

Alterations in Gut Microbiota Composition in Patients with COVID-19: A Pilot Study of Whole Hypervariable 16S rRNA Gene Sequencing
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subject and Design
2.2. Metaprofiling and Data Processing
2.2.1. DNA Extraction
2.2.2. Next-Generation Sequencing (NGS)
2.2.3. Bioinformatic Analysis
2.3. Statistical Analysis
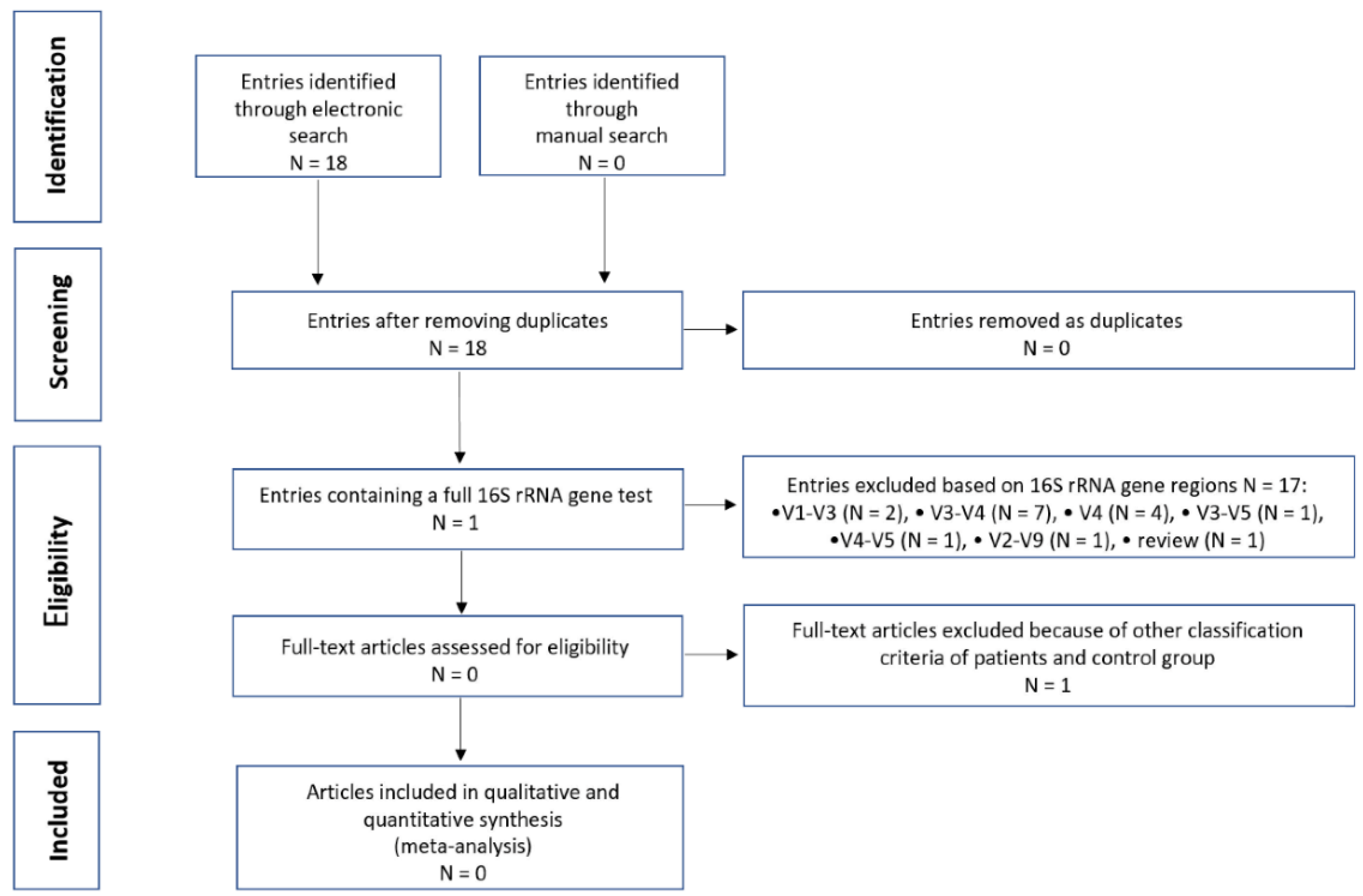
2.4. Search and Selection of Studies for Integrative Research
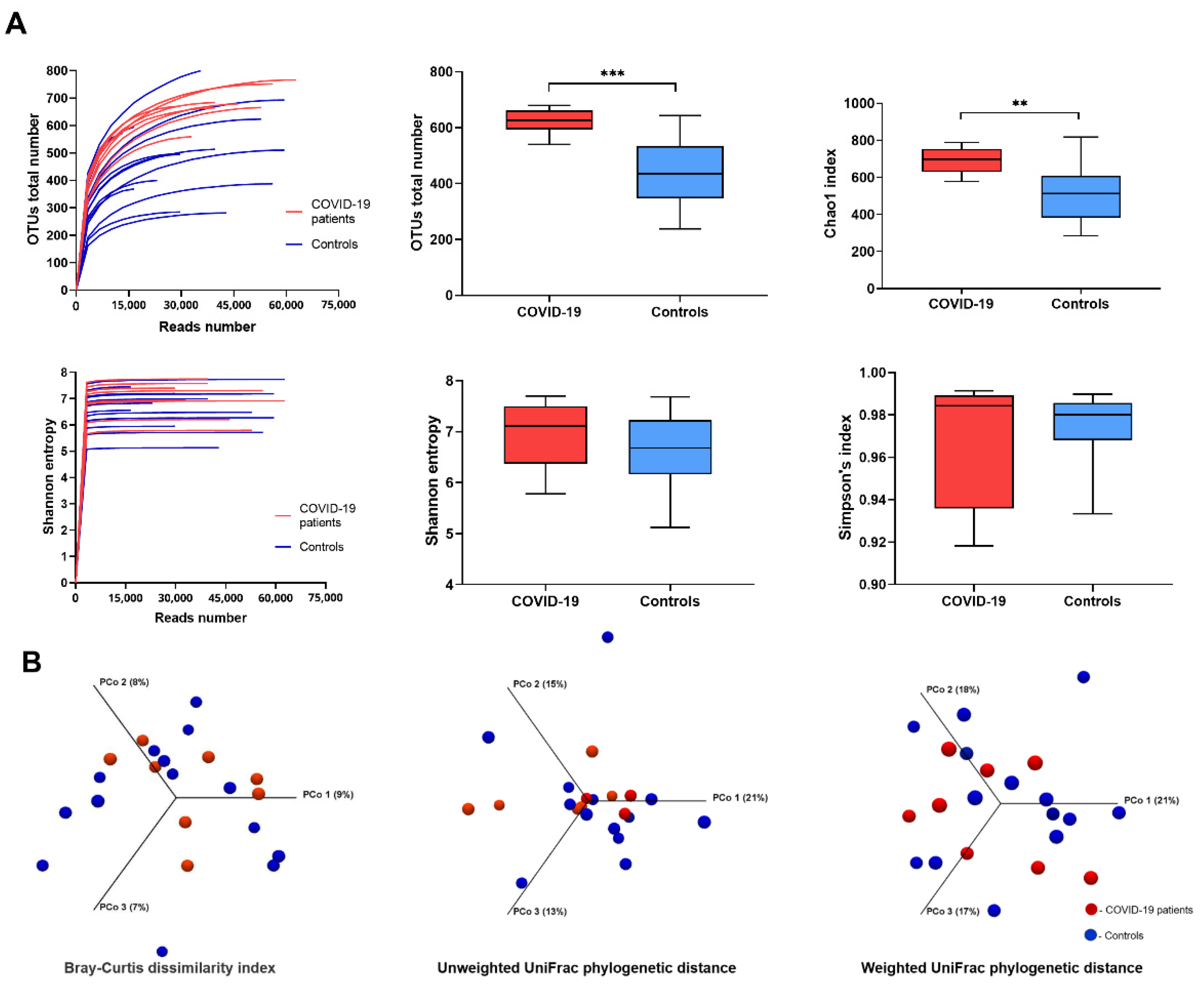
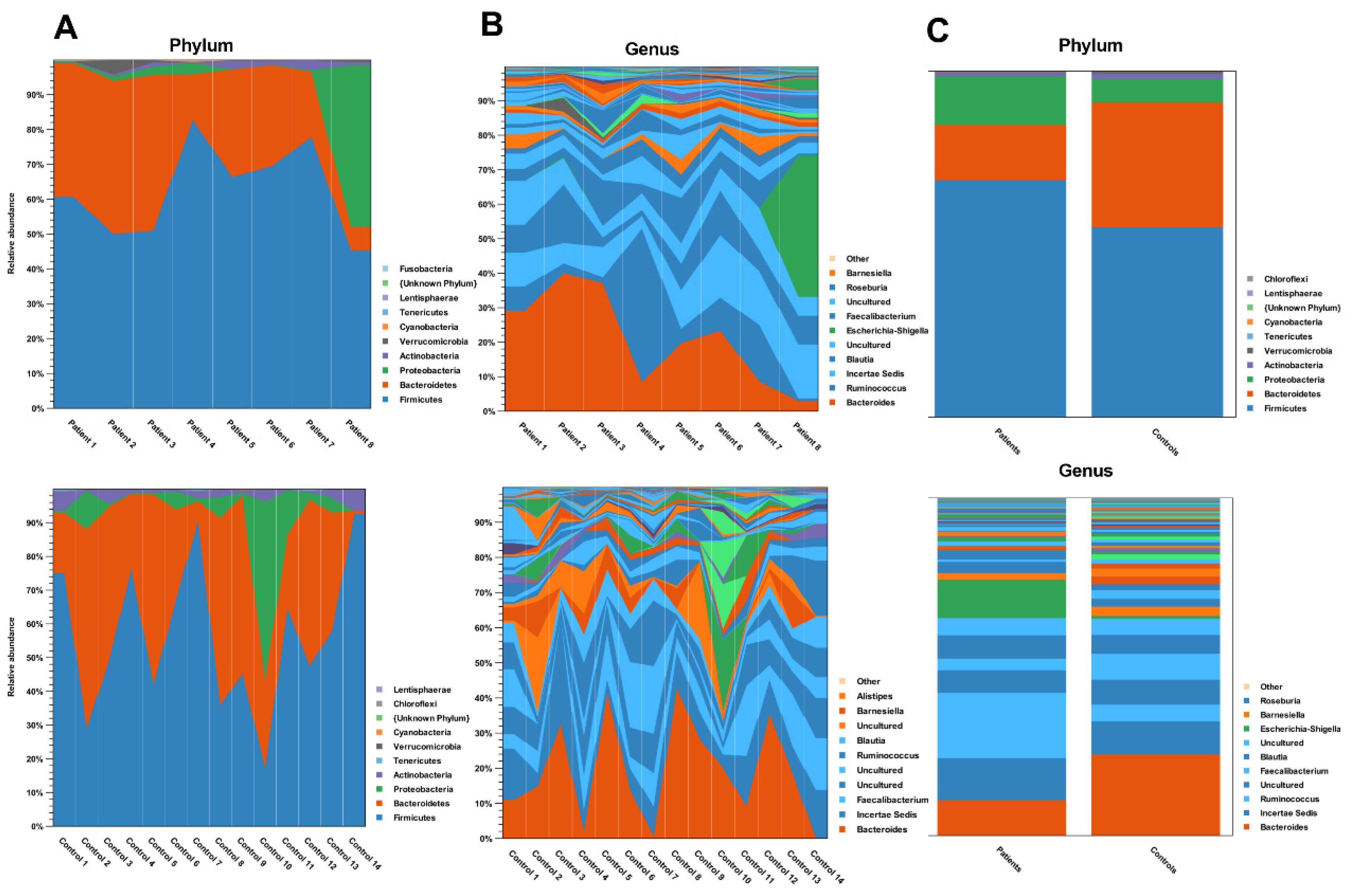
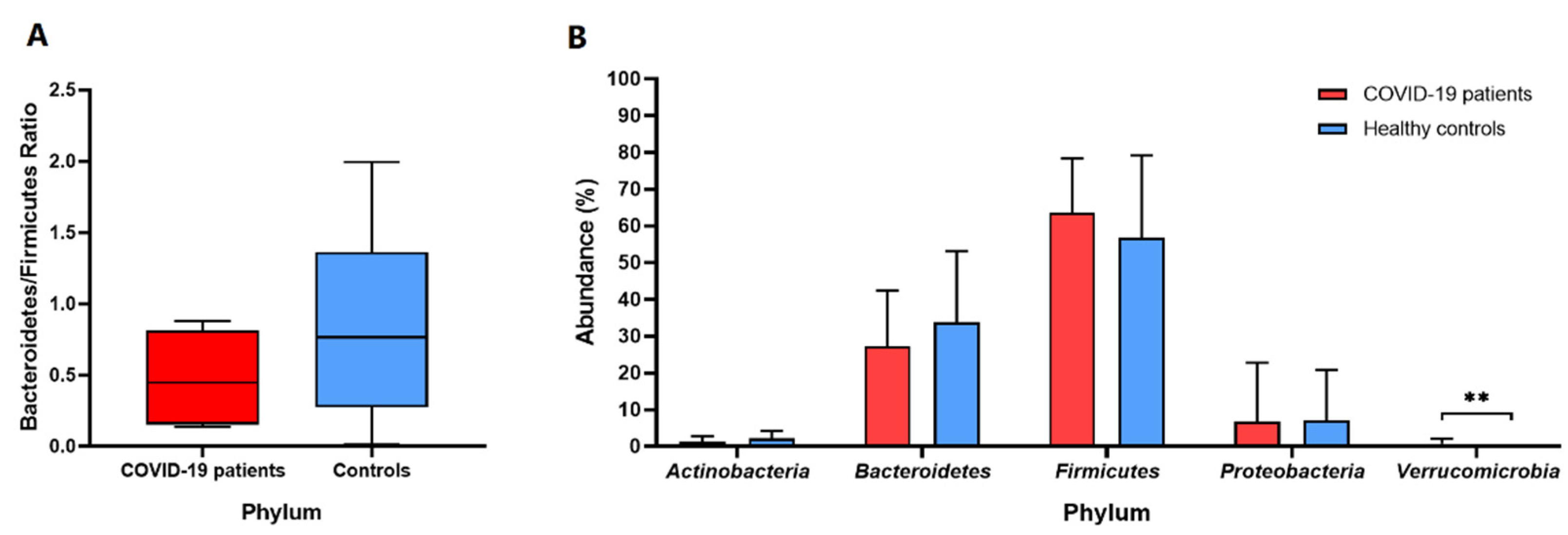
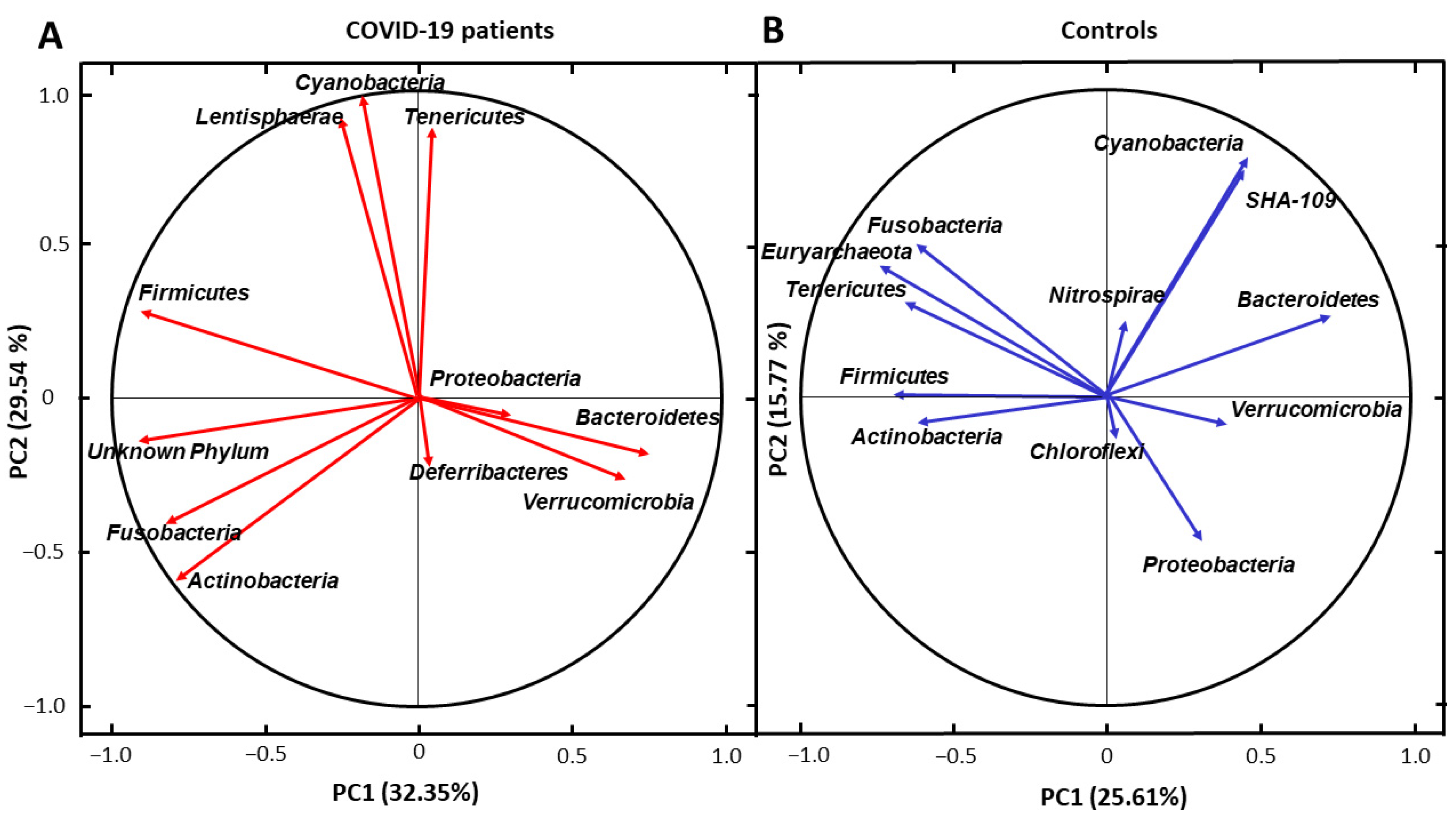
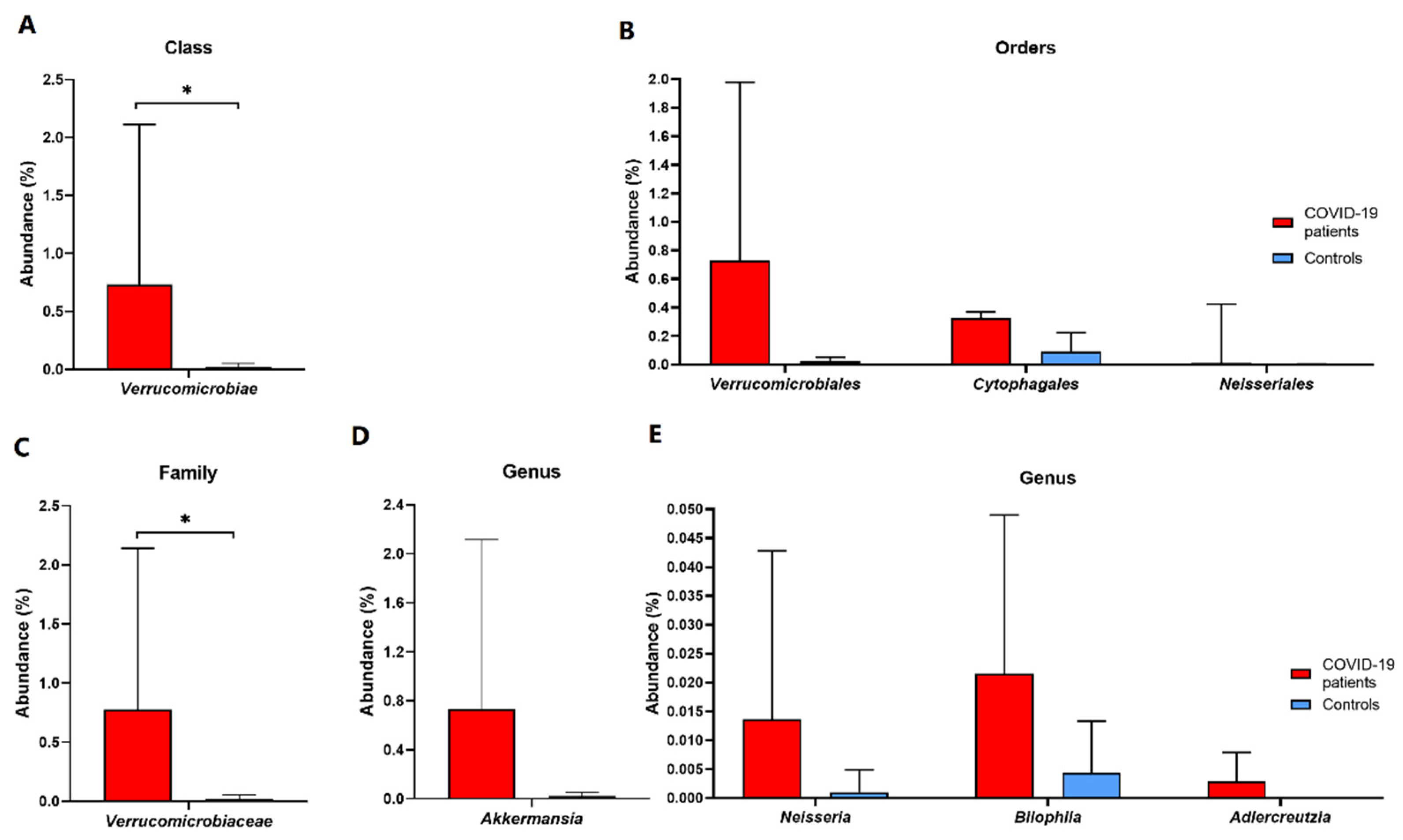
