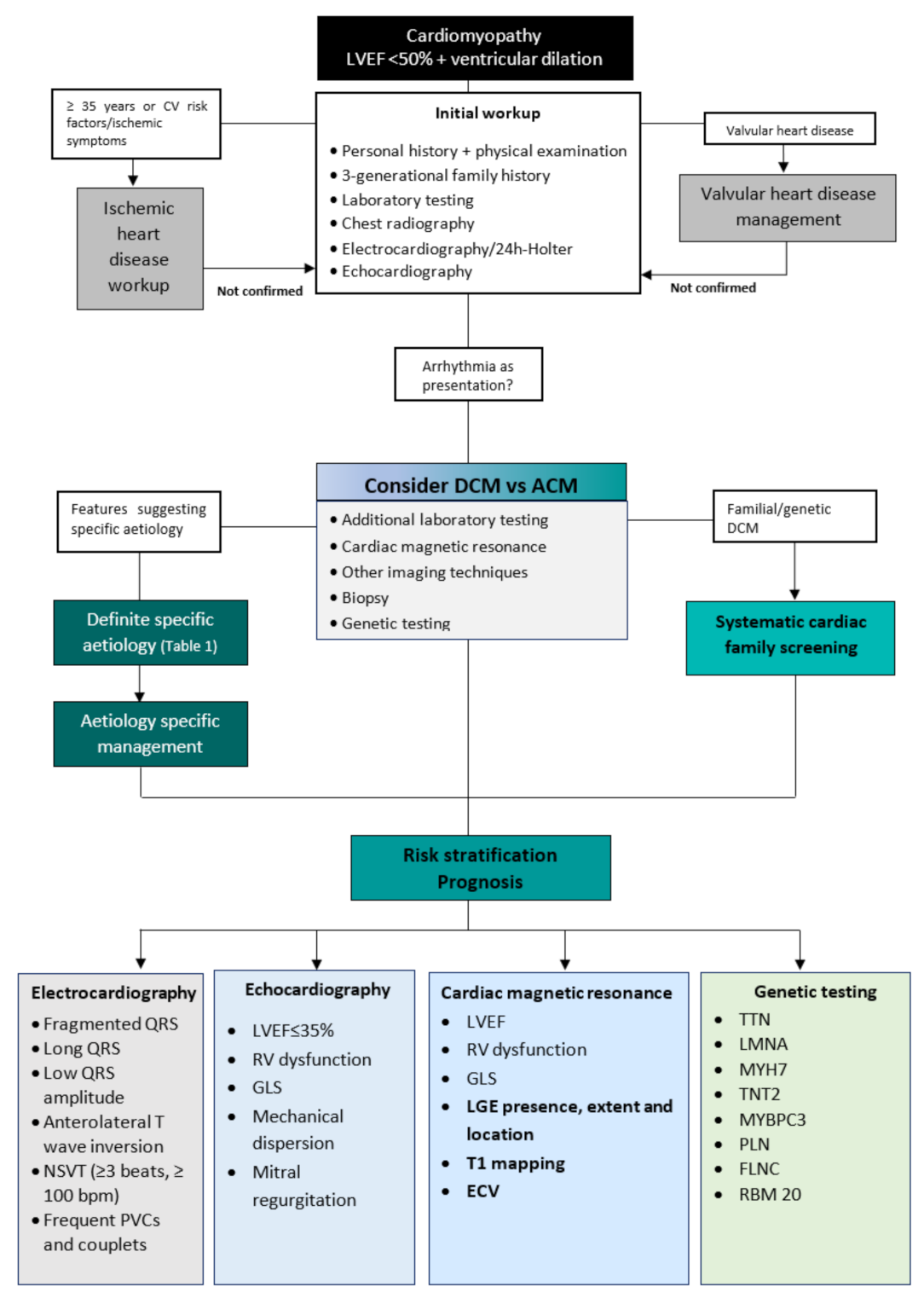
2. Diagnostic Workup
3. Differential Diagnosis
Differential Diagnosis of Dilated and Arrhythmogenic Cardiomyopathy
4. Aetiologies
5. Risk Stratification and Prognosis
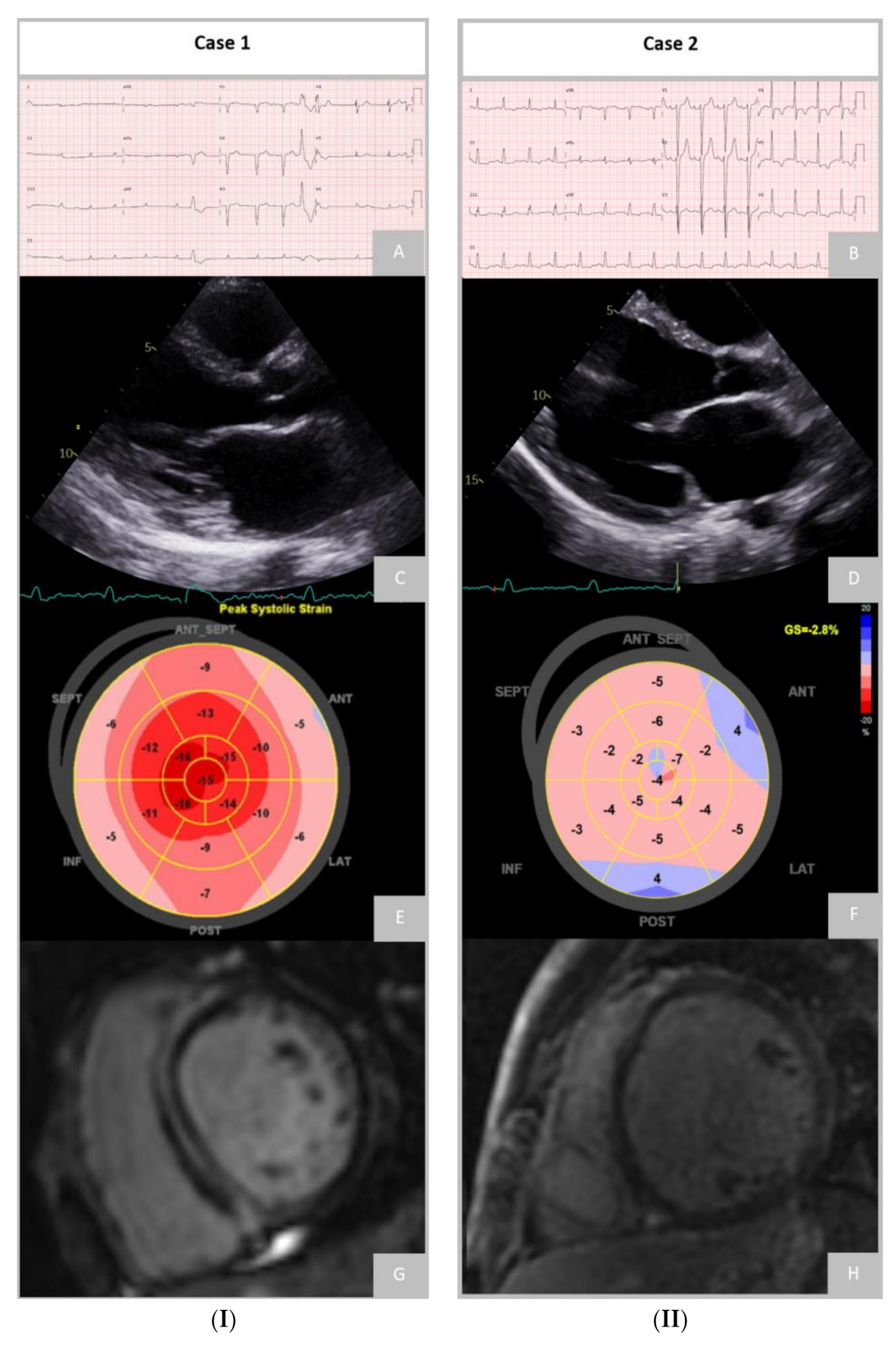
5.1. Electrocardiogram
5.2. Echocardiography
5.3. Cardiac Magnetic Resonance
5.3.1. Late Gadolinium Enhancement
Extension
Location
Pattern
5.3.2. T1 and Extracellular Volume
5.3.3. Feature-Tracking Strain Analysis
5.4. Genetic Testing
5.4.1. Titin
5.4.2. Lamin A/C
5.4.3. Desmossomal
5.4.4. Filamin C
5.4.5. RBM20
5.4.6. Phospholamban
5.4.7. Desmoplakin
5.4.8. Sodium Voltage-Gated Channel Alpha Subunit 5 (SCN5A)
6. Prediction of Left Ventricle Reverse Remodelling
7. Patient Selection for Device Implantation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.H.; Edwards, L.B.; Dipchand, A.I.; Goldfarb, S.; Kucheryavaya, A.Y.; Levvey, B.J.; Meiser, B.; Rossano, J.W.; Yusen, R.D.; Stehlik, J. The Registry of the International Society for Heart and Lung Transplantation: Thirty-third Adult Heart Transplantation Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant. J. Heart Lung Transplant. 2016, 35, 1158–1169. [Google Scholar] [CrossRef]
- Halliday, B.P.; Gulati, A.; Ali, A.; Newsome, S.; Lota, A.; Tayal, U.; Vassiliou, V.S.; Arzanauskaite, M.; Izgi, C.; Krishnathasan, K.; et al. Sex- and age-based differences in the natural history and outcome of dilated cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 1392–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donal, E.; Delgado, V.; Bucciarelli-Ducci, C.; Galli, E.; Haugaa, K.H.; Charron, P.; Voigt, J.U.; Cardim, N.; Masci, P.G.; Galderisi, M.; et al. Multimodality imaging in the diagnosis, risk stratification, and management of patients with dilated cardiomyopathies: An expert consensus document from the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 1075–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.P.; Charron, P.; Gimeno-Blanes, J.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2012, 34, 1448–1458. [Google Scholar] [CrossRef] [Green Version]
- Grimm, W.; Christ, M.; Bach, J.; Müller, H.H.; Maisch, B. Noninvasive arrhythmia risk stratification in idiopathic dilated cardiomyopathy: Results of the Marburg Cardiomyopathy Study. Circulation 2003, 108, 2883–2891. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.R.; Kramer, C.M. Role of Cardiac Magnetic Resonance in the Diagnosis and Prognosis of Nonischemic Cardiomyopathy. JACC Cardiovasc. Imaging 2017, 10, 1180–1193. [Google Scholar] [CrossRef]
- Guigui, S.A.; Horvath, S.A.; Arenas, I.A.; Mihos, C.G. Cardiac geometry, function and mechanics in left ventricular non-compaction cardiomyopathy with preserved ejection fraction. J. Echocardiogr. 2022, 20, 144–150. [Google Scholar] [CrossRef]
