
Hypertrophic, Dilated, and Arrhythmogenic Cardiomyopathy: Where Are We?


Abstract
:1. Introduction
2. Hypertrophic Cardiomyopathy
2.1. Etiology and Pathophysiology
2.2. Clinical Manifestation
2.3. Diagnosis
2.3.1. Electrocardiogram and Dynamic Holter
2.3.2. Echocardiography
2.3.3. Cardiac Magnetic Resonance Imaging
2.3.4. Genetic Counselling and Testing
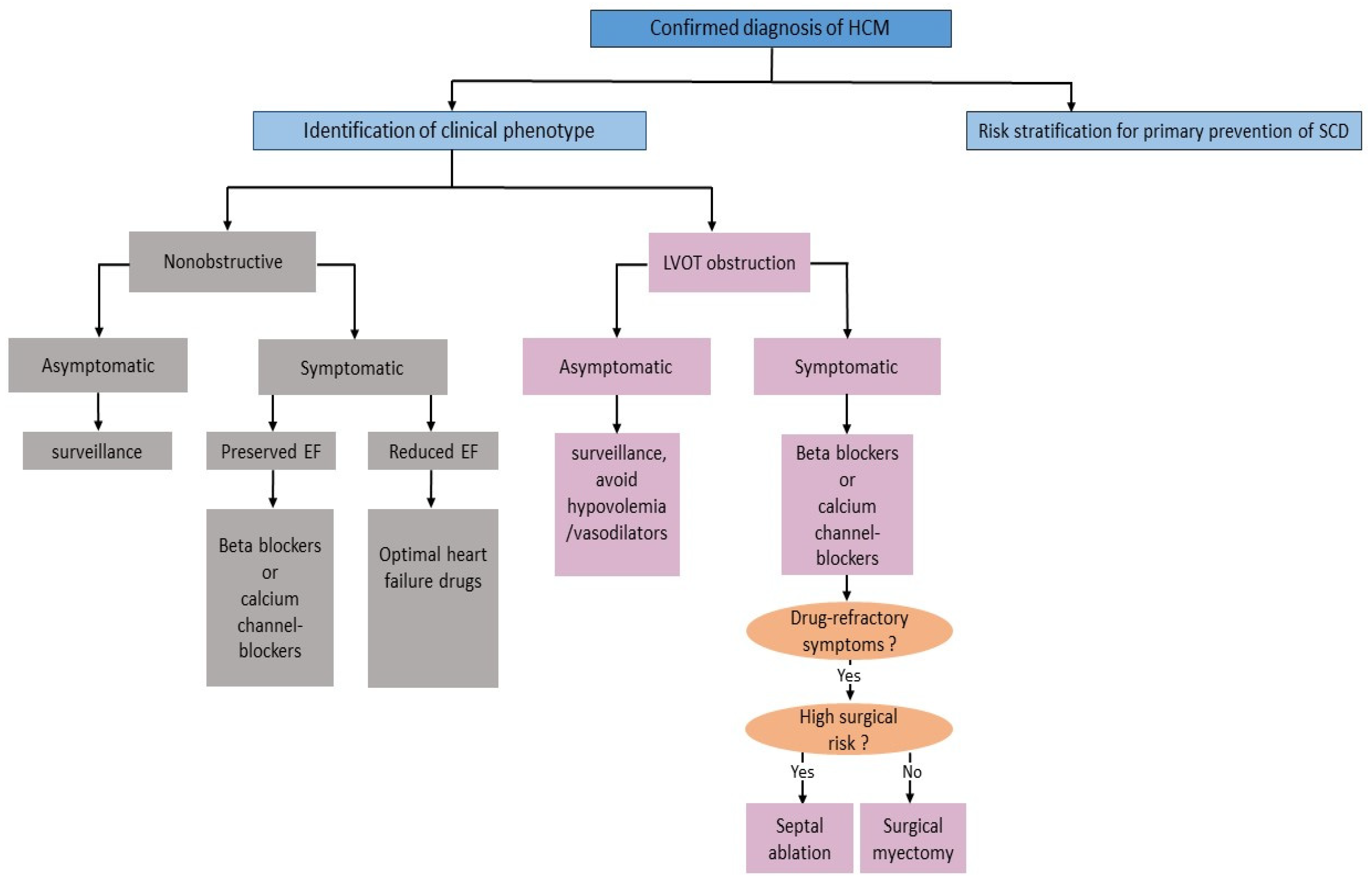
2.4. Management Aspects
2.4.1. Lifestyle Modification
2.4.2. Pharmacologic Therapy
2.4.3. Septal Reduction Therapy
2.4.4. Risk Stratification and Primary Prevention of SCD
2.5. Emerging Therapeutic Approaches for HCM
2.5.1. Modulators of Cardiac Myosin
2.5.2. Ion Channel Inhibitors
2.5.3. Myocardial Metabolism Modulators
3. Dilated Cardiomyopathy
3.1. Etiology and Pathophysiology
3.1.1. Genetic Causes of DCM
3.1.2. Nongenetic Causes of DCM
Myocarditis
Drugs and Cardiotoxins
Peripartum Cardiomyopathy
3.2. Clinical Manifestation
3.3. Diagnosis
3.3.1. Echocardiography
3.3.2. CMR
3.3.3. Coronary Angiography and Endomyocardial Biopsy
3.3.4. Genetic Counselling and Testing
3.4. Management Aspects
3.4.1. Pharmacological Therapy
3.4.2. Primary Prevention of SCD
3.4.3. Approach to End-Stage Heart Failure
3.5. Emerging Pharmacological Approaches in Non-Ischemic DCM
4. Arrhythmogenic Cardiomyopathy
4.1. Definition
4.2. Etiology and Pathophysiology
4.2.1. Genetic Hallmarks
4.2.2. Inflammation
4.3. Clinical Manifestation
4.4. Diagnosis
4.4.1. Electrocardiogram
4.4.2. Echocardiography
4.4.3. CMR
4.4.4. EMB
4.4.5. Genetic Counselling and Testing
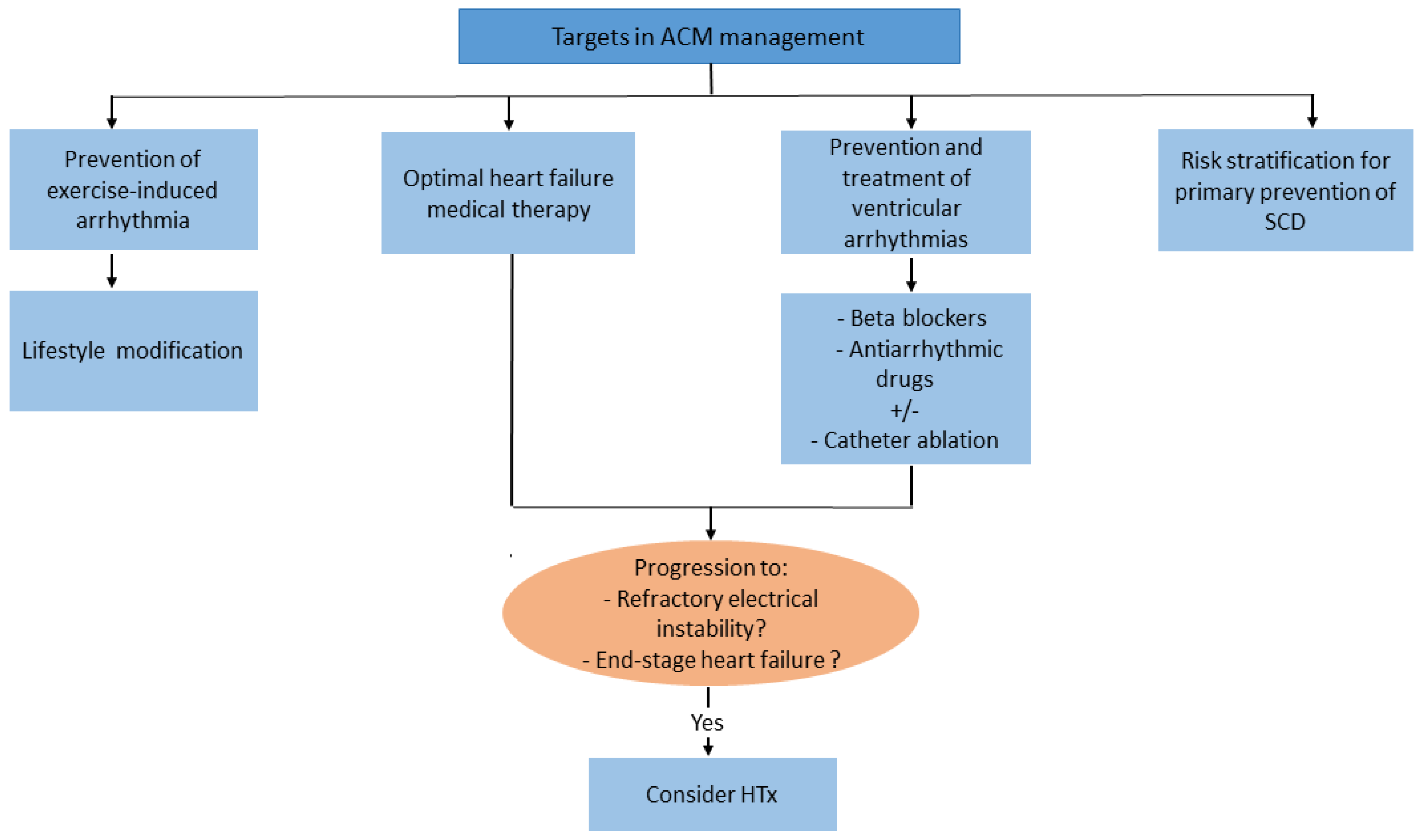
4.5. Management Aspects
4.5.1. Lifestyle Changes and Pharmacological Therapy
4.5.2. Primary Prevention of SCD
4.6. Promising Pharmacological Therapies in ACM
5. Conclusions and Gaps in Knowledge
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| ACTC1 | actin-alpha cardiac muscle |
| ACE | angiotensin-converting enzyme |
| ACM | arrhythmogenic cardiomyopathy |
| ACTC | cardiac actin |
| ATPase | adenosine triphosphatase |
| ARBs | angiotensin receptor blockers |
| ARNI | angiotensin receptor-neprilysin inhibitor |
| ACC | American College of Cardiology |
| AHA | American Heart Association |
| BMCs | marrow-derived cells |
| BM-MSCs | bone marrow mesenchymal stem cells |
| CAD | coronary artery disease |
| CMR | cardiac magnetic resonance |
| COVID-19 | coronavirus disease 2019 |
| DCM | dilated cardiomyopathy |
| DES | desmin |
| DMD | dystrophin |
| DSC2 | desmocollin |
| DSG2 | desmoglein |
| DSP | desmoplakin |
| ESC | European Society of Cardiology |
| EDMD | muscular dystrophy Emery-Dreyfus |
| EMB | endomyocardial biopsy |
| ECV | extracellular volume |
| FDA | U.S. Food and Drug administration |
| FWHM | full-width at half maximum |
| FLNC | filamin C |
| G-CSF | granulocyte colony-stimulating factor |
| HbA1c | haemoglobin A1c |
| HCM | hypertrophic cardiomyopathy |
| HTx | orthotopic heart transplantation |
| IL-1β | Interleukine-1β |
| ICD | implantable cardioverter-defibrillator |
| LVEF | left ventricular ejection of fraction |
| LVEDVi | left ventricular end-diastolic volume index |
| LVEDD | left ventricular end-diastolic diameter |
| LGE | late gadolinium enhancement |
| LMNA | Lamin A/C |
| LRP6 | low density lipoprotein receptor-related protein 6 |
| LVAD | left ventricular assist devices |
| LVOT | left ventricular outflow tract |
