
Biomedical Advances in ABCA1 Transporter: From Bench to Bedside

Abstract
:1. Brief History of ABCA1, Tangier Disease
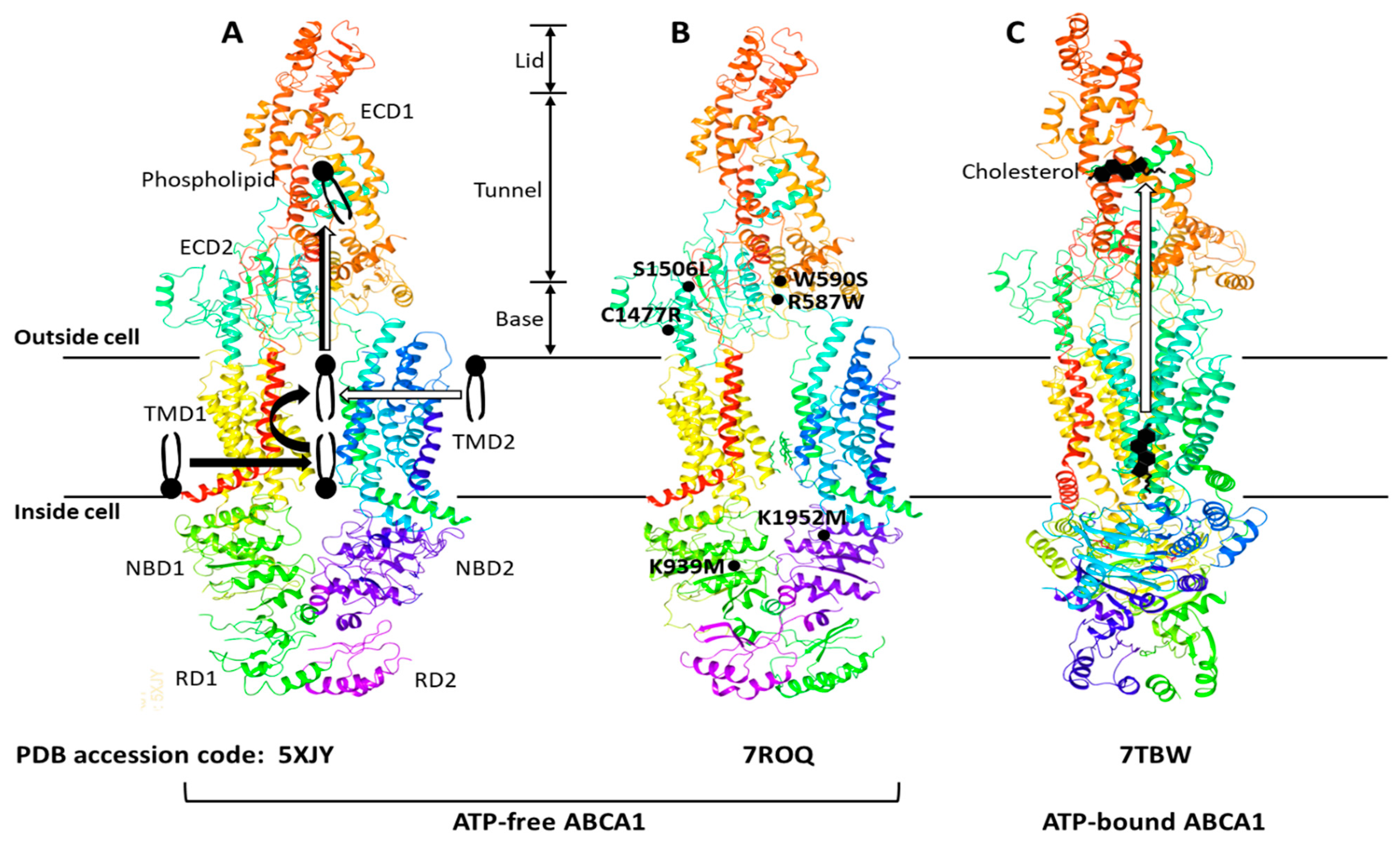
2. The Structure and Function of ABCA1

3. ABCA1 and Biogenesis of HDL
4. Regulation of ABCA1
5. Translational Biology and Drug Trials
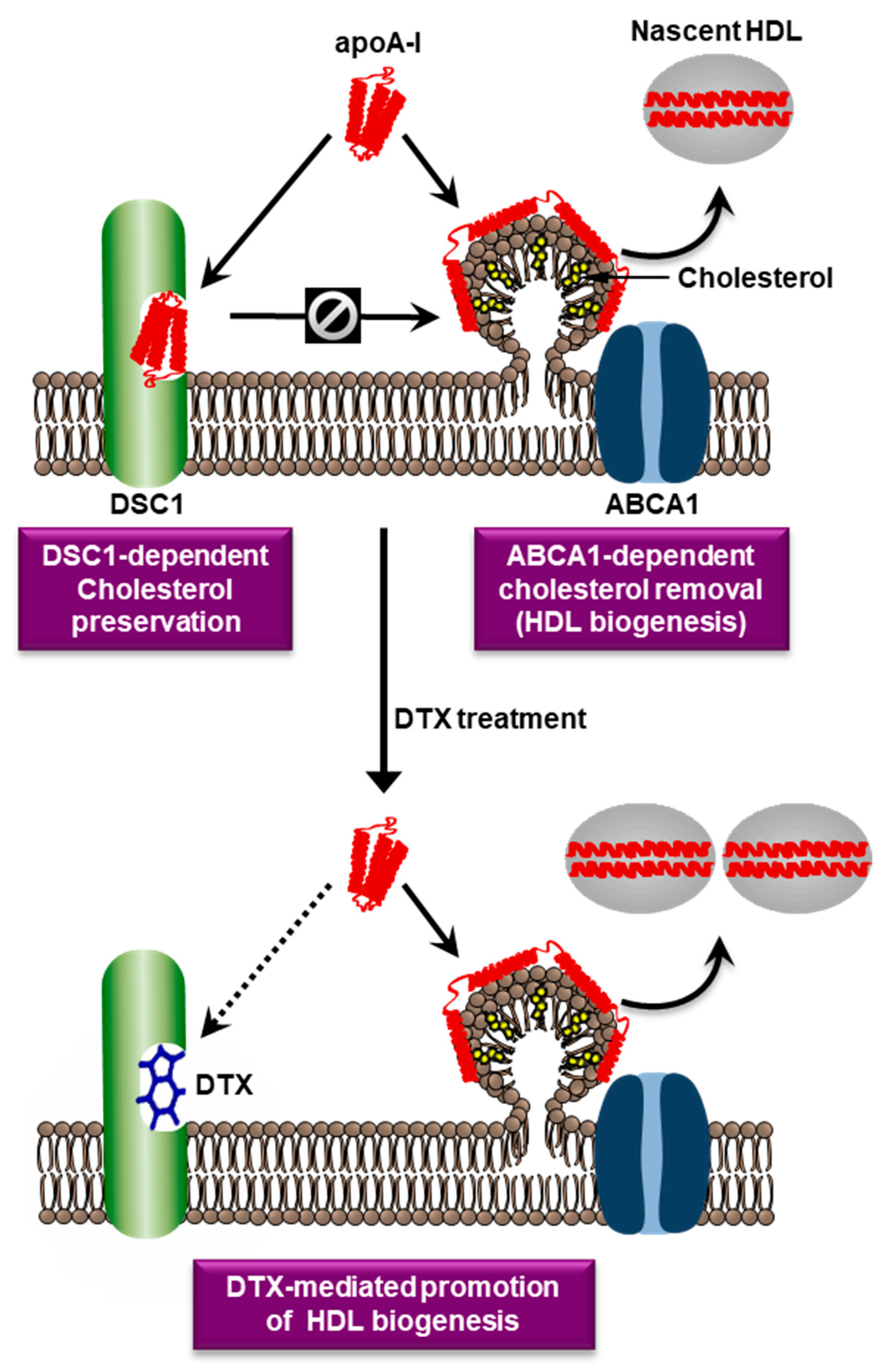
6. A Novel Druggable Target in the ABCA1 Pathway
7. Targeting of DSC1 with Docetaxel
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Bloch, K.E. Evolutionary perfection of a small molecule. In Blondes in Venitian Paintings, the Nine-Banded Armadillo and Other Assays in Biochemistry; Yale University Press: New Haven, CT, USA, 1994; pp. 14–36. ISBN 0-300-05881. [Google Scholar]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oram, J.F. HDL apolipoproteins and ABCA1: Partners in the removal of excess cellular cholesterol. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Mott, S.; Yu, L.; Marcil, M.; Boucher, B.; Rondeau, C.; Genest, J., Jr. Decreased cellular cholesterol efflux is a common cause of familial hypoalphalipoproteinemia: Role of the ABCA1 gene mutations. Atherosclerosis 2000, 152, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Luciani, M.F.; Denizot, F.; Savary, S.; Mattei, M.G.; Chimini, G. Cloning of two novel ABC transporters mapping on human chromosome 9. Genomics 1994, 21, 150–159. [Google Scholar] [CrossRef]
- Becq, F.; Hamon, Y.; Bajetto, A.; Gola, M.; Verrier, B.; Chimini, G. ABC1, an ATP binding cassette transporter required for phagocytosis of apoptotic cells, generates a regulated anion flux after expression in Xenopus laevis oocytes. J. Biol. Chem. 1997, 272, 2695–2699. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, G.; Kaminski, W.E.; Porsch-Ozcurumez, M.; Klucken, J.; Orso, E.; Bodzioch, M.; Buchler, C.; Drobnik, W. ATP-binding cassette transporter A1 (ABCA1) in macrophages: A dual function in inflammation and lipid metabolism? Pathobiology 1999, 67, 236–240. [Google Scholar] [CrossRef]
- Brooks-Wilson, A.; Marcil, M.; Clee, S.M.; Zhang, L.H.; Roomp, K.; van Dam, M.; Yu, L.; Brewer, C.; Collins, J.A.; Molhuizen, H.O.; et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 1999, 22, 336–345. [Google Scholar] [CrossRef]
- Rust, S.; Rosier, M.; Funke, H.; Real, J.; Amoura, Z.; Piette, J.C.; Deleuze, J.F.; Brewer, H.B.; Duverger, N.; Denefle, P.; et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 1999, 22, 352–355. [Google Scholar] [CrossRef]
- Bodzioch, M.; Orso, E.; Klucken, J.; Langmann, T.; Bottcher, A.; Diederich, W.; Drobnik, W.; Barlage, S.; Buchler, C.; Porsch-Ozcurumez, M.; et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999, 22, 347–351. [Google Scholar] [CrossRef]