- Srivastava, S.; Yavari, M.; Al-Abcha, A.; Banga, S.; Abela, G. Ventricular non-compaction review. Heart Fail. Rev. 2022, 27, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V.; Roos-Hesselink, J.W.; Bauersachs, J.; Blomström-Lundqvist, C.; Cífková, R.; De Bonis, M.; Iung, B.; Johnson, M.R.; Kintscher, U.; Kranke, P.; et al. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur. Heart J. 2018, 39, 3165–3241. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.B.; Arany, Z.; McNamara, D.M.; Goland, S.; Elkayam, U. Peripartum Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Miles, C.; Finocchiaro, G.; Papadakis, M.; Gray, B.; Westaby, J.; Ensam, B.; Basu, J.; Parry-Williams, G.; Papatheodorou, E.; Paterson, C.; et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation 2019, 139, 1786–1797. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019, 16, e301–e372. [Google Scholar] [CrossRef] [Green Version]
- Hoorntje, E.T.; Te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Chun, K.H.; Oh, J.; Hong, Y.J.; Yu, H.T.; Lee, C.J.; Kim, T.H.; Joung, B.; Pak, H.N.; Lee, M.H.; Kim, Y.J.; et al. Prognostic Cardiac Magnetic Resonance Markers of Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy for Predicting Heart Failure Outcomes. J. Am. Heart Assoc. 2022, 11, e023167. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef] [PubMed]
- Segura-Rodríguez, D.; Bermúdez-Jiménez, F.J.; Carriel, V.; López-Fernández, S.; González-Molina, M.; Oyonarte Ramírez, J.M.; Fernández-Navarro, L.; García-Roa, M.D.; Cabrerizo, E.M.; Durand-Herrera, D.; et al. Myocardial fibrosis in arrhythmogenic cardiomyopathy: A genotype-phenotype correlation study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Mattesi, G.; Bariani, R.; Cecere, A.; Martini, N.; De Michieli, L.; Da Pozzo, S.; Corradin, S.; De Conti, G.; Zorzi, A.; et al. Cardiac magnetic resonance imaging of arrhythmogenic cardiomyopathy: Evolving diagnostic perspectives. Eur. Radiol. 2023, 33, 270–282. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am. J. Cardiol. 2013, 112, 1197–1206. [Google Scholar] [CrossRef]
- Bondue, A.; Arbustini, E.; Bianco, A.; Ciccarelli, M.; Dawson, D.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.F.; et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303. [Google Scholar] [CrossRef] [Green Version]
- Goldberger, J.J.; Subačius, H.; Patel, T.; Cunnane, R.; Kadish, A.H. Sudden cardiac death risk stratification in patients with nonischemic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 1879–1889. [Google Scholar] [CrossRef] [Green Version]
- Marume, K.; Noguchi, T.; Tateishi, E.; Morita, Y.; Kamakura, T.; Ishibashi, K.; Noda, T.; Miura, H.; Nishimura, K.; Nakai, M.; et al. Mortality and Sudden Cardiac Death Risk Stratification Using the Noninvasive Combination of Wide QRS Duration and Late Gadolinium Enhancement in Idiopathic Dilated Cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2018, 11, e006233. [Google Scholar] [CrossRef]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving concepts in dilated cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Daubert, J.P.; Zareba, W.; Hall, W.J.; Schuger, C.; Corsello, A.; Leon, A.R.; Andrews, M.L.; McNitt, S.; Huang, D.T.; Moss, A.J. Predictive value of ventricular arrhythmia inducibility for subsequent ventricular tachycardia or ventricular fibrillation in Multicenter Automatic Defibrillator Implantation Trial (MADIT) II patients. J. Am. Coll. Cardiol. 