| MAACE | mortality and major adverse arrhythmic cardiac events |
| MYH7 | beta-myosin heavy chain |
| NT-proBNP | N-terminal pro-brain natriuretic peptide |
| NF-κB | nuclear factor-κB |
| NYHA | New York Heart Association |
| PKP2 | plakophilin |
| PLN | phospholamban |
| PPCM | peripartum cardiomyopathy |
| SAM | systolic anterior motion of the mitral valve |
| SCD | sudden cardiac death |
| SGCD | delta-sarcoglycan |
| SGLT2 | sodium-glucose co-transporter 2 |
| TDI | tissue Doppler imaging |
| TNNI3 | troponin I |
| TNNT2 | troponin T |
| TTN | titin |
| TDI | tissue Doppler imaging |
| TNFα | tumor necrosis factor alpha |
| ↑ | increase |
| ↓ | decrease |
| ↔ | no change |
References
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Vulpian, A. Contribution à l’étude des rétrécissements de l’orifice ventriculo-aortique. Arch. Physiol. 1868, 3, 456–457. [Google Scholar]
- Teare, D. Asymmetrical hypertrophy of the heart in young adults. Br. Heart J. 1958, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brockenbrough, E.C.; Braunwald, E.; Morrow, A.G. A Hemodynamic Technic for the Detection of Hypertrophic Subaortic Stenosis. Circulation 1961, 23, 189–194. [Google Scholar] [CrossRef]
- Maron, B.J. Clinical course and management of hypertrophic cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Ware, S.M. Importance of genetic evaluation and testing in pediatric cardiomyopathy. World J. Cardiol. 2014, 6, 1156–1165. [Google Scholar] [CrossRef]
- Vakrou, S.; Abraham, M.R. Hypertrophic cardiomyopathy: A heart in need of an energy bar? Front. Physiol. 2014, 5, 309. [Google Scholar] [CrossRef]
- Ashrafian, H.; Redwood, C.; Blair, E.; Watkins, H. Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet. 2003, 19, 263–268. [Google Scholar] [CrossRef]
- Güçlü, A.; Knaapen, P.; Harms, H.J.; Parbhudayal, R.Y.; Michels, M.; Lammertsma, A.A.; van Rossum, A.C.; Germans, T.; van der Velden, J. Disease Stage-Dependent Changes in Cardiac Contractile Performance and Oxygen Utilization Underlie Reduced Myocardial Efficiency in Human Inherited Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Imaging 2017, 10, e005604. [Google Scholar] [CrossRef]
- Aoyama, R.; Takano, H.; Kobayashi, Y.; Kitamura, M.; Asai, K.; Amano, Y.; Kumita, S.I.; Shimizu, W. Evaluation of myocardial glucose metabolism in hypertrophic cardiomyopathy using 18F-fluorodeoxyglucose positron emission tomography. PLoS ONE 2017, 12, e0188479. [Google Scholar] [CrossRef]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2022, 118, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Malahfji, M.; Senapati, A.; Debs, D.; Angulo, C.; Zhan, Y.; Nagueh, S.F.; Shah, D.J. Examining the impact of inducible ischemia on myocardial fibrosis and exercise capacity in hypertrophic cardiomyopathy. Sci. Rep. 2020, 10, 15977. [Google Scholar] [CrossRef] [PubMed]
- Baxi, A.J.; Restrepo, C.S.; Vargas, D.; Marmol-Velez, A.; Ocazionez, D.; Murillo, H. Hypertrophic Cardiomyopathy from A to Z: Genetics, Pathophysiology, Imaging, and Management. Radiographics 2016, 36, 335–354. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Gimeno, J.R.; Tomé, M.T.; Shah, J.; Ward, D.; Thaman, R.; Mogensen, J.; McKenna, W.J. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2006, 27, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Olivotto, I.; Zenovich, A.G.; Link, M.S.; Pandian, N.G.; Kuvin, J.T.; Nistri, S.; Cecchi, F.; Udelson, J.E.; Maron, B.J. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006, 114, 2232–2239. [Google Scholar] [CrossRef]
- Marstrand, P.; Han, L.; Day, S.M.; Olivotto, I.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Wittekind, S.G.; Helms, A.; Saberi, S.; et al. SHaRe Investigators. Hypertrophic Cardiomyopathy With Left Ventricular Systolic Dysfunction: Insights From the SHaRe Registry. Circulation 2020, 141, 1371–1383. [Google Scholar] [CrossRef]
- Adamczak, D.M.; Oko-Sarnowska, Z. Sudden Cardiac Death in Hypertrophic Cardiomyopathy. Cardiol. Rev. 2018, 26, 145–151. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Sheikh, N.; Biagini, E.; Papadakis, M.; Maurizi, N.; Sinagra, G.; Pelliccia, A.; Rapezzi, C.; Sharma, S.; Olivotto, I. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020, 17, 142–151. [Google Scholar] [CrossRef]
- Pelliccia, A.; Sharma, S.; Gati, S.; Bäck, M.; Börjesson, M.; Caselli, S.; Collet, J.P.; Corrado, D.; Drezner, J.A.; Halle, M.; et al. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur. Heart J. 2021, 42, 17–96. [Google Scholar] [CrossRef]
- Roşca, M.; Călin, A.; Beladan, C.C.; Enache, R.; Mateescu, A.D.; Gurzun, M.M.; Varga, P.; Băicuş, C.; Coman, I.M.; Jurcuţ, R.; et al. Right ventricular remodeling, its correlates, and its clinical impact in hypertrophic cardiomyopathy. J. Am. Soc. Echocardiogr. 2015, 28, 1329–1338. [Google Scholar] [CrossRef]
- Serri, K.; Reant, P.; Lafitte, M.; Berhouet, M.; Le Bouffos, V.; Roudaut, R.; Lafitte, S. Global and regional myocardial function quantification by two-dimensional strain: Application in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2006, 47, 1175–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Pozios, I.; Haileselassie, B.; Nowbar, A.; Sorensen, L.L.; Phillip, S.; Lu, D.Y.; Ventoulis, I.; Luo, H.; Abraham, M.R.; et al. Role of Global Longitudinal Strain in Predicting Outcomes in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2017, 120, 670–675. [Google Scholar] [CrossRef]
- Rakowski, H.; Hoss, S.; Williams, L.K. Echocardiography in the Diagnosis and Management of Hypertrophic Cardiomyopathy. Cardiol Clin. 2019, 37, 11–26. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [PubMed]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar]
- Tower-Rader, A.; Kramer, C.M.; Neubauer, S.; Nagueh, S.F.; Desai, M.Y. Multimodality Imaging in Hypertrophic Cardiomyopathy for Risk Stratification. Circ. Cardiovasc. Imaging 2020, 13, e009026. [Google Scholar] [CrossRef]
- Raman, B.; Ariga, R.; Spartera, M.; Sivalokanathan, S.; Chan, K.; Dass, S.; Petersen, S.E.; Daniels, M.J.; Francis, J.; Smillie, R.; et al. Progression of myocardial fibrosis in hypertrophic cardiomyopathy: Mechanisms and clinical implications. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Yao, J.; Chan, R.H.; He, J.; Yang, X.; Zhou, Y.; He, Y. Prognostic Value of LGE-CMR in HCM: A Meta-Analysis. JACC Cardiovasc. Imaging 2016, 12, 1392–1402. [Google Scholar] [CrossRef]
- Mentias, A.; Raeisi-Giglou, P.; Smedira, N.G.; Feng, K.; Sato, K.; Wazni, O.; Kanj, M.; Flamm, S.D.; Thamilarasan, M.; Popovic, Z.B.; et al. Late Gadolinium Enhancement in Patients With Hypertrophic Cardiomyopathy and Preserved Systolic Function. J. Am. Coll. Cardiol. 2018, 72, 857–870. [Google Scholar] [CrossRef]