- Remaley, A.T.; Rust, S.; Rosier, M.; Knapper, C.; Naudin, L.; Broccardo, C.; Peterson, K.M.; Koch, C.; Arnould, I.; Prades, C.; et al. Human ATP-binding cassette transporter 1 (ABC1): Genomic organization and identification of the genetic defect in the original Tangier disease kindred. Proc. Natl. Acad. Sci. USA 1999, 96, 12685–12690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, A.J.; Hegele, R.A.; Burnett, J.R. Tangier disease: Update for 2020. Curr. Opin. Lipidol. 2020, 31, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Oram, J.F. The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim. Biophys. Acta 2009, 1791, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L.; Terasaka, N.; Pagler, T.; Wang, N. HDL, ABC transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008, 7, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frikke-Schmidt, R.; Nordestgaard, B.G.; Stene, M.C.; Sethi, A.A.; Remaley, A.T.; Schnohr, P.; Grande, P.; Tybjaerg-Hansen, A. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 2008, 299, 2524–2532. [Google Scholar] [CrossRef]
- Tall, A.R.; Rader, D.J. Trials and Tribulations of CETP Inhibitors. Circ. Res. 2018, 122, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. HDL in Morbidity and Mortality: A 40+ Year Perspective. Clin. Chem. 2021, 67, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.; Rosolowski, M.; Boettner, J.; Desch, S.; Jobs, A.; Thiele, H.; Buettner, P. High-density lipoprotein cholesterol efflux capacity and incidence of coronary artery disease and cardiovascular mortality: A systematic review and meta-analysis. Lipids Health Dis. 2022, 21, 47. [Google Scholar] [CrossRef]
- Groenen, A.G.; Halmos, B.; Tall, A.R.; Westerterp, M. Cholesterol efflux pathways, inflammation, and atherosclerosis. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 426–439. [Google Scholar] [CrossRef]
- Albrecht, C.; Viturro, E. The ABCA subfamily–gene and protein structures, functions and associated hereditary diseases. Pflugers. Arch. 2007, 453, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.C. Is ABCA1 a lipid transfer protein? J. Lipid Res. 2018, 59, 749–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239.e1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardetzky, O. Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970. [Google Scholar] [CrossRef]
- Segrest, J.P.; Tang, C.; Song, H.D.; Jones, M.K.; Davidson, W.S.; Aller, S.G.; Heinecke, J.W. ABCA1 is an extracellular phospholipid translocase. Nat. Commun. 2022, 13, 4812. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Zhang, Z.; Fang, Q.; Du, B.; Gong, X. Structural basis of substrate recognition and translocation by human ABCA4. Nat. Commun. 2021, 12, 3853. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.; Chen, J. Molecular structures of the eukaryotic retinal importer ABCA4. Elife 2021, 10, e63524. [Google Scholar] [CrossRef]