2006, 47, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Bricknell, K.; Hanekom, L.; Marwick, T.H. Reproducibility and accuracy of echocardiographic measurements of left ventricular parameters using real-time three-dimensional echocardiography. J. Am. Coll. Cardiol. 2004, 44, 878–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venner, C.; Selton-Suty, C.; Huttin, O.; Erpelding, M.L.; Aliot, E.; Juillière, Y. Right ventricular dysfunction in patients with idiopathic dilated cardiomyopathy: Prognostic value and predictive factors. Arch. Cardiovasc. Dis. 2016, 109, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Merlo, M.; Pyxaras, S.A.; Pinamonti, B.; Barbati, G.; Di Lenarda, A.; Sinagra, G. Prevalence and prognostic significance of left ventricular reverse remodeling in dilated cardiomyopathy receiving tailored medical treatment. J. Am. Coll. Cardiol. 2011, 57, 1468–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popescu, B.A.; Beladan, C.C.; Calin, A.; Muraru, D.; Deleanu, D.; Rosca, M.; Ginghina, C. Left ventricular remodelling and torsional dynamics in dilated cardiomyopathy: Reversed apical rotation as a marker of disease severity. Eur. J. Heart Fail. 2009, 11, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignot, A.; Donal, E.; Zaroui, A.; Reant, P.; Salem, A.; Hamon, C.; Monzy, S.; Roudaut, R.; Habib, G.; Lafitte, S. Global longitudinal strain as a major predictor of cardiac events in patients with depressed left ventricular function: A multicenter study. J. Am. Soc. Echocardiogr. 2010, 23, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Sengeløv, M.; Jørgensen, P.G.; Jensen, J.S.; Bruun, N.E.; Olsen, F.J.; Fritz-Hansen, T.; Nochioka, K.; Biering-Sørensen, T. Global Longitudinal Strain Is a Superior Predictor of All-Cause Mortality in Heart Failure With Reduced Ejection Fraction. JACC Cardiovasc. Imaging 2015, 8, 1351–1359. [Google Scholar] [CrossRef] [Green Version]
- Vîjîiac, A.; Onciul, S.; Guzu, C.; Verinceanu, V.; Bătăilă, V.; Deaconu, S.; Scărlătescu, A.; Zamfir, D.; Petre, I.; Onuţ, R.; et al. The prognostic value of right ventricular longitudinal strain and 3D ejection fraction in patients with dilated cardiomyopathy. Int. J. Cardiovasc. Imaging 2021, 37, 3233–3244. [Google Scholar] [CrossRef]
- Lancellotti, P.; Pellikka, P.A.; Budts, W.; Chaudhry, F.A.; Donal, E.; Dulgheru, R.; Edvardsen, T.; Garbi, M.; Ha, J.W.; Kane, G.C.; et al. The clinical use of stress echocardiography in non-ischaemic heart disease: Recommendations from the European Association of Cardiovascular Imaging and the American Society of Echocardiography. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 1191–1229. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.A.J.; Cornel, J.H.; van de Ven, P.M.; van Rossum, A.C.; Allaart, C.P.; Germans, T. The Prognostic Value of Late Gadolinium-Enhanced Cardiac Magnetic Resonance Imaging in Nonischemic Dilated Cardiomyopathy: A Review and Meta-Analysis. JACC Cardiovasc. Imaging 2018, 11, 1274–1284. [Google Scholar] [CrossRef]