- Ko, C.; Arscott, P.; Concannon, M.; Saberi, S.; Day, S.M.; Yashar, B.M.; Helms, A.S. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet. Med. 2018, 20, 69–75. [Google Scholar] [CrossRef]
- Hoss, S.; Habib, M.; Silver, J.; Care, M.; Chan, R.H.; Hanneman, K.; Morel, C.F.; Iwanochko, R.M.; Gollob, M.H.; Rakowski, H.; et al. Genetic Testing for Diagnosis of Hypertrophic Cardiomyopathy Mimics: Yield and Clinical Significance. Circ. Genom. Precis. Med. 2020, 13, e002748. [Google Scholar] [CrossRef]
- Dybro, A.M.; Rasmussen, T.B.; Nielsen, R.R.; Andersen, M.J.; Jensen, M.K.; Poulsen, S.H. Randomized Trial of Metoprolol in Patients With Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2021, 78, 2505–2517. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [PubMed]
- Cui, H.; Schaff, H.V.; Wang, S.; Lahr, B.D.; Rowin, E.J.; Rastegar, H.; Hu, S.; Eleid, M.F.; Dearani, J.A.; Kimmelstiel, C.; et al. Survival Following Alcohol Septal Ablation or Septal Myectomy for Patients With Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 1647–1655. [Google Scholar] [CrossRef]
- Mentias, A.; Smedira, N.G.; Krishnaswamy, A.; Reed, G.W.; Ospina, S.; Thamilarasan, M.; Popovic, Z.B.; Xu, B.; Kapadia, S.R.; Desai, M.Y. Survival After Septal Reduction in Patients > 65 Years Old With Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 105–115. [Google Scholar] [CrossRef]
- Norrish, G.; Qu, C.; Field, E.; Cervi, E.; Khraiche, D.; Klaassen, S.; Ojala, T.H.; Sinagra, G.; Yamazawa, H.; Marrone, C.; et al. External validation of the HCM Risk-Kids model for predicting sudden cardiac death in childhood hypertrophic cardiomyopathy. Eur. J. Prev. Cardiol. 2022, 29, 678–686. [Google Scholar] [CrossRef]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten Treatment for Obstructive Hypertrophic Cardiomyopathy: A Clinical Trial. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Mealiffe, M.E.; Bach, R.G.; Bhattacharya, M.; Choudhury, L.; Edelberg, J.M.; Hegde, S.M.; Jacoby, D.; Lakdawala, N.K.; Lester, S.J.; et al. Evaluation of Mavacamten in Symptomatic Patients With Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Owens, A.; Geske, J.B.; Wolski, K.; Naidu, S.S.; Smedira, N.G.; Cremer, P.C.; Schaff, H.; McErlean, E.; Sewell, C.; et al. Myosin Inhibition in Patients With Obstructive Hypertrophic Cardiomyopathy Referred for Septal Reduction Therapy. J. Am. Coll. Cardiol. 2022, 80, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.; Collibee, S.; Ashcraft, L.; Wang, W.; Vander Wal, M.; Wang, X.; Hwee, D.T.; Wu, Y.; Wang, J.; Chin, E.R.; et al. Discovery of Aficamten (CK-274), a Next-Generation Cardiac Myosin Inhibitor for the Treatment of Hypertrophic Cardiomyopathy. J. Med. Chem. 2021, 64, 14142–14152. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Choudhury, L.; Olivotto, I.; Saberi, S.; Wang, A.; Garcia-Pavia, P.; Lakdawala, N.K.; Nagueh, S.F.; Rader, F.; et al. Phase 2 Study of Aficamten in Patients With Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 34–45. [Google Scholar] [CrossRef] [PubMed]
- CY 6031 Study Will Evaluate the Effects of Treatment with Aficamten (CK-3773274) over a 24-Week Period on Cardiopulmonary Exercise Capacity and Health Status in Patients with Symptomatic oHCM (SEQUOIA-HCM). Available online: https://www.clinicaltrials.gov/ct2/show/NCT05186818 (accessed on 20 December 2022).
- Coppini, R.; Ferrantini, C.; Yao, L.; Fan, P.; Del Lungo, M.; Stillitano, F.; Sartiani, L.; Tosi, B.; Suffredini, S.; Tesi, C.; et al. Late Sodium Current Inhibition Reverses Electromechanical Dysfunction in Human Hypertrophic Cardiomyopathy. Circulation 2013, 127, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Coppini, R.; Mazzoni, L.; Ferrantini, C.; Gentile, F.; Pioner, J.M.; Laurino, A.; Santini, L.; Bargelli, V.; Rotellini, M.; Bartolucci, G.; et al. Ranolazine Prevents Phenotype Development in a Mouse Model of Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2017, 10, 003565. [Google Scholar] [CrossRef] [PubMed]
- Tamargo, J.; Lopez-Sendon, J. Ranolazine: A better understanding of its pathophysiology and patient profile to guide treatment of chronic stable angina. Future Cardiol. 2022, 18, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Camici, P.G.; Merlini, P.A.; Rapezzi, C.; Patten, M.; Climent, V.; Sinagra, G.; Tomberli, B.; Marin, F.; Ehlermann, P.; et al. Efficacy of Ranolazine in Patients With Symptomatic Hypertrophic Cardiomyopathy: The RESTYLE-HCM Randomized, Double-Blind, Placebo-Controlled Study. Circ. Heart Fail. 2018, 11, e004124. [Google Scholar] [CrossRef]
- Olivotto, I.; Hellawell, J.L.; Farzaneh-Far, R.; Blair, C.; Coppini, R.; Myers, J.; Belardinelli, L.; Maron, M.S. Novel Approach Targeting the Complex Pathophysiology of Hypertrophic Cardiomyopathy: The Impact of Late Sodium Current Inhibition on Exercise Capacity in Subjects with Symptomatic Hypertrophic Cardiomyopathy (LIBERTY-HCM) Trial. Circ. Heart Fail. 2016, 9, e002764. [Google Scholar] [CrossRef]
- Lee, L.; Campbell, R.; Scheuermann-Freestone, M.; Taylor, R.; Gunaruwan, P.; Williams, L.; Ashrafian, H.; Horowitz, J.; Fraser, A.G.; Clarke, K.; et al. Metabolic modulation with perhexiline in chronic heart failure: A randomized, controlled trial of short-term use of a novel treatment. Circulation 2005, 112, 3280–3288. [Google Scholar] [CrossRef] [PubMed]
- Abozguia, K.; Elliott, P.; McKenna, W.; Phan, T.T.; Nallur-Shivu, G.; Ahmed, I.; Maher, A.R.; Kaur, K.; Taylor, J.; Henning, A.; et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 2010, 122, 1562–1569. [Google Scholar] [CrossRef]
- A Study to Evaluate the Safety, Tolerability, and Efficacy of IMB-1018972 in Patients with Non-Obstructive Hypertrophic Cardiomyopathy Trial (IMPROVE-HCM). Available online: https://clinicaltrials.gov/ct2/show/NCT04826185 (accessed on 20 November 2022).
- Tremaine, L.; Al-Fayoumi, S.; Wetering, J.; Van Iersel, T.; Patel, J.; Chamberlin, P. A clinical drug-drug interaction study of Imb-1018972, a novel investigational cardiac mitotrope in phase 2 development for the treatment of myocardial ischemia and hypertrophic cardiomyopathy. Circulation. 2021, 144, A10372. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving concepts in dilated cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Tayal, U.; Ware, J.S.; Lakdawala, N.K.; Heymans, S.; Prasad, S.K. Understanding the genetics of adult-onset dilated cardiomyopathy: What a clinician needs to know. Eur. Heart J. 2021, 42, 2384–2396. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef]
- McNally, E.M.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Tharp, C.A.; Haywood, M.E.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. The Giant Protein Titin’s Role in Cardiomyopathy: Genetic, Transcriptional, and Post-translational Modifications of TTN and Their Contribution to Cardiac Disease. Front. Physiol. 2019, 10, 1436. [Google Scholar] [CrossRef] [PubMed]
- Crasto, S.; My, I.; Di Pasquale, E. The Broad Spectrum of LMNA Cardiac Diseases: From Molecular Mechanisms to Clinical Phenotype. Front. Physiol. 2020, 11, 761. [Google Scholar] [CrossRef]
- Dal Ferro, M.; Severini, G.M.; Gigli, M.; Mestroni, L.; Sinagra, G. Genetics of Dilated Cardiomyopathy: Current Knowledge and Future Perspectives. In Dilated Cardiomyopathy; Sinagra, G., Merlo, M., Pinamonti, B., Eds.; Springer: Cham, Switzerland, 2019; ISBN 978-3-030-13864-6. [Google Scholar]
- Chen, Z.; Li, Y.; Wang, Y.; Qian, J.; Ma, H.; Wang, X.; Jiang, G.; Liu, M.; An, Y.; Ma, L.; et al. Cardiomyocyte-Restricted Low Density Lipoprotein Receptor-Related Protein 6 (LRP6) Deletion Leads to Lethal Dilated Cardiomyopathy Partly Through Drp1 Signaling. Theranostics 2018, 8, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.P.; Malipiero, G.; Marcolongo, R.; Iliceto, S. Myocarditis: A Clinical Overview. Curr. Cardiol. Rep. 2017, 19, 63. [Google Scholar] [CrossRef]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and Outcomes of 21 Critically Ill Patients With COVID-19 in Washington State. JAMA 2020, 323, 1612–1614. [Google Scholar] [CrossRef]
- Komiyama, M.; Hasegawa, K.; Matsumori, A. Dilated Cardiomyopathy Risk in Patients with Coronavirus Disease 2019: How to Identify and Characterise it Early? Eur. Cardiol. 2020, 15, e49. [Google Scholar] [CrossRef] [PubMed]
- Laonigro, I.; Correale, M.; Di Biase, M.; Altomare, E. Alcohol abuse and heart failure. Eur. J. Heart Fail. 2009, 11, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Mirijello, A.; Tarli, C.; Vassallo, G.A.; Sestito, L.; Antonelli, M.; d’Angelo, C.; Ferrulli, A.; De Cosmo, S.; Gasbarrini, A.; Addolorato, G. Alcoholic cardiomyopathy: What is known and what is not known. Eur. J. Intern. Med. 2017, 43, 1–5. [Google Scholar] [CrossRef]
- Mladěnka, P.; Applová, L.; Patočka, J.; Costa, V.M.; Remiao, F.; Pourová, J.; Mladěnka, A.; Karlíčková, J.; Jahodář, L.; Vopršalová, M.; et al. TOX-OER and CARDIOTOX Hradec Králové Researchers and Collaborators. Comprehensive review of cardiovascular toxicity of drugs and related agents. Med. Res. Rev. 2018, 38, 1332–1403. [Google Scholar] [CrossRef]
- Davis, M.B.; Arany, Z.; McNamara, D.M.; Goland, S.; Elkayam, U. Peripartum Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, J.; König, T.; van der Meer, P.; Petrie, M.C.; Hilfiker-Kleiner, D.; Mbakwem, A.; Hamdan, R.; Jackson, A.M.; Forsyth, P.; de Boer, R.A.; et al. Pathophysiology, diagnosis and management of peripartum cardiomyopathy: A position statement from the Heart Failure Association of the European Society of Cardiology Study Group on peripartum cardiomyopathy. Eur. J. Heart Fail. 2019, 21, 827–843. [Google Scholar] [CrossRef]
- Puwanant, S.; Priester, T.C.; Mookadam, F.; Bruce, C.J.; Redfield, M.M.; Chandrasekaran, K. Right ventricular function in patients with preserved and reduced ejection fraction heart failure. Eur. J. Echocardiogr. 2009, 10, 733–737. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Merken, J.J.; Brunner-La Rocca, H.P.; Hazebroek, M.R.; Eurlings, C.G.M.J.; Thijssen, E.; Wang, P.; Weerts, J.; van Empel, V.; Schummers, G.; et al. Value of Speckle Tracking-Based Deformation Analysis in Screening Relatives of Patients With Asymptomatic Dilated Cardiomyopathy. JACC Cardiovasc. Imaging 2020, 13 2 Pt 2, 549–558. [Google Scholar] [CrossRef]
- Brown, P.F.; Miller, C.; Di Marco, A.; Schmitt, M. Towards cardiac MRI based risk stratification in idiopathic dilated cardiomyopathy. Heart 2019, 105, 270–275. [Google Scholar] [CrossRef]
- Wang, J.; Yang, F.; Wan, K.; Mui, D.; Han, Y.; Chen, Y. Left ventricular midwall fibrosis as a predictor of sudden cardiac death in non-ischaemic dilated cardiomyopathy: A meta-analysis. ESC Heart Fail. 2020, 7, 2184–2192. [Google Scholar] [CrossRef]
- Guaricci, A.I.; Masci, P.G.; Muscogiuri, G.; Guglielmo, M.; Baggiano, A.; Fusini, L.; Lorenzoni, V.; Martini, C.; Andreini, D.; Pavon, A.G.; et al. CarDiac magnEtic Resonance for prophylactic Implantable-cardioVerter defibrillAtor ThErapy in Non-Ischaemic dilated CardioMyopathy: An international Registry. Europace 2021, 23, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Mathew, T.; Williams, L.; Navaratnam, G.; Rana, B.; Wheeler, R.; Collins, K.; Harkness, A.; Jones, R.; Knight, D.; O’Gallagher, K.; et al. Diagnosis and assessment of dilated cardiomyopathy: A guideline protocol from the British Society of Echocardiography. Echo. Res. Pract. 2017, 4, G1–G13. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm. 2022, 19, e1–e60. [Google Scholar] [CrossRef]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, B.; Du, J.; Wang, J.; Jing, X.; Liu, Y.; Deng, S.; Du, J.; She, Q. SGLT2i versus ARNI in heart failure with reduced ejection fraction: A systematic review and meta-analysis. ESC Heart Fail. 2021, 8, 2210–2219. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Beggs, S.A.S.; Jhund, P.S.; Jackson, C.E.; McMurray, J.J.V.; Gardner, R.S. Non-ischaemic cardiomyopathy, sudden death and implantable defibrillators: A review and meta-analysis. Heart 2018, 104, 144–150. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef]
- Elming, M.B.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Signorovitch, J.; Olesen, L.L.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Age and Outcomes of Primary Prevention Implantable Cardioverter-Defibrillators in Patients With Nonischemic Systolic Heart Failure. Circulation 2017, 136, 1772–1780. [Google Scholar] [CrossRef]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef]
- Crespo-Leiro, M.G.; Barge-Caballero, E. Advanced Heart Failure: Definition, Epidemiology, and Clinical Course. Heart Fail Clin. 2021, 17, 533–545. [Google Scholar] [CrossRef]
- Fischer, S.A.; Doree, C.; Mathur, A.; Taggart, D.P.; Martin-Rendon, E. Stem cell therapy for chronic ischaemic heart disease and congestive heart failure. Cochrane Database Syst. Rev. 2016, 12, CD007888. [Google Scholar] [CrossRef]
- Fischer-Rasokat, U.; Assmus, B.; Seeger, F.H.; Honold, J.; Leistner, D.; Fichtlscherer, S.; Schächinger, V.; Tonn, T.; Martin, H.; Dimmeler, S.; et al. A pilot trial to assess potential effects of selective intracoronary bone marrow-derived progenitor cell infusion in patients with nonischemic dilated cardiomyopathy: Final 1-year results of the transplantation of progenitor cells and functional regeneration enhancement pilot trial in patients with nonischemic dilated cardiomyopathy. Circ. Heart Fail. 2009, 2, 417–423. [Google Scholar]
- Seth, S.; Bhargava, B.; Narang, R.; Ray, R.; Mohanty, S.; Gulati, G.; Kumar, L.; Airan, B.; Venugopal, P. AIIMS Stem Cell Study Group. The ABCD (Autologous Bone Marrow Cells in Dilated Cardiomyopathy) trial a long-term follow-up study. J. Am. Coll. Cardiol. 2010, 55, 1643–1644. [Google Scholar] [CrossRef]
- Hamshere, S.; Arnous, S.; Choudhury, T.; Choudry, F.; Mozid, A.; Yeo, C.; Barrett, C.; Saunders, N.; Gulati, A.; Knight, C.; et al. Randomized trial of combination cytokine and adult autologous bone marrow progenitor cell administration in patients with non-ischaemic dilated cardiomyopathy: The REGENERATE-DCM clinical trial. Eur. Heart J. 2015, 36, 3061–3069. [Google Scholar] [CrossRef]
- Hare, J.M.; DiFede, D.L.; Rieger, A.C.; Florea, V.; Landin, A.M.; El-Khorazaty, J.; Khan, A.; Mushtaq, M.; Lowery, M.H.; Byrnes, J.J.; et al. Randomized Comparison of Allogeneic Versus Autologous Mesenchymal Stem Cells for Nonischemic Dilated Cardiomyopathy: POSEIDON-DCM Trial. J. Am. Coll. Cardiol. 2017, 69, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Te Rijdt, W.P.; Hoorntje, E.T.; de Brouwer, R.; Oomen, A.; Amin, A.; van der Heijden, J.F.; Karper, J.C.; Westenbrink, B.D.; Silljé, H.H.W.; Te Riele, A.S.J.M.; et al. Rationale and design of the PHOspholamban RElated CArdiomyopathy intervention STudy (i-PHORECAST). Neth. Heart J. 2022, 30, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Champion, S.; Lapidus, N.; Cherié, G.; Spagnoli, V.; Oliary, J.; Solal, A.C. Pentoxifylline in heart failure: A meta-analysis of clinical trials. Cardiovasc. Ther. 2014, 32, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Akdis, D.; Saguner, A.M.; Shah, K.; Wei, C.; Medeiros-Domingo, A.; von Eckardstein, A.; Lüscher, T.F.; Brunckhorst, C.; Chen, H.S.V.; Duru, F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: From a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur. Heart J. 2017, 38, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Cerrone, M.; Saguner, A.M.; Brunckhorst, C.; Delmar, M.; Duru, F. Arrhythmogenic cardiomyopathy: An in-depth look at molecular mechanisms and clinical correlates. Trends Cardiovasc. Med. 2021, 31, 395–402. [Google Scholar] [CrossRef]
- Basso, C.; Czarnowska, E.; Della Barbera, M.; Bauce, B.; Beffagna, G.; Wlodarska, E.K.; Pilichou, K.; Ramondo, A.; Lorenzon, A.; Wozniek, O.; et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: An electron microscopy investigation on endomyocardial biopsies. Eur. Heart J. 2006, 27, 1847–1854. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Calore, M.; Lorenzon, A.; De Bortoli, M.; Poloni, G.; Rampazzo, A. Arrhythmogenic cardiomyopathy: A disease of intercalated discs. Cell Tissue Res. 2015, 360, 491–500. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, R.; Huang, Y.; Yang, Z.; Xian, J.; Huang, J.; Qiu, Z.; Lin, X.; Zhang, M.; Chen, H.; et al. Reactivation of PPARα alleviates myocardial lipid accumulation and cardiac dysfunction by improving fatty acid β-oxidation in Dsg2-deficient arrhythmogenic cardiomyopathy. Acta Pharm. Sin. B 2022, 13, 192–203. [Google Scholar] [CrossRef]
- Asatryan, B.; Asimaki, A.; Landstrom, A.P.; Khanji, M.Y.; Odening, K.E.; Cooper, L.T.; Marchlinski, F.E.; Gelzer, A.R.; Semsarian, C.; Reichlin, T.; et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation 2021, 144, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Campian, M.E.; Verberne, H.J.; Hardziyenka, M.; de Groot, E.A.; van Moerkerken, A.F.; van Eck-Smit, B.L.; Tan, H.L. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef] [Green Version]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, G.D.; De Luca, A.; Cappelletto, C.; Raimondi, F.; Bianco, F.; Botto, N.; Lesizza, P.; Grigoratos, C.; Minati, M.; Dell’Omodarme, M.; et al. Prognostic Value of Magnetic Resonance Phenotype in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2753–2765. [Google Scholar] [CrossRef]
- Perazzolo Marra, M.; Cipriani, A.; Rizzo, S.; De Lazzari, M.; De Gaspari, M.; Akrami, N.; Bariani, R.; Zorzi, A.; Migliore, F.; Rigato, I.; et al. Myocardial Tissue Characterization in Arrhythmogenic Cardiomyopathy: Comparison Between Endomyocardial Biopsy and Cardiac Magnetic Resonance. JACC Cardiovasc. Imaging 2021, 14, 1675–1678. [Google Scholar] [CrossRef]
- James, C.A.; Syrris, P.; van Tintelen, J.P.; Calkins, H. The role of genetics in cardiovascular disease: Arrhythmogenic cardiomyopathy. Eur Heart J. 2020, 41, 1393–1400. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Prondzynski, M.; Carrier, L.; Pu, W.T. Gene therapy for inherited arrhythmias. Cardiovasc. Res. 2020, 116, 1635–1650. [Google Scholar] [CrossRef]
- Scalco, A.; Liboni, C.; Angioni, R.; Di Bona, A.; Albiero, M.; Bertoldi, N.; Fadini, G.P.; Thiene, G.; Chelko, S.P.; Basso, C.; et al. Arrhythmogenic Cardiomyopathy Is a Multicellular Disease Affecting Cardiac and Bone Marrow Mesenchymal Stromal Cells. J. Clin. Med. 2021, 10, 1871. [Google Scholar] [CrossRef]
- Quarta, G.; Aquaro, G.D.; Pedrotti, P.; Pontone, G.; Dellegrottaglie, S.; Iacovoni, A.; Brambilla, P.; Pradella, S.; Todiere, G.; Rigo, F.; et al. Cardiovascular magnetic resonance imaging in hypertrophic cardiomyopathy: The importance of clinical context. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Pathak, R.K.; Sanders, P.; Deo, R. Primary prevention implantable cardioverter-defibrillator and opportunities for sudden cardiac death risk assessment in non-ischaemic cardiomyopathy. Eur. Heart J. 2018, 39, 2859–2866. [Google Scholar] [CrossRef]
- C-MRI Guidance of Implantable CRT-Implantation in Non-Ischemic Dilated Cardiomyopathy (CMR-ICD). Available online: https://clinicaltrials.gov/ct2/show/NCT04558723 (accessed on 25 October 2022).
- Agrimi, J.; Scalco, A.; Agafonova, J.; Williams Iii, L.; Pansari, N.; Keceli, G.; Jun, S.; Wang, N.; Mastorci, F.; Tichnell, C.; et al. Psychosocial Stress Hastens Disease Progression and Sudden Death in Mice with Arrhythmogenic Cardiomyopathy. J. Clin. Med. 2020, 9, 3804. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Protein | Cardiac Phenotype |
|---|---|---|
| MYBPC | Myosin-binding protein C | HCM |
| MYH7 | Myosin heavy chain 7 | HCM, DCM |
| TNNI3, TNNT2 | Cardiac troponin I and T | HCM, DCM |
| LMNA | Lamin A/C | DCM, ACM |
| PLN | Phospolamban | DCM, ACM |
| FLNC | Filamin C | DCM, ACM |
| DES | Desmin | DCM; ACM |
| ACTC | Actin alpha cardiac muscle 1 | DCM |
| SGCD | Delta-sarcoglycan | DCM |
| DSP | Desmoplakin | ACM |
| PKP2 | Plakophilin-2 | ACM |
| DSC2 | Desmocollin-2 | ACM |
| DSG2 | Desmoglein-2 | ACM |
| TMEM43 | Transmembrane protein 43 | ACM |
| TGF-β3 | Transforming growth factor-β3 | ACM |
| SCN5A | Cardiac sodium channel | ACM |
| Trial | Phase | Agent | No. of Patients | Patient Category | Key Findings |
|---|---|---|---|---|---|
| PIONEER-HCM (prospective, open label) | 2 | Mavacamten | 20 | Obstructive HCM with resting LVOT gradient ≥ 30 mmHg | ↓ Exercise-induced LVOT gradient ↓ Dyspnea scores ↑ Peak oxygen consumption |
| Explorer-HCM (prospective, randomized, double blind) | 3 | Mavacamten | 251 | Obstructive HCM with NYHA class II-III | ↓ Exercise-induced LVOT gradient ↑ Peak oxygen consumption ↓ NYHA class |
| VALOR-HCM (prospective, randomized, double blind) | 3 | Mavacamten | 112 | Obstructive HCM referred for septal reduction procedure | ↓ Exercise-induced LVOT gradient ↓ NYHA class ↓ need septal reduction therapy |
| MAVERICK-HCM (prospective, randomized, double blind) | 2 | Mavacamten | 59 | Nonobstructive HCM with NYHA class II-III | No significant change in Peak oxygen consumption or NYHA class ↓ NT-proBNP ↓ High-sensitivity troponin |
| SEQUOIA-HCM (prospective, randomized, double blind) | 3 | Aficamten | 270 (estimated) | Symptomatic obstructive HCM | Ongoing study, aims to: Evaluate changes in peak oxygen consumption, NYHA class, post-valsalva LVOT gradient |
| REDWOOD-HCM (prospective, randomized, double blind) | 2 | Aficamten | 41 | Obstructive HCM with NYHA class II-III | ↓ Resting and provocable LVOT gradient (dose-dependent) ↓ NYHA class |
| RESTYLE-HCM (prospective, randomized, double blind) | 2 | Ranolazine | 80 | Nonobstructive HCM | No significant improvement in peak oxygen consumption, exercise tolerance or quality of life ↓ arrhythmic profile |
| LIBERTY-HCM (prospective, randomized, double blind) | 2 | Eleclazine | 172 | Obstructive HCM NYHA class I-IV | No significant improvement in exercise tolerance |
| Trial | Phase | Agent | No. of Patients | Follow-Up (Months) | Patient Category | Key Findings |
|---|---|---|---|---|---|---|
| iPHORECAST (prospective, randomized, open-label) | III | Eplerenone | 60 | 36 | Phospholamban R14del Carriers with NYHA II and LVEF ≥ 40% | Results pending, aims to: assess CMR parameters (functional and morphological), changes in electrocardiographic indicators and NYHA class, cardiovascular death |
| TOPCARE-DCM (prospective, open-label) | II | Intracoronary infusion of BMCs | 33 (33 treated, no control) | 12 | NYHA I-III and LVEF ≤ 40% | ↑ LVEF |
| ABCD trial (prospective, randomized, open-label) | II | Intracoronary infusion of BMCs | 85 (45 treated, 40 control) | 6 | NYHA ≥ II and LVEF ≤ 40% | ↑ LVEF ↑ Quality of life ↔ Mortality |
| REGENERATE-DCM (prospective, randomized, placebo-controlled, double-blind) | II | Intracoronary infusion of BMCs and peripheral G-CSF | 60 (30 treated, 30 control) | 12 | NYHA ≥ II and LVEF ≤ 45% | ↑ LVEF ↓ NYHA class ↑ exercise capacity |
| POSEIDON-DCM (prospective, randomized, open-label) | I/II | autologous or allogeneic BMCs | 37 (19 allo BMC, 18 auto BMC) | 12 | LVEF < 40% and LVEDD > 5.9 cm in males and > 5.6 cm in females | ↑ LVEF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Hadi, H.; Freund, A.; Desch, S.; Thiele, H.; Majunke, N. Hypertrophic, Dilated, and Arrhythmogenic Cardiomyopathy: Where Are We? Biomedicines 2023, 11, 524. https://doi.org/10.3390/biomedicines11020524
El Hadi H, Freund A, Desch S, Thiele H, Majunke N. Hypertrophic, Dilated, and Arrhythmogenic Cardiomyopathy: Where Are We? Biomedicines. 2023; 11(2):524. https://doi.org/10.3390/biomedicines11020524
Chicago/Turabian StyleEl Hadi, Hamza, Anne Freund, Steffen Desch, Holger Thiele, and Nicolas Majunke. 2023. "Hypertrophic, Dilated, and Arrhythmogenic Cardiomyopathy: Where Are We?" Biomedicines 11, no. 2: 524. https://doi.org/10.3390/biomedicines11020524