
The Bidirectional Relationship of NPY and Mitochondria in Energy Balance Regulation


Abstract
:1. Introduction
2. NPY as the Master Regulator of Energy Balance
2.1. NPY Regulation and Feeding Regulation—Ghrelin and Leptin Take the Control
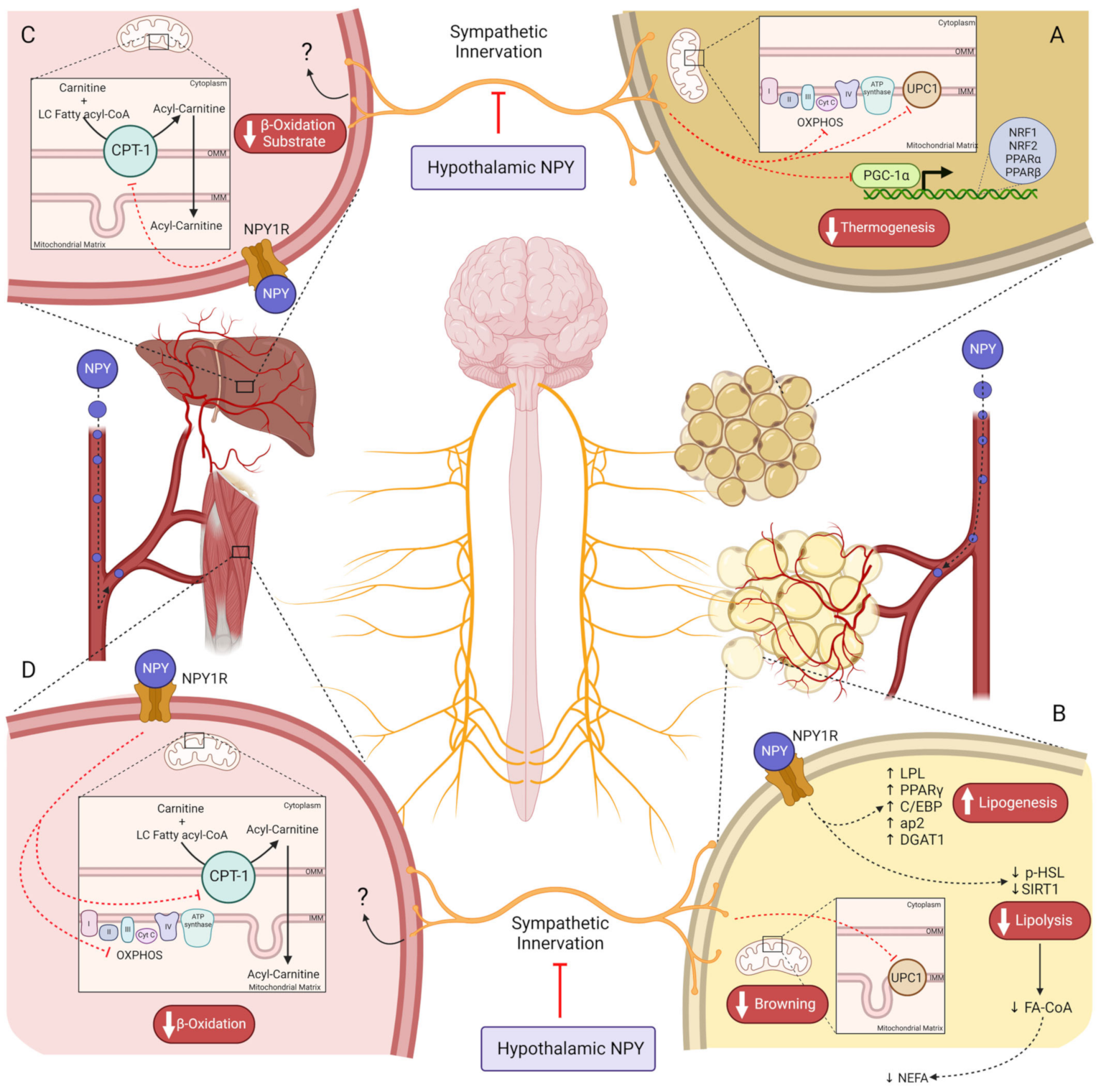
2.2. NPY Regulates Energy Balance through Energy Expenditure Inhibition
2.2.1. Central NPY Effects
2.2.2. Peripheral NPY Effects
3. Mitochondria as a Regulator of Energy Balance and Metabolism
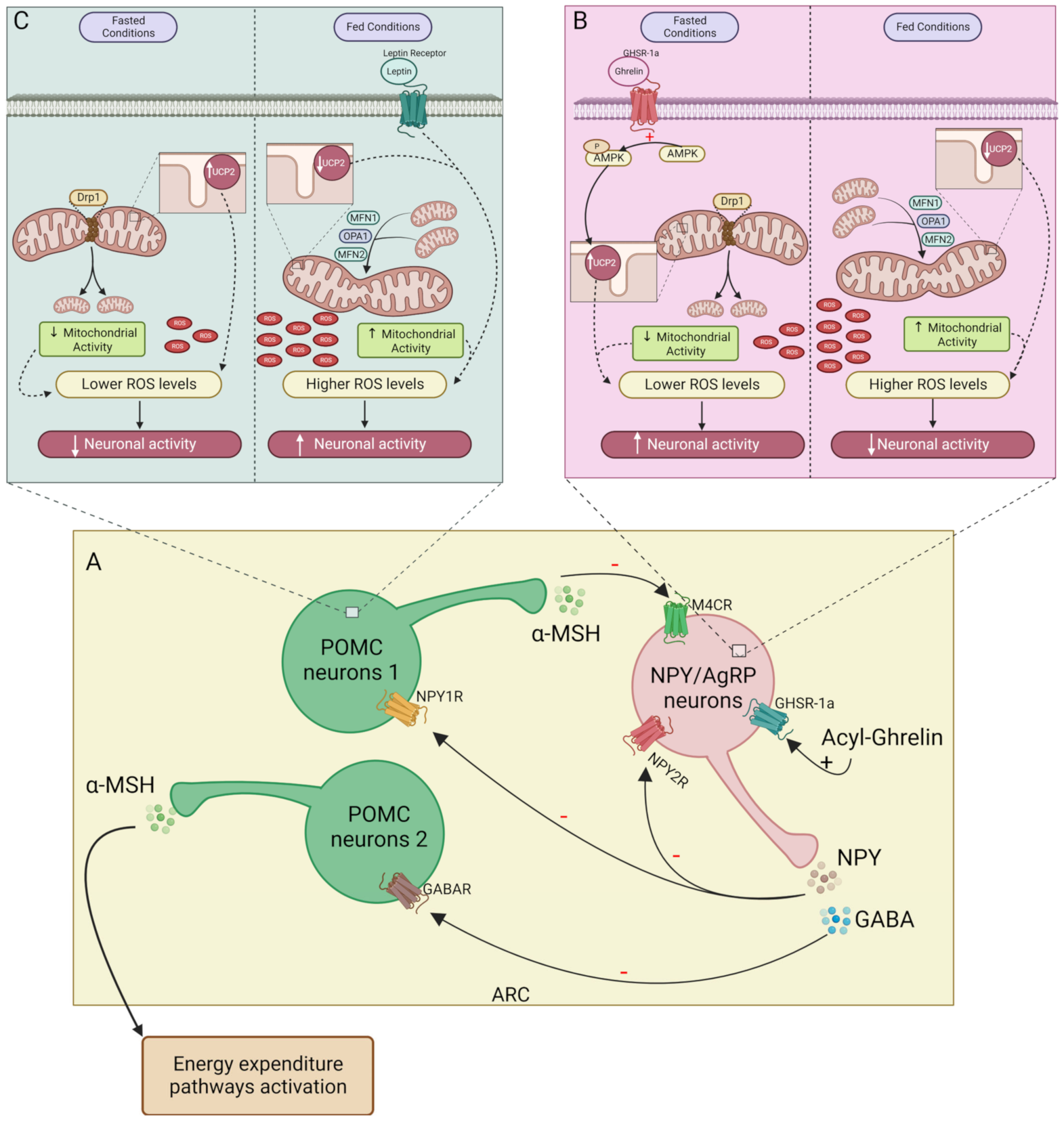
3.1. Mitochondria’s Role in Central Control of Energy Balance
3.2. Obesogenic Diets-Induced Mitochondrial Alterations on Feeding Circuitry
4. NPY as a Regulator of Mitochondrial (dys) Function
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, L.; Bijker, M.S.; Herzog, H. The neuropeptide Y system: Pathophysiological and therapeutic implications in obesity and cancer. Pharmacol. Ther. 2011, 131, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Gumbs, M.C.; Heuvel, J.K.V.D.; la Fleur, S.E. The effect of obesogenic diets on brain Neuropeptide Y. Physiol. Behav. 2016, 162, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Gyengesi, E.; Paxinos, G.; Andrews, Z.B. Oxidative Stress in the Hypothalamus: The Importance of Calcium Signaling and Mitochondrial ROS in Body Weight Regulation. Curr. Neuropharmacol. 2012, 10, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, C.M.; Horvath, T.L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014, 10, 650–658. [Google Scholar] [CrossRef]
- Valassi, E.; Scacchi, M.; Cavagnini, F. Neuroendocrine control of food intake. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 158–168. [Google Scholar] [CrossRef]
- Costa, J.; Moreira, A.; Moreira, P.; Delgado, L.; Silva, D. Effects of weight changes in the autonomic nervous system: A systematic review and meta-analysis. Clin. Nutr. 2019, 38, 110–126. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hinder, L.M.; Callaghan, B.C.; Feldman, E.L. Neurological consequences of obesity. Lancet Neurol. 2017, 16, 465–477. [Google Scholar] [CrossRef]
- Thorp, A.A.; Schlaich, M.P. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J. Diabetes Res. 2015, 2015, 341583. [Google Scholar] [CrossRef]
- Li, R.; Guan, H.; Yang, K. Neuropeptide Y potentiates beta-adrenergic stimulation of lipolysis in 3T3-L1 adipocytes. Regul. Pept. 2012, 178, 16–20. [Google Scholar] [CrossRef]
- Cho, Y.K.; La Lee, Y.; Jung, C.H. Pathogenesis, Murine Models, and Clinical Implications of Metabolically Healthy Obesity. Int. J. Mol. Sci. 2022, 23, 9614. [Google Scholar] [CrossRef]
- Tang, H.-N.; Xiao, F.; Chen, Y.-R.; Zhuang, S.-Q.; Guo, Y.; Wu, H.-X.; Zhou, H.-D. Higher Serum Neuropeptide y Levels Are Associated with Metabolically Unhealthy Obesity in Obese Chinese Adults: A Cross-Sectional Study. Mediat. Inflamm. 2020, 2020, 7903140. [Google Scholar] [CrossRef]
- Patel, H.R.; Qi, Y.; Hawkins, E.J.; Hileman, S.M.; Elmquist, J.K.; Imai, Y.; Ahima, R.S. Neuropeptide Y deficiency attenuates responses to fasting and high-fat diet in obesity-prone mice. Diabetes 2006, 55, 3091–3098. [Google Scholar] [CrossRef]
- Jackson, E.K.; Gillespie, D.G.; Tofovic, S.P. DPP4 inhibition, NPY1-36, PYY1-36, SDF-1α, and a hypertensive genetic background conspire to augment cell proliferation and collagen production: Effects that are abolished by low concentrations of 2-methoxyestradiol. J. Pharmacol. Exp. Ther. 2020, 373, 135–148. [Google Scholar] [CrossRef]
- Wahlestedt, C.; Grundemar, L.; Håkanson, R.; Heilig, M.; Shen, G.H.; Zukowska-Grojec, Z.; Reis, N.J. Neuropeptide Y Receptor Subtypes, Y1 and Y2. Ann. N. Y. Acad. Sci. 1990, 611, 7–26. [Google Scholar] [CrossRef]
- Shen, G.H.; Grundemar, L.; Zukowska-Grojec, Z.; Håkanson, R.; Wahlestedt, C. C-terminal neuropeptide Y fragments are mast cell-dependent vasodepressor agents. Eur. J. Pharmacol. 1991, 204, 249–256. [Google Scholar] [CrossRef]