- Di Marco, A.; Anguera, I.; Schmitt, M.; Klem, I.; Neilan, T.G.; White, J.A.; Sramko, M.; Masci, P.G.; Barison, A.; McKenna, P.; et al. Late Gadolinium Enhancement and the Risk for Ventricular Arrhythmias or Sudden Death in Dilated Cardiomyopathy: Systematic Review and Meta-Analysis. JACC Heart Fail. 2017, 5, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Halliday, B.P.; Baksi, A.J.; Gulati, A.; Ali, A.; Newsome, S.; Izgi, C.; Arzanauskaite, M.; Lota, A.; Tayal, U.; Vassiliou, V.S.; et al. Outcome in Dilated Cardiomyopathy Related to the Extent, Location, and Pattern of Late Gadolinium Enhancement. JACC Cardiovasc. Imaging 2019, 12, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.; Jabbour, A.; Ismail, T.F.; Guha, K.; Khwaja, J.; Raza, S.; Morarji, K.; Brown, T.D.; Ismail, N.A.; Dweck, M.R.; et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013, 309, 896–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alba, A.C.; Gaztañaga, J.; Foroutan, F.; Thavendiranathan, P.; Merlo, M.; Alonso-Rodriguez, D.; Vallejo-García, V.; Vidal-Perez, R.; Corros-Vicente, C.; Barreiro-Pérez, M.; et al. Prognostic Value of Late Gadolinium Enhancement for the Prediction of Cardiovascular Outcomes in Dilated Cardiomyopathy: An International, Multi-Institutional Study of the MINICOR Group. Circ. Cardiovasc. Imaging 2020, 13, e010105. [Google Scholar] [CrossRef]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef]
- aus dem Siepen, F.; Buss, S.J.; Messroghli, D.; Andre, F.; Lossnitzer, D.; Seitz, S.; Keller, M.; Schnabel, P.A.; Giannitsis, E.; Korosoglou, G.; et al. T1 mapping in dilated cardiomyopathy with cardiac magnetic resonance: Quantification of diffuse myocardial fibrosis and comparison with endomyocardial biopsy. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puntmann, V.O.; Carr-White, G.; Jabbour, A.; Yu, C.Y.; Gebker, R.; Kelle, S.; Hinojar, R.; Doltra, A.; Varma, N.; Child, N.; et al. T1-Mapping and Outcome in Nonischemic Cardiomyopathy: All-Cause Mortality and Heart Failure. JACC Cardiovasc. Imaging 2016, 9, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Sohal, M.; Voigt, T.; Sammut, E.; Tobon-Gomez, C.; Child, N.; Jackson, T.; Shetty, A.; Bostock, J.; Cooklin, M.; et al. Myocardial tissue characterization by cardiac magnetic resonance imaging using T1 mapping predicts ventricular arrhythmia in ischemic and non-ischemic cardiomyopathy patients with implantable cardioverter-defibrillators. Heart Rhythm 2015, 12, 792–801. [Google Scholar] [CrossRef]
- Vita, T.; Gräni, C.; Abbasi, S.A.; Neilan, T.G.; Rowin, E.; Kaneko, K.; Coelho-Filho, O.; Watanabe, E.; Mongeon, F.P.; Farhad, H.; et al. Comparing CMR Mapping Methods and Myocardial Patterns Toward Heart Failure Outcomes in Nonischemic Dilated Cardiomyopathy. JACC Cardiovasc. Imaging 2019, 12, 1659–1669. [Google Scholar] [CrossRef]
- Romano, S.; Judd, R.M.; Kim, R.J.; Kim, H.W.; Klem, I.; Heitner, J.F.; Shah, D.J.; Jue, J.; White, B.E.; Indorkar, R.; et al. Feature-Tracking Global Longitudinal Strain Predicts Death in a Multicenter Population of Patients With Ischemic and Nonischemic Dilated Cardiomyopathy Incremental to Ejection Fraction and Late Gadolinium Enhancement. JACC Cardiovasc. Imaging 2018, 11, 1419–1429. [Google Scholar] [CrossRef]
- Fatkin, D.; Huttner, I.G.; Kovacic, J.C.; Seidman, J.G.; Seidman, C.E. Precision Medicine in the Management of Dilated Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 2921–2938. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135a. [Google Scholar] [CrossRef] [PubMed]
- Verdonschot, J.A.J.; Hazebroek, M.R.; Krapels, I.P.C.; Henkens, M.; Raafs, A.; Wang, P.; Merken, J.J.; Claes, G.R.F.; Vanhoutte, E.K.; van den Wijngaard, A.; et al. Implications of Genetic Testing in Dilated Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Lopez, L.; Ochoa, J.P.; Royuela, A.; Verdonschot, J.A.J.; Dal Ferro, M.; Espinosa, M.A.; Sabater-Molina, M.; Gallego-Delgado, M.; Larrañaga-Moreira, J.M.; Garcia-Pinilla, J.M.; et al. Clinical Risk Score to Predict Pathogenic Genotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 80, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm 2022, 19, e1–e60. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchía, J.; García-Pinilla, J.M.; Pascual-Figal, D.A.; Nuñez, J.; Guzzo-Merello, G.; et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J. Am. Coll. Cardiol. 2018, 71, 2293–2302. [Google Scholar] [CrossRef]
- Jansweijer, J.A.; Nieuwhof, K.; Russo, F.; Hoorntje, E.T.; Jongbloed, J.D.; Lekanne Deprez, R.H.; Postma, A.V.; Bronk, M.; van Rijsingen, I.A.; de Haij, S.; et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 512–521. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Hazebroek, M.R.; Derks, K.W.J.; Barandiarán Aizpurua, A.; Merken, J.J.; Wang, P.; Bierau, J.; van den Wijngaard, A.; Schalla, S.M.; Abdul Hamid, M.A.; et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur. Heart J. 2018, 39, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef]
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307. [Google Scholar] [CrossRef]
- Hasselberg, N.E.; Edvardsen, T.; Petri, H.; Berge, K.E.; Leren, T.P.; Bundgaard, H.; Haugaa, K.H. Risk prediction of ventricular arrhythmias and myocardial function in Lamin A/C mutation positive subjects. Europace 2014, 16, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Castelletti, S.; Vischer, A.S.; Syrris, P.; Crotti, L.; Spazzolini, C.; Ghidoni, A.; Parati, G.; Jenkins, S.; Kotta, M.C.; McKenna, W.J.; et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. Int. J. Cardiol. 2017, 249, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Parikh, V.N.; Caleshu, C.; Reuter, C.; Lazzeroni, L.C.; Ingles, J.; Garcia, J.; McCaleb, K.; Adesiyun, T.; Sedaghat-Hamedani, F.; Kumar, S.; et al. Regional Variation in RBM20 Causes a Highly Penetrant Arrhythmogenic Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005371. [Google Scholar] [CrossRef]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- Zegkos, T.; Panagiotidis, T.; Parcharidou, D.; Efthimiadis, G. Emerging concepts in arrhythmogenic dilated cardiomyopathy. Heart Fail. Rev. 2021, 26, 1219–1229. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Ayala, J.M.; Gómez-Milanés, I.; Sánchez Muñoz, J.J.; Ruiz-Espejo, F.; Ortíz, M.; González-Carrillo, J.; López-Cuenca, D.; Oliva-Sandoval, M.J.; Monserrat, L.; Valdés, M.; et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: Characterizing a phenotype. Europace 2014, 16, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; Saal, S.; Amarouch, M.Y.; Béziau, D.M.; Marsman, R.F.; Faivre, L.; Barc, J.; Dina, C.; Bertaux, G.; Barthez, O.; et al. Multifocal ectopic Purkinje-related premature contractions: A new SCN5A-related cardiac channelopathy. J. Am. Coll. Cardiol. 2012, 60, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Merlo, M.; Stolfo, D.; Anzini, M.; Negri, F.; Pinamonti, B.; Barbati, G.; Ramani, F.; Lenarda, A.D.; Sinagra, G. Persistent recovery of normal left ventricular function and dimension in idiopathic dilated cardiomyopathy during long-term follow-up: Does real healing exist? J. Am. Heart Assoc. 2015, 4, e001504. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewska-Koperska, J.; Franaszczyk, M.; Bilińska, Z.; Truszkowska, G.; Karczmarz, M.; Szumowski, Ł.; Zieliński, T.; Płoski, R.; Bilińska, M. Rapid and effective response of the R222Q SCN5A to quinidine treatment in a patient with Purkinje-related ventricular arrhythmia and familial dilated cardiomyopathy: A case report. BMC Med. Genet. 2018, 19, 94. [Google Scholar] [CrossRef]
- Rubiś, P.; Wiśniowska-Śmialek, S.; Wypasek, E.; Biernacka-Fijalkowska, B.; Rudnicka-Sosin, L.; Dziewiecka, E.; Faltyn, P.; Khachatryan, L.; Karabinowska, A.; Kozanecki, A.; et al. Fibrosis of extracellular matrix is related to the duration of the disease but is unrelated to the dynamics of collagen metabolism in dilated cardiomyopathy. Inflamm. Res. 2016, 65, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Okumura, T.; Morimoto, R.; Kazama, S.; Shibata, N.; Oishi, H.; Araki, T.; Mizutani, T.; Kuwayama, T.; Hiraiwa, H.; et al. A clinical score for predicting left ventricular reverse remodelling in patients with dilated cardiomyopathy. ESC Heart Fail. 2021, 8, 1359–1368. [Google Scholar] [CrossRef]
- Cannatà, A.; De Angelis, G.; Boscutti, A.; Normand, C.; Artico, J.; Gentile, P.; Zecchin, M.; Heymans, S.; Merlo, M.; Sinagra, G. Arrhythmic risk stratification in non-ischaemic dilated cardiomyopathy beyond ejection fraction. Heart 2020, 106, 656–664. [Google Scholar] [CrossRef]
- Neilan, T.G.; Coelho-Filho, O.R.; Danik, S.B.; Shah, R.V.; Dodson, J.A.; Verdini, D.J.; Tokuda, M.; Daly, C.A.; Tedrow, U.B.; Stevenson, W.G.; et al. CMR quantification of myocardial scar provides additive prognostic information in nonischemic cardiomyopathy. JACC Cardiovasc. Imaging 2013, 6, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Barison, A.; Aimo, A.; Ortalda, A.; Todiere, G.; Grigoratos, C.; Passino, C.; Camici, P.G.; Aquaro, G.D.; Emdin, M. Late gadolinium enhancement as a predictor of functional recovery, need for defibrillator implantation and prognosis in non-ischemic dilated cardiomyopathy. Int. J. Cardiol. 2018, 250, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Merlo, M.; Masè, M.; Vitrella, G.; Belgrano, M.; Faganello, G.; Di Giusto, F.; Boscutti, A.; Gobbo, M.; Gigli, M.; Altinier, A.; et al. Usefulness of Addition of Magnetic Resonance Imaging to Echocardiographic Imaging to Predict Left Ventricular Reverse Remodeling in Patients With Nonischemic Cardiomyopathy. Am. J. Cardiol. 2018, 122, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Höke, U.; Khidir, M.J.; van der Geest, R.J.; Schalij, M.J.; Bax, J.J.; Delgado, V.; Ajmone Marsan, N. Relation of Myocardial Contrast-Enhanced T(1) Mapping by Cardiac Magnetic Resonance to Left Ventricular Reverse Remodeling After Cardiac Resynchronization Therapy in Patients With Nonischemic Cardiomyopathy. Am. J. Cardiol. 2017, 119, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Tobita, T.; Nomura, S.; Fujita, T.; Morita, H.; Asano, Y.; Onoue, K.; Ito, M.; Imai, Y.; Suzuki, A.; Ko, T.; et al. Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci. Rep. 2018, 8, 1998. [Google Scholar] [PubMed] [Green Version]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [Green Version]
- Kadish, A.; Dyer, A.; Daubert, J.P.; Quigg, R.; Estes, N.A.; Anderson, K.P.; Calkins, H.; Hoch, D.; Goldberger, J.; Shalaby, A.; et al. Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N. Engl. J. Med. 2004, 350, 2151–2158. [Google Scholar] [CrossRef] [Green Version]
- Elming, M.B.; Hammer-Hansen, S.; Voges, I.; Nyktari, E.; Raja, A.A.; Svendsen, J.H.; Pehrson, S.; Signorovitch, J.; Køber, L.; Prasad, S.K.; et al. Myocardial fibrosis and the effect of primary prophylactic defibrillator implantation in patients with non-ischemic systolic heart failure-DANISH-MRI. Am. Heart J. 2020, 221, 165–176. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef] [Green Version]
- Beggs, S.A.S.; Jhund, P.S.; Jackson, C.E.; McMurray, J.J.V.; Gardner, R.S. Non-ischaemic cardiomyopathy, sudden death and implantable defibrillators: A review and meta-analysis. Heart 2018, 104, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Gutman, S.J.; Costello, B.T.; Papapostolou, S.; Voskoboinik, A.; Iles, L.; Ja, J.; Hare, J.L.; Ellims, A.; Kistler, P.M.; Marwick, T.H.; et al. Reduction in mortality from implantable cardioverter-defibrillators in non-ischaemic cardiomyopathy patients is dependent on the presence of left ventricular scar. Eur. Heart J. 2019, 40, 542–550. [Google Scholar] [CrossRef]
- Halliday, B.P.; Gulati, A.; Ali, A.; Guha, K.; Newsome, S.; Arzanauskaite, M.; Vassiliou, V.S.; Lota, A.; Izgi, C.; Tayal, U.; et al. Association Between Midwall Late Gadolinium Enhancement and Sudden Cardiac Death in Patients With Dilated Cardiomyopathy and Mild and Moderate Left Ventricular Systolic Dysfunction. Circulation 2017, 135, 2106–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Genetic | Toxicity and Overload | Antineoplastic Drugs | |
|---|---|---|---|
| Cardiac phenotype | Neuromuscular diseases | Alcohol | Anthracycline |
| Titin (TTN) | Duchenne muscular dystrophy | Cocaine | Trastuzumab |
| Lamin A/C (LMNA) | Becker muscular dystrophy | Amphetamines | Antimetabolites |
| Myosin heavy chain (MYH7) | Steinert Emery–Dreifuss muscular dystrophy | Ecstasy | Alkylating agents |
| Troponin T (TNT2) | Hemochromatosis | Monoclonal antibodies | |
| Myosin-binding protein C (MYBPC3) | Amyloidosis | Tyrosine kinase inhibitors | |
| Phosholamban (PLN) | Lead | Immunomodulating agents | |
| Mitochondrial diseases | Psychiatric drugs | ||
| Infectious | Clozapine | ||
| Viruses | Others | Risperidone | |
| Post-myocarditis (Viral) | HIV | Lithium | |
| Enteroviruses | Chagas | Tricyclic antidepressants | |
| Parvovirus B19 | Lyme disease | ||
| Adenoviruses | |||
| Herpes Viruses | |||
| Echoviruses | |||
| Hepatitis C Virus | |||
| Systemic immune-mediated disease | Endocrine/Metabolic | Nutritional deficiency | |
| Autoimmune | Autoinflammatory | Acromegaly | Selenium deficiency |
| Giant-cell myocarditis | Chron’s disease | Pheochromocytoma | Thiamine deficiency |
| Rheumatoid arthritis | Ulcerative colitis | Thyroid dysfunction | |
| Coeliac disease | Gout | ||
| Systemic lupus erythematosus | Reactive arthritis | ||
| Dermatomyositis | |||
| Polymyositis | |||
| Systemic sclerosis | |||
| Primary biliary cirrhosis | |||
| Vasculitis | |||
| Myasthenia gravis | |||
| Pemphigus | |||
| Peripartum | |||
| Tachycardia-induced cardiomyopathy | |||
| Stress-induced cardiomyopathy (Takotsubo) | |||
| Level of Suspicion | Low | Intermediate | High |
|---|---|---|---|
| ECG | Low QRS amplitude Anterolateral T-wave inversion | Fragmented QRS Long QRS NSVT Frequent PVCs | Aborted SCD VT/VF |
| LVEF | ≥50% | 35–50% | <35% |
| GLS 1 | Normal |  |  |
| RV dysfunction | No | Mild impaired | Moderate to severe impairment |
| LGE location | No LGE or free-wall | Septal | Septal + free-wall |
| LGE pattern | Linear mid-wall or focal | Sub-epicardial | Multiple |
| T1 mapping 1 | Normal |  |  |
| ECV | Normal |  |  |
| Genetic panel | TTNtv | PLN RBM 20 SCN5A | LMNA FLNC |
| LVRR Negative Predictors | |
|---|---|
| Clinical | Family history of DCM Symptom duration >90 days |
| ECG | LBBB QRS duration > 120 ms |
| Echocardiography | Very dilated LV (LVDD > 65mm or LVDD/BSA > 35 mm/m2) RV dysfunction unimprovement (at 6–12 months) Mitral regurgitation  GLS1 |
| CMR | LGE presence |
| T2 mapping 1 | Higher T2-mapping value |
| ECV | Higher ECV value |
| Genetic panel | LMNA Structural cytoskeleton Z-disk variants |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, A.; Ferreira, V.; Antunes, M.M.; Lousinha, A.; Pereira-da-Silva, T.; Antunes, D.; Cunha, P.S.; Oliveira, M.; Ferreira, R.C.; Rosa, S.A. Dilated Cardiomyopathy: A Comprehensive Approach to Diagnosis and Risk Stratification. Biomedicines 2023, 11, 834. https://doi.org/10.3390/biomedicines11030834
Ferreira A, Ferreira V, Antunes MM, Lousinha A, Pereira-da-Silva T, Antunes D, Cunha PS, Oliveira M, Ferreira RC, Rosa SA. Dilated Cardiomyopathy: A Comprehensive Approach to Diagnosis and Risk Stratification. Biomedicines. 2023; 11(3):834. https://doi.org/10.3390/biomedicines11030834
Chicago/Turabian StyleFerreira, André, Vera Ferreira, Miguel Marques Antunes, Ana Lousinha, Tiago Pereira-da-Silva, Diana Antunes, Pedro Silva Cunha, Mário Oliveira, Rui Cruz Ferreira, and Sílvia Aguiar Rosa. 2023. "Dilated Cardiomyopathy: A Comprehensive Approach to Diagnosis and Risk Stratification" Biomedicines 11, no. 3: 834. https://doi.org/10.3390/biomedicines11030834