- Aller, S.G.; Segrest, J.P. The regulatory domains of the lipid exporter ABCA1 form domain swapped latches. PLoS ONE 2022, 17, e0262746. [Google Scholar] [CrossRef]
- Sun, Y.; Li, X. Cholesterol efflux mechanism revealed by structural analysis of human ABCA1 conformational states. Nat. Cardiovasc. Res. 2022, 1, 238–245. [Google Scholar] [CrossRef]
- Wang, S.; Smith, J.D. ABCA1 and nascent HDL biogenesis. Biofactors 2014, 40, 547–554. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, Z.W.; Zeng, P.H.; Zhou, Y.J.; Yin, W.J. Molecular mechanisms for ABCA1-mediated cholesterol efflux. Cell Cycle 2022, 21, 1121–1139. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.O.; Nakada, C.; Kasai, R.S.; Kusumi, A.; Ueda, K. ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc. Natl. Acad. Sci. USA 2013, 110, 5034–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigami, M.; Ogasawara, F.; Nagao, K.; Hashimoto, H.; Kimura, Y.; Kioka, N.; Ueda, K. Temporary sequestration of cholesterol and phosphatidylcholine within extracellular domains of ABCA1 during nascent HDL generation. Sci. Rep. 2018, 8, 6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedhachalam, C.; Ghering, A.B.; Davidson, W.S.; Lund-Katz, S.; Rothblat, G.H.; Phillips, M.C. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1603–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segrest, J.P.; Jones, M.K.; Catte, A.; Manchekar, M.; Datta, G.; Zhang, L.; Zhang, R.; Li, L.; Patterson, J.C.; Palgunachari, M.N.; et al. Surface Density-Induced Pleating of a Lipid Monolayer Drives Nascent High-Density Lipoprotein Assembly. Structure 2015, 23, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- Timmins, J.M.; Lee, J.Y.; Boudyguina, E.; Kluckman, K.D.; Brunham, L.R.; Mulya, A.; Gebre, A.K.; Coutinho, J.M.; Colvin, P.L.; Smith, T.L.; et al. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J. Clin. Investig. 2005, 115, 1333–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunham, L.R.; Kruit, J.K.; Iqbal, J.; Fievet, C.; Timmins, J.M.; Pape, T.D.; Coburn, B.A.; Bissada, N.; Staels, B.; Groen, A.K.; et al. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Investig. 2006, 116, 1052–1062. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yeung, A.W.K.; Atanasov, A.G. A Review: Molecular Mechanism of Regulation of ABCA1 Expression. Curr. Protein Pept. Sci. 2022, 23, 170–191. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, Q.; Wang, L.; Wang, Y.; Wang, D.; Ding, H. Role of ABCA1 in Cardiovascular Disease. J. Pers. Med. 2022, 12, 1010. [Google Scholar] [CrossRef]
- Infante, T.; Franzese, M.; Ruocco, A.; Schiano, C.; Affinito, O.; Pane, K.; Memoli, D.; Rizzo, F.; Weisz, A.; Bontempo, P.; et al. ABCA1, TCF7, NFATC1, PRKCZ, and PDGFA DNA methylation as potential epigenetic-sensitive targets in acute coronary syndrome via network analysis. Epigenetics 2022, 17, 547–563. [Google Scholar] [CrossRef]
- Tachibana, K.; Kusumoto, K.; Ogawa, M.; Ando, H.; Shimizu, T.; Ishima, Y.; Ishida, T.; Okuhira, K. FTY720 Reduces Lipid Accumulation by Upregulating ABCA1 through Liver X Receptor and Sphingosine Kinase 2 Signaling in Macrophages. Int. J. Mol. Sci. 2022, 23, 14617. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.J.; Mooradian, A.D. Potential Therapeutic Agents That Target ATP Binding Cassette A1 (ABCA1) Gene Expression. Drugs 2022, 82, 1055–1075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, L.; Wang, J.; Zhang, T.; Ye, T.; Wang, S.; Xing, D.; Chen, W. Recent advances in the regulation of ABCA1 and ABCG1 by lncRNAs. Clin. Chim. Acta 2021, 516, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Arakawa, R.; Wu, C.A.; Iwamoto, N.; Lu, R.; Tsujita, M.; Abe-Dohmae, S. Calpain-mediated ABCA1 degradation: Post-translational regulation of ABCA1 for HDL biogenesis. Biochim. Biophys. Acta 2012, 1821, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Hayashi, H.; Kusuhara, H. Cellular Cholesterol Accumulation Facilitates Ubiquitination and Lysosomal Degradation of Cell Surface-Resident ABCA1. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1347–1356. [Google Scholar] [CrossRef]
- Genest, J.; Choi, H.Y. Novel Approaches for HDL-Directed Therapies. Curr. Atheroscler. Rep. 2017, 19, 55. [Google Scholar] [CrossRef]
- Arakawa, R.; Tsujita, M.; Iwamoto, N.; Ito-Ohsumi, C.; Lu, R.; Wu, C.A.; Shimizu, K.; Aotsuka, T.; Kanazawa, H.; Abe-Dohmae, S.; et al. Pharmacological inhibition of ABCA1 degradation increases HDL biogenesis and exhibits antiatherogenesis. J. Lipid Res. 2009, 50, 2299–2305. [Google Scholar] [CrossRef] [Green Version]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [Green Version]
- Group, H.T.C. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: Trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur. Heart J. 2013, 34, 1279–1291. [Google Scholar] [CrossRef] [Green Version]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Group, A.S.; Ginsberg, H.N.; Elam, M.B.; Lovato, L.C.; Crouse, J.R., 3rd; Leiter, L.A.; Linz, P.; Friedewald, W.T.; Buse, J.B.; Gerstein, H.C.; et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 2010, 362, 1563–1574. [Google Scholar] [CrossRef]
- Jun, M.; Foote, C.; Lv, J.; Neal, B.; Patel, A.; Nicholls, S.J.; Grobbee, D.E.; Cass, A.; Chalmers, J.; Perkovic, V. Effects of fibrates on cardiovascular outcomes: A systematic review and meta-analysis. Lancet 2010, 375, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Das Pradhan, A.; Glynn, R.J.; Fruchart, J.C.; MacFadyen, J.G.; Zaharris, E.S.; Everett, B.M.; Campbell, S.E.; Oshima, R.; Amarenco, P.; Blom, D.J.; et al. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N. Engl. J. Med. 2022, 387, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, E.; Giaccari, A.; Gallo, M.; Targher, G.; Pintaudi, B.; Candido, R.; Monami, M.; SID-AMD Joint Panel for Italian Guidelines on Treatment of Type 2 Diabetes. Effects of pioglitazone on cardiovascular events and all-cause mortality in patients with type 2 diabetes: A meta-analysis of randomized controlled trials. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Munehira, Y.; Ohnishi, T.; Kawamoto, S.; Furuya, A.; Shitara, K.; Imamura, M.; Yokota, T.; Takeda, S.; Amachi, T.; Matsuo, M.; et al. Alpha1-syntrophin modulates turnover of ABCA1. J. Biol. Chem. 2004, 279, 15091–15095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, R.; Yokoyama, S. Helical apolipoproteins stabilize ATP-binding cassette transporter A1 by protecting it from thiol protease-mediated degradation. J. Biol. Chem. 2002, 277, 22426–22429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Lay, S.; Robichon, C.; Le Liepvre, X.; Dagher, G.; Ferre, P.; Dugail, I. Regulation of ABCA1 expression and cholesterol efflux during adipose differentiation of 3T3-L1 cells. J. Lipid. Res. 2003, 44, 1499–1507. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.Y.; Ruel, I.; Choi, S.; Genest, J. New Strategies to Promote Macrophage Cholesterol Efflux. Front. Cardiovasc. Med. 2021, 8, 795868. [Google Scholar] [CrossRef]
- Choi, H.Y.; Rahmani, M.; Wong, B.W.; Allahverdian, S.; McManus, B.M.; Pickering, J.G.; Chan, T.; Francis, G.A. ATP-binding cassette transporter A1 expression and apolipoprotein A-I binding are impaired in intima-type arterial smooth muscle cells. Circulation 2009, 119, 3223–3231. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef]
- Choi, H.Y.; Ruel, I.; Malina, A.; Garrod, D.R.; Oda, M.N.; Pelletier, J.; Schwertani, A.; Genest, J. Desmocollin 1 is abundantly expressed in atherosclerosis and impairs high-density lipoprotein biogenesis. Eur. Heart J. 2018, 39, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Genest, J.; Schwertani, A.; Choi, H.Y. Membrane microdomains and the regulation of HDL biogenesis. Curr. Opin. Lipidol. 2018, 29, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Ruel, I.; Genest, J. Identification of Docetaxel as a Potential Drug to Promote HDL Biogenesis. Front. Pharmacol. 2021, 12, 679456. [Google Scholar] [CrossRef] [PubMed]
- Xiang, P.; Blanchard, V.; Francis, G.A. Smooth Muscle Cell-Macrophage Interactions Leading to Foam Cell Formation in Atherosclerosis: Location, Location, Location. Front. Physiol. 2022, 13, 921597. [Google Scholar] [CrossRef]
- Kenmotsu, H.; Tanigawara, Y. Pharmacokinetics, dynamics and toxicity of docetaxel: Why the Japanese dose differs from the Western dose. Cancer Sci. 2015, 106, 497–504. [Google Scholar] [CrossRef]
- Baker, S.D.; Zhao, M.; Lee, C.K.; Verweij, J.; Zabelina, Y.; Brahmer, J.R.; Wolff, A.C.; Sparreboom, A.; Carducci, M.A. Comparative pharmacokinetics of weekly and every-three-weeks docetaxel. Clin. Cancer Res. 2004, 10, 1976–1983. [Google Scholar] [CrossRef] [Green Version]
- Matesanz, R.; Barasoain, I.; Yang, C.G.; Wang, L.; Li, X.; de Ines, C.; Coderch, C.; Gago, F.; Barbero, J.J.; Andreu, J.M.; et al. Optimization of taxane binding to microtubules: Binding affinity dissection and incremental construction of a high-affinity analog of paclitaxel. Chem. Biol. 2008, 15, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Canales, A.; Rodriguez-Salarichs, J.; Trigili, C.; Nieto, L.; Coderch, C.; Andreu, J.M.; Paterson, I.; Jimenez-Barbero, J.; Diaz, J.F. Insights into the interaction of discodermolide and docetaxel with tubulin. Mapping the binding sites of microtubule-stabilizing agents by using an integrated NMR and computational approach. ACS Chem. Biol. 2011, 6, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Bayet-Robert, M.; Morvan, D.; Chollet, P.; Barthomeuf, C. Pharmacometabolomics of docetaxel-treated human MCF7 breast cancer cells provides evidence of varying cellular responses at high and low doses. Breast Cancer Res. Treat. 2010, 120, 613–626. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Class | Drug | Mechanism of Action | Clinical Trial | ASCVD Outcomes |
|---|---|---|---|---|
| Niacin | Niacin | LXR | AIM-HIGH [49], HPS2-THRIVE [50] | Neutral/Harm |
| Glitazones | Pioglitazone | PPARγ | Meta-analysis [55] | Neutral/Harm |
| Fibric acid derivatives | Fenofibrate, Pemafibrate | PPARα | FIELD [51], ACCORD [52], PROMINENT [54] | Neutral |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.Y.; Choi, S.; Iatan, I.; Ruel, I.; Genest, J. Biomedical Advances in ABCA1 Transporter: From Bench to Bedside. Biomedicines 2023, 11, 561. https://doi.org/10.3390/biomedicines11020561
Choi HY, Choi S, Iatan I, Ruel I, Genest J. Biomedical Advances in ABCA1 Transporter: From Bench to Bedside. Biomedicines. 2023; 11(2):561. https://doi.org/10.3390/biomedicines11020561
Chicago/Turabian StyleChoi, Hong Y., Senna Choi, Iulia Iatan, Isabelle Ruel, and Jacques Genest. 2023. "Biomedical Advances in ABCA1 Transporter: From Bench to Bedside" Biomedicines 11, no. 2: 561. https://doi.org/10.3390/biomedicines11020561