- Jacques, D.; Sader, S.; Perreault, C.; Abdel-Samad, D.; Provost, C. Roles of nuclear NPY and NPY receptors in the regulation of the endocardial endothelium and heart function. Can. J. Physiol. Pharmacol. 2006, 84, 695–705. [Google Scholar] [CrossRef]
- Sainsbury, A.; Schwarzer, C.; Couzens, M.; Herzog, H. Y2 receptor deletion attenuates the type 2 diabetic syndrome of ob/ob mice. Diabetes 2002, 51, 3420–3427. [Google Scholar] [CrossRef]
- Shi, Y.-C.; Baldock, P.A. Central and peripheral mechanisms of the NPY system in the regulation of bone and adipose tissue. Bone 2012, 50, 430–436. [Google Scholar] [CrossRef]
- Chatree, S.; Sitticharoon, C.; Maikaew, P.; Uawithya, P.; Chearskul, S. Adipose Y5R mRNA is higher in obese than non-obese humans and is correlated with obesity parameters. Exp. Biol. Med. 2018, 243, 786–795. [Google Scholar] [CrossRef]
- Fukasaka, Y.; Nambu, H.; Tanioka, H.; Obata, A.; Tonomura, M.; Okuno, T.; Yukioka, H. An insurmountable NPY Y5 receptor antagonist exhibits superior anti-obesity effects in high-fat diet-induced obese mice. Neuropeptides 2018, 70, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Rosmaninho-Salgado, J.; Cortez, V.; Estrada, M.; Santana, M.M.; Gonçalves, A.; Marques, A.P.; Cavadas, C. Intracellular mechanisms coupled to NPY Y2 and Y5 receptor activation and lipid accumulation in murine adipocytes. Neuropeptides 2012, 46, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Tanday, N.; Flatt, P.R.; Irwin, N. Pancreatic polypeptide revisited: Potential therapeutic effects in obesity-diabetes. Peptides 2023, 160, 170923. [Google Scholar] [CrossRef] [PubMed]
- Wieland, H.A.; Hamilton, B.S.; Krist, B.; Doods, H.N. The role of NPY in metabolic homeostasis: Implications for obesity therapy. Expert Opin. Investig. Drugs 2000, 9, 1327–1346. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Nomura, T.; Momose, K.; Ikeda, Y.; Kondou, Y.; Akiho, H.; Togami, J.; Kimura, Y.; Okada, M.; Yamaguchi, T. Inactivation of a novel neuropeptide Y/peptide YY receptor gene in primate species. J. Biol. Chem. 1996, 271, 27217–27220. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Tan, Q.; Han, S.; Diemar, A.; Löbner, K.; Wang, H.; Schüß, C.; Behr, V.; Mörl, K.; Wang, M.; et al. Receptor-specific recognition of NPY peptides revealed by structures of NPY receptors. Sci. Adv. 2022, 8, eabm1232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cline, M.A.; Gilbert, E.R. Hypothalamus-adipose tissue crosstalk: Neuropeptide y and the regulation of energy metabolism. Nutr. Metab. 2014, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Lindner, J.; Beck-Sickinger, A.G.; Hey-Hawkins, E.; Bellmann-Sickert, K. Carbaboranylation of Truncated C-Terminal Neuropeptide Y Analogue Leads to Full hY1 Receptor Agonism. Chembiochem 2018, 19, 2300–2306. [Google Scholar] [CrossRef]
- Ruipan, Z.; Xiangzhi, M.; Li, L.; Ying, Z.; Mingliang, Q.; Peng, J.; Jingwei, L.; Zijun, Z.; Yan, G. Differential expression and localization of neuropeptide y peptide in pancreatic islet of diabetic and high fat fed rats. Peptides 2014, 54, 33–38. [Google Scholar] [CrossRef]
- Kos, K.; Harte, A.L.; James, S.; Snead, D.R.; O’Hare, J.P.; McTernan, P.G.; Kumar, S. Secretion of neuropeptide Y in human adipose tissue and its role in maintenance of adipose tissue mass. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Guan, H.; Arany, E.; Hill, D.J.; Cao, X. Neuropeptide Y is produced in visceral adipose tissue and promotes proliferation of adipocyte precursor cells via the Y1 receptor. FASEB J. 2008, 22, 2452–2464. [Google Scholar] [CrossRef]
- Paulo Matafome, R.S.; Matafome, P.; Seiça, R. Function and Dysfunction of Adipose Tissue. In Obesity and Brain Function; Springer: Berlin/Heidelberg, Germany, 2017; Volume 19, Chapter 1; pp. 3–31. [Google Scholar] [CrossRef]
- Coutinho, E.A.; Okamoto, S.; Ishikawa, A.W.; Yokota, S.; Wada, N.; Hirabayashi, T.; Saito, K.; Sato, T.; Takagi, K.; Wang, C.-C.; et al. Activation of SF1 neurons in the ventromedial hypothalamus by DREADD technology increases insulin sensitivity in peripheral tissues. Diabetes 2017, 66, 2372–2386. [Google Scholar] [CrossRef]
- Mercer, R.E.; Chee, M.J.S.; Colmers, W.F. The role of NPY in hypothalamic mediated food intake. Front. Neuroendocrinol. 2011, 32, 398–415. [Google Scholar] [CrossRef]
- Myrsén-Axcrona, U.; Ekblad, E.; Sundler, F. Developmental expression of NPY, PYY and PP in the rat pancreas and their coexistence with islet hormones. Regul. Pept. 1997, 68, 165–175. [Google Scholar] [CrossRef]
- Sohn, J.-W.; Elmquist, J.K.; Williams, K.W. Neuronal circuits that regulate feeding behavior and metabolism. Trends Neurosci. 2013, 36, 504–512. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, D.; Sweeney, P.; Yang, Y. An excitatory ventromedial hypothalamus to paraventricular thalamus circuit that suppresses food intake. Nat. Commun. 2020, 11, 6326. [Google Scholar] [CrossRef]
- Rossi, M.A.; Stuber, G.D. Overlapping Brain Circuits for Homeostatic and Hedonic Feeding. Cell Metab. 2018, 27, 42–56. [Google Scholar] [CrossRef]
- Paulo Matafome, R.S.; Matafome, P.; Seiça, R. The Role of Brain in Energy Balance. In Obesity and Brain Function; Springer: Berlin/Heidelberg, Germany, 2017; Volume 19, Chapter 1; pp. 33–48. [Google Scholar] [CrossRef]
- Claret, M.; Smith, M.; Batterham, R.; Selman, C.; Choudhury, A.I.; Fryer, L.G.; Clements, M.; Al-Qassab, H.; Heffron, H.; Xu, A.W.; et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Investig. 2007, 117, 2325–2336. [Google Scholar] [CrossRef] [Green Version]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.-B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- Lage, R.; Vázquez, M.J.; Varela, L.; Saha, A.K.; Vidal-Puig, A.; Nogueiras, R.; Diéguez, C.; López, M. Ghrelin effects on neuropeptides in the rat hypothalamus depend on fatty acid metabolism actions on BSX but not on gender. FASEB J. 2010, 24, 2670–2679. [Google Scholar] [CrossRef]
- Toda, C.; Santoro, A.; Kim, J.D.; Diano, S. POMC Neurons: From Birth to Death. Annu. Rev. Physiol. 2017, 79, 209–236. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C. POMC Neurons: Feeding, Energy Metabolism, and Beyond; Springer: Berlin/Heidelberg, Germany, 2018; pp. 17–29. [Google Scholar] [CrossRef]
- Sohn, J.W. Network of hypothalamic neurons that control appetite. BMB Rep BMB Rep. 2015, 48, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.P.; Dube, M.G.; Pu, S.; Xu, B.; Horvath, T.L.; Kalra, P.S. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 1999, 20, 68–100. [Google Scholar] [CrossRef]
- Sutton, A.K.; Myers, M.G.; Olson, D.P. The Role of PVH Circuits in Leptin Action and Energy Balance. Annu. Rev. Physiol. 2016, 78, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Biglari, N.; Gaziano, I.; Schumacher, J.; Radermacher, J.; Paeger, L.; Klemm, P.; Chen, W.; Corneliussen, S.; Wunderlich, C.M.; Sue, M.; et al. Functionally distinct POMC-expressing neuron subpopulations in hypothalamus revealed by intersectional targeting. Nat. Neurosci. 2021, 24, 913–929. [Google Scholar] [CrossRef]
- Quarta, C.; Claret, M.; Zeltser, L.M.; Williams, K.W.; Yeo, G.S.H.; Tschöp, M.H.; Diano, S.; Brüning, J.C.; Cota, D. POMC neuronal heterogeneity in energy balance and beyond: An integrated view. Nat. Metab. 2021, 3, 299–308. [Google Scholar] [CrossRef]
- Nunez-Salces, M.; Li, H.; Christie, S.; Page, A.J. The effect of high-fat diet-induced obesity on the expression of nutrient chemosensors in the mouse stomach and the gastric ghrelin cell. Nutrients 2020, 12, 2493. [Google Scholar] [CrossRef]
- Poher, A.-L.; Tschöp, M.H.; Müller, T.D. Ghrelin regulation of glucose metabolism. Peptides 2018, 100, 236–242. [Google Scholar] [CrossRef]
- Kirchner, H.; Gutierrez, J.A.; Solenberg, P.J.; Pfluger, P.T.; Czyzyk, T.A.; Willency, J.A.; Schürmann, A.; Joost, H.-G.; Jandacek, R.J.; Hale, J.E.; et al. GOAT links dietary lipids with the endocrine control of energy balance. Nat. Med. 2009, 15, 741–745. [Google Scholar] [CrossRef]
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef]
- Yanagi, S.; Sato, T.; Kangawa, K.; Nakazato, M. The Homeostatic Force of Ghrelin. Cell Metab. 2018, 27, 786–804. [Google Scholar] [CrossRef]
- Resh, M.D. Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 2016, 63, 120–131. [Google Scholar] [CrossRef]
- Barazzoni, R.; Zanetti, M.; Ferreira, C.; Vinci, P.; Pirulli, A.; Mucci, M.; Dore, F.; Fonda, M.; Ciocchi, B.; Cattin, L.; et al. Relationships between desacylated and acylated ghrelin and insulin sensitivity in the metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 3935–3940. [Google Scholar] [CrossRef]
- Delhanty, P.J.; Neggers, S.J.; van der Lely, A.J. Des-acyl ghrelin: A metabolically active peptide. Endocr. Dev. 2013, 25, 112–121. [Google Scholar] [CrossRef]
- Jéquier, E. Leptin signaling, adiposity, and energy balance. Ann. New York Acad. Sci. 2002, 967, 379–388. [Google Scholar] [CrossRef]
- Cui, H.; López, M.; Rahmouni, K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat. Rev. Endocrinol. 2017, 13, 338–351. [Google Scholar] [CrossRef]
- Kohno, D.; Yada, T. Arcuate NPY neurons sense and integrate peripheral metabolic signals to control feeding. Neuropeptides 2012, 46, 315–319. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Heymsfield, S.B.; Haft, C.; Kahn, B.B.; Laughlin, M.; Leibel, R.L.; Tschöp, M.H.; Yanovski, J.A. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012, 15, 150–156. [Google Scholar] [CrossRef]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Baldini, G.; Phelan, K.D. The melanocortin pathway and control of appetite-progress and therapeutic implications. J. Endocrinol. 2019, 241, R1–R33. [Google Scholar] [CrossRef]
- Mitchell, C.S.; Begg, D.P. The regulation of food intake by insulin in the central nervous system. J. Neuroendocr. 2021, 33, e12952. [Google Scholar] [CrossRef] [PubMed]
- Péterfi, Z.; Szilvásy-Szabó, A.; Farkas, E.; Ruska, Y.; Pyke, C.; Knudsen, L.B.; Fekete, C. GLP-1 regulates the POMC neurons of the arcuate nucleus both directly and indirectly via presynaptic action. Neuroendocrinology 2020, 111, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Konturek, S.J.; Konturek, J.W.; Pawlik, T.; Brzozowski, T. Brain-gut axis and its role in the control of food intake. J. Physiol. Pharmacol. 2004, 55, 137–154. [Google Scholar] [PubMed]
- Monteiro, M.P.; Batterham, R.L. The Importance of the Gastrointestinal Tract in Controlling Food Intake and Regulating Energy Balance. Gastroenterology 2017, 152, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Ballaz, S. The unappreciated roles of the cholecystokinin receptor CCK(1) in brain functioning. Rev. Neurosci. 2017, 28, 573–585. [Google Scholar] [CrossRef]
- Parker, R.M.C.; Herzog, H. Regional distribution of Y-receptor subtype mRNAs in rat brain. Eur. J. Neurosci. 1999, 11, 1431–1448. [Google Scholar] [CrossRef]
- Suzuki, K.; Simpson, K.A.; Minnion, J.S.; Shillito, J.C.; Bloom, S.R. The role of gut hormones and the hypothalamus in appetite regulation. Endocr. J. 2010, 57, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Dubey, V.; Ghosh, A.R. Obesity: An overview of possible role(s) of gut hormones, lipid sensing and gut microbiota. Metabolism 2016, 65, 48–65. [Google Scholar] [CrossRef]
- Asakawa, A.; Inui, A.; Yuzuriha, H.; Ueno, N.; Katsuura, G.; Fujimiya, M.; Fujino, M.A.; Niijima, A.; Meguid, M.M.; Kasuga, M. Characterization of the effects of pancreatic polypeptide in the regulation of energy balance. Gastroenterology 2003, 124, 1325–1336. [Google Scholar] [CrossRef]
- Zeng, W.; Yang, F.; Shen, W.L.; Zhan, C.; Zheng, P.; Hu, J. Interactions between central nervous system and peripheral metabolic organs. Sci. China Life Sci. 2022, 65, 1929–1958. [Google Scholar] [CrossRef]
- Minokoshi, Y. Neural Control of Homeostatic Feeding and Food Selection. In New Insights into Metabolic Syndrome; Intechopen: London, UK, 2020. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Z.; Liu, Z.; Zhou, H.; Xu, X.; Li, Z.; Chen, H.; Wang, Y.; Zhou, Z.; Wang, M.; et al. Ventromedial Hypothalamus Activation Aggravates Hypertension Myocardial Remodeling Through the Sympathetic Nervous System. Front. Cardiovasc. Med. 2021, 8, 737135. [Google Scholar] [CrossRef]
- Chee, M.; Myers, M.G.; Price, C.J.; Colmers, W.F. Neuropeptide Y suppresses anorexigenic output from the ventromedial nucleus of the hypothalamus. J. Neurosci. 2010, 30, 3380–3390. [Google Scholar] [CrossRef]
- Castillo-Armengol, J.; Barquissau, V.; Geller, S.; Ji, H.; Severi, I.; Venema, W.; Fenandez, E.A.; Moret, C.; Huber, K.; Leal-Esteban, L.C.; et al. Hypothalamic CDK 4 regulates thermogenesis by modulating sympathetic innervation of adipose tissues. EMBO Rep. 2020, 21, e49807. [Google Scholar] [CrossRef]
- Monda, M.; Sullo, A.; De Luca, V.; Viggiano, A.; Pellicano, M. Acute lesions of the ventromedial hypothalamus reduce sympathetic activation and thermogenic changes induced by PGE1. J. Physiol. Paris 1997, 91, 285–290. [Google Scholar] [CrossRef]
- Kondo, H.; Kondo, M.; Hayashi, K.; Kusafuka, S.; Hamamura, K.; Tanaka, K.; Kodama, D.; Hirai, T.; Sato, T.; Ariji, Y.; et al. Orthodontic tooth movement-activated sensory neurons contribute to enhancing osteoclast activity and tooth movement through sympathetic nervous signalling. Eur. J. Orthod. 2021, 44, 404–411. [Google Scholar] [CrossRef]
- Gavini, C.K.; Britton, S.L.; Koch, L.G.; Novak, C.M. Inherently Lean Rats Have Enhanced Activity and Skeletal Muscle Response to Central Melanocortin Receptors. Obesity 2018, 26, 885–894. [Google Scholar] [CrossRef]
- Billington, C.J.; Briggs, J.E.; Grace, M.; Levine, A.S. Effects of intracerebroventricular injection of neuropeptide Y on energy metabolism. Am. J. Physiol. Integr. Comp. Physiol. 1991, 260, R321–R327. [Google Scholar] [CrossRef]
- Shi, Y.-C.; Lau, J.; Lin, Z.; Zhang, H.; Zhai, L.; Sperk, G.; Heilbronn, R.; Mietzsch, M.; Weger, S.; Huang, X.-F.; et al. Arcuate NPY controls sympathetic output and BAT function via a relay of tyrosine hydroxylase neurons in the PVN. Cell Metab. 2013, 17, 236–248. [Google Scholar] [CrossRef]
- Shimada, K.; Ohno, Y.; Okamatsu-Ogura, Y.; Suzuki, M.; Kamikawa, A.; Terao, A.; Kimura, K. Neuropeptide y activates phosphorylation of ERK and STAT3 in stromal vascular cells from brown adipose tissue, but fails to affect thermogenic function of brown adipocytes. Peptides 2012, 34, 336–342. [Google Scholar] [CrossRef]
- Egawa, M.; Yoshimatsu, H.; Bray, G.A. Neuropeptide Y suppresses sympathetic activity to interscapular brown adipose tissue in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1991, 260, R328–R334. [Google Scholar] [CrossRef]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Michael, N.J.; Simonds, S.E.; Top, M.V.D.; Cowley, M.A.; Spanswick, D. Mitochondrial uncoupling in the melanocortin system differentially regulates NPY and POMC neurons to promote weight-loss. Mol. Metab. 2017, 6, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Kataoka, N.; Nakamura, K. An oxytocinergic neural pathway that stimulates thermogenic and cardiac sympathetic outflow. Cell Rep. 2022, 40, 111380. [Google Scholar] [CrossRef] [PubMed]
- Baldock, P.A.; Allison, S.J.; Lundberg, P.; Lee, N.J.; Slack, K.; Lin, E.-J.D.; Enriquez, R.F.; McDonald, M.M.; Zhang, L.; During, M.J.; et al. Novel role of Y1 receptors in the coordinated regulation of bone and energy homeostasis. J. Biol. Chem. 2007, 282, 19092–19102. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, D. NPY Y1 and Y5 Receptor Selective Antagonists as Anti-Obesity Drugs. Curr. Top. Med. Chem. 2007, 7, 1721–1733. [Google Scholar] [CrossRef]
- Hwa, J.J.; Witten, M.B.; Williams, P.; Ghibaudi, L.; Gao, J.; Salisbury, B.G.; Mullins, D.; Hamud, F.; Strader, C.D.; Parker, E.M. Activation of the NPY Y5 receptor regulates both feeding and energy expenditure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999, 277, R1428–R1434. [Google Scholar] [CrossRef]
- Shi, Y.-C.; Lin, S.; Wong, I.P.L.; Baldock, P.A.; Aljanova, A.; Enriquez, R.F.; Castillo, L.; Mitchell, N.F.; Ye, J.-M.; Zhang, L.; et al. NPY neuron-specific Y2 receptors regulate adipose tissue and trabecular bone but not cortical bone homeostasis in mice. PLoS ONE 2010, 5, e11361. [Google Scholar] [CrossRef]
- Turtzo, L.C.; Marx, R.; Lane, M.D. Cross-talk between sympathetic neurons and adipocytes in coculture. Proc. Natl. Acad. Sci. USA 2001, 98, 12385–12390. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Wang, Z.L.; Wang, R.M.; Smith, D.M.; Ghatei, M.A.; Bloom, S.R. Glibenclamide but not other sulphonylureas stimulates release of neuropeptide Y from perifused rat islets and hamster insulinoma cells. J. Endocrinol. 2000, 165, 509–518. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Pham, H.G.; Dang, T.T.H.; Yun, J.W. Salvianolic acid B induces browning in 3T3-L1 white adipocytes via activation of β3-AR and ERK signaling pathways. J. Funct. Foods 2021, 81, 104475. [Google Scholar] [CrossRef]
- Kuo, L.E.; Kitlinska, J.B.; Tilan, J.U.; Li, L.; Baker, S.B.; Johnson, M.D.; Lee, E.; Burnett, M.S.; Fricke, S.T.; Kvetnansky, R.; et al. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 2007, 13, 803–811. [Google Scholar] [CrossRef]
- Park, S.; Fujishita, C.; Komatsu, T.; Kim, S.E.; Chiba, T.; Mori, R.; Shimokawa, I. NPY antagonism reduces adiposity and attenuates age-related imbalance of adipose tissue metabolism. FASEB J. 2014, 28, 5337–5348. [Google Scholar] [CrossRef]
- Zhang, L.; Macia, L.; Turner, N.; Enriquez, R.F.; Riepler, S.J.; Nguyen, A.D.; Lin, S.; Lee, N.J.; Shi, Y.C.; Yulyaningsih, E.; et al. Peripheral neuropeptide y Y1 receptors regulate lipid oxidation and fat accretion. Int. J. Obes. 2010, 34, 357–373. [Google Scholar] [CrossRef]
- Mashiko, S.; Ishihara, A.; Iwaasa, H.; Sano, H.; Ito, J.; Gomori, A.; Oda, Z.; Moriya, R.; Matsushita, H.; Jitsuoka, M.; et al. A pair-feeding study reveals that a Y5 antagonist causes weight loss in diet-induced obese mice by modulating food intake and energy expenditure. Mol. Pharmacol. 2007, 71, 602–608. [Google Scholar] [CrossRef]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’H, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.O.; Liu, Z.-W.; Horvath, T.L. Mitochondrial Dynamics Controlled by Mitofusins Regulate Agrp Neuronal Activity and Diet-Induced Obesity. Cell 2013, 155, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Yoon, N.A.; Liu, Z.-W.; Song, J.E.; Horvath, T.L.; Kim, J.D.; Diano, S. Drp1 is required for AgRP neuronal activity and feeding. Elife 2021, 10, e64351. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Campolo, M.; Liu, C.; Sesaki, H.; Meli, R.; Liu, Z.-W.; Kim, J.D.; Diano, S. DRP1 Suppresses Leptin and Glucose Sensing of POMC Neurons. Cell Metab. 2017, 25, 647–660. [Google Scholar] [CrossRef]
- Drougard, A.; Fournel, A.; Valet, P.; Knauf, C. Impact of hypothalamic reactive oxygen species in the regulation of energy metabolism and food intake. Front. Neurosci. 2015, 9, 56. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Liu, Z.-W.; Walllingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschöp, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.I.; et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 2008, 454, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Coppola, A.; Liu, Z.-W.; Andrews, Z.B.; Paradis, E.; Roy, M.-C.; Friedman, J.M.; Ricquier, D.; Richard, D.; Horvath, T.L.; Gao, X.-B.; et al. A Central Thermogenic-like Mechanism in Feeding Regulation: An Interplay between Arcuate Nucleus T3 and UCP2. Cell Metab. 2007, 5, 21–33. [Google Scholar] [CrossRef]
- Parton, L.E.; Ye, C.P.; Coppari, R.; Enriori, P.J.; Choi, B.; Zhang, C.-Y.; Xu, C.; Vianna, C.R.; Balthasar, N.; Lee, C.E.; et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 2007, 449, 228–232. [Google Scholar] [CrossRef]
- Diano, S.; Liu, Z.-W.; Jeong, J.K.; Dietrich, M.O.; Ruan, H.-B.; Kim, E.; Suyama, S.; Kelly, K.; Gyengesi, E.; Arbiser, J.L.; et al. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat. Med. 2011, 17, 1121–1128. [Google Scholar] [CrossRef]
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC Neurons Connects ER Stress with Leptin Resistance and Energy Imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef]
- Hirooka, Y.; Kishi, T.; Sakai, K.; Takeshita, A.; Sunagawa, K. Imbalance of central nitric oxide and reactive oxygen species in the regulation of sympathetic activity and neural mechanisms of hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R818–R826. [Google Scholar] [CrossRef]
- Duparc, T.; Naslain, D.; Colom, A.; Muccioli, G.G.; Massaly, N.; Delzenne, N.M.; Valet, P.; Cani, P.D.; Knauf, C. Jejunum inflammation in obese and diabetic mice impairs enteric glucose detection and modifies nitric oxide release in the hypothalamus. Antioxid. Redox Signal. 2011, 14, 415–423. [Google Scholar] [CrossRef]
- Ono, H. Molecular mechanisms of hypothalamic insulin resistance. Int. J. Mol. Sci. 2019, 20, 1317. [Google Scholar] [CrossRef]
- Newton, A.J.; Hess, S.; Paeger, L.; Vogt, M.C.; Lascano, J.F.; Nillni, E.A.; Brüning, J.C.; Kloppenburg, P.; Xu, A.W. AgRP Innervation onto POMC Neurons Increases with Age and Is Accelerated with Chronic High-Fat Feeding in Male Mice. Endocrinology 2013, 154, 172–183. [Google Scholar] [CrossRef]
- Paeger, L.; Pippow, A.; Hess, S.; Paehler, M.; Klein, A.C.; Husch, A.; Pouzat, C.; Brüning, J.C.; Kloppenburg, P. Energy imbalance alters Ca2+ handling and excitability of POMC neurons. Elife 2017, 6, e25641. [Google Scholar] [CrossRef]
- Kaipio, K.; Kallio, J.; Pesonen, U. Mitochondrial targeting signal in human neuropeptide Y gene. Biochem. Biophys. Res. Commun. 2005, 337, 633–640. [Google Scholar] [CrossRef]
- Kaipio, K.; Pesonen, U. The intracellular mobility of NPY and a putative mitochondrial form of NPY in neuronal cells. Neurosci. Lett. 2009, 450, 181–185. [Google Scholar] [CrossRef]
- Karvonen, M.K.; Pesonen, U.; Koulu, M.; Niskanen, L.; Laakso, M.; Rissanen, A.; Dekker, J.M.; Hart, L.M.; Valve, R.; Uusitupa, M.I. Association of a leucine(7)-to-proline(7) polymorphism in the signal peptide of neuropeptide Y with high serum cholesterol and LDL cholesterol levels. Nat. Med. 1998, 4, 1434–1437. [Google Scholar] [CrossRef]
- Pesonen, U. NPY L7P polymorphism and metabolic diseases. Regul. Pept. 2008, 149, 51–55. [Google Scholar] [CrossRef]
- Kallio, J. Altered intracellular processing and release of neuropeptide Y due to leucine 7 to proline 7 polymorphism in the signal peptide of preproneuropeptide Y in humans. FASEB J. 2001, 15, 1242–1244. [Google Scholar] [CrossRef]
- Van Rossum, C.T.M.; Pijl, H.; Adan, R.A.H.; Hoebee, B.; Seidell, J. Polymorphisms in the NPY and AGRP genes and body fatness in Dutch adults. Int. J. Obes. 2006, 30, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Dhamad, A.; Zampiga, M.; Greene, E.S.; Sirri, F.; Dridi, S. Neuropeptide Y and its receptors are expressed in chicken skeletal muscle and regulate mitochondrial function. Gen. Comp. Endocrinol. 2021, 310, 113798. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Xu, X.; Guo, W.; Luo, C.; Wang, H.; Meng, X.; Zhu, S.; Wei, Y. Neuropeptide y damages the integrity of mitochondrial structure and disrupts energy metabolism in cultured neonatal rat cardiomyocytes. Peptides 2015, 71, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Xue, R.; Wang, Y.; Zhang, Q.; Huang, S.; Wu, W.; Ye, H.; Zhang, Z.; Li, Y. The effect of neuropeptide y on brown-like adipocyte’s differentiation and activation. Peptides 2015, 63, 126–133. [Google Scholar] [CrossRef]
- Beck, B.; Burlet, A.; Bazin, R.; Nicolas, J.P.; Burlet, C. Elevated neuropeptide Y in the arcuate nucleus of young obese Zucker rats may contribute to the development of their overeating. J. Nutr. 1993, 123, 1168–1172. [Google Scholar] [CrossRef]
- Gøtzsche, C.; Woldbye, D. The role of NPY in learning and memory. Neuropeptides 2016, 55, 79–89. [Google Scholar] [CrossRef]
- Yulyaningsih, E.; Zhang, L.; Herzog, H.; Sainsbury, A. NPY receptors as potential targets for anti-obesity drug development. Br. J. Pharmacol. 2011, 163, 1170–1202. [Google Scholar] [CrossRef]
- Williams, D.M.; Nawaz, A.; Evans, M. Drug Therapy in Obesity: A Review of Current and Emerging Treatments. Diabetes Ther. 2020, 11, 1199–1216. [Google Scholar] [CrossRef]
- Kamiji, M.M.; Inui, A. NPY Y2 and Y4 Receptors Selective Ligands: Promising Anti-Obesity Drugs? Curr. Top. Med. Chem. 2007, 7, 1734–1742. [Google Scholar] [CrossRef]
- Ishiguchi, T.; Amano, T.; Matsubayashi, H.; Tada, H.; Fujita, M.; Takahashi, T. Centrally administered neuropeptide Y delays gastric emptying via Y2 receptors in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R1522–R1530. [Google Scholar] [CrossRef]
- Kask, A.; Rägo, L.; Harro, J. Anxiolytic-like effect of neuropeptide Y (NPY) and NPY13-36 microinjected into vicinity of locus coeruleus in rats. Brain Res. 1998, 788, 345–348. [Google Scholar] [CrossRef]
- Chen, Q.-C.; Zhang, Y. The Role of NPY in the Regulation of Bone Metabolism. Front. Endocrinol. 2022, 13, 833485. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Castrejón-Téllez, V.; Soto, M.E.; Rubio-Ruiz, M.E.; Manzano-Pech, L.; Guarner-Lans, V. Oxidative Stress, Plant Natural Antioxidants, and Obesity. Int. J. Mol. Sci. 2021, 22, 1786. [Google Scholar] [CrossRef]
- Fasciolo, G.; Napolitano, G.; Aprile, M.; Cataldi, S.; Costa, V.; Ciccodicola, A.; Di Meo, S.; Venditti, P. Hepatic Insulin Resistance in Hyperthyroid Rat Liver: Vitamin E Supplementation Highlights a Possible Role of ROS. Antioxidants 2022, 11, 1295. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Receptor | Localization | Outcome |
|---|---|---|
| NPY1R | Hypothalamus | ↑ Feeding |
| WAT | ↑ Lipogenesis ↓ Lipolysis | |
| NPY2R | Hypothalamus | ↓ NPY release |
| WAT | ↑ Adipogenesis ↑ Angiogenesis | |
| NPY5R | Hypothalamus | ↑ Feeding |
| Hypothalamus/WAT? | ↑ Adiposity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, D.; Lopes, E.; Rosendo-Silva, D.; Matafome, P. The Bidirectional Relationship of NPY and Mitochondria in Energy Balance Regulation. Biomedicines 2023, 11, 446. https://doi.org/10.3390/biomedicines11020446
Sousa D, Lopes E, Rosendo-Silva D, Matafome P. The Bidirectional Relationship of NPY and Mitochondria in Energy Balance Regulation. Biomedicines. 2023; 11(2):446. https://doi.org/10.3390/biomedicines11020446
Chicago/Turabian StyleSousa, Diana, Eduardo Lopes, Daniela Rosendo-Silva, and Paulo Matafome. 2023. "The Bidirectional Relationship of NPY and Mitochondria in Energy Balance Regulation" Biomedicines 11, no. 2: 446. https://doi.org/10.3390/biomedicines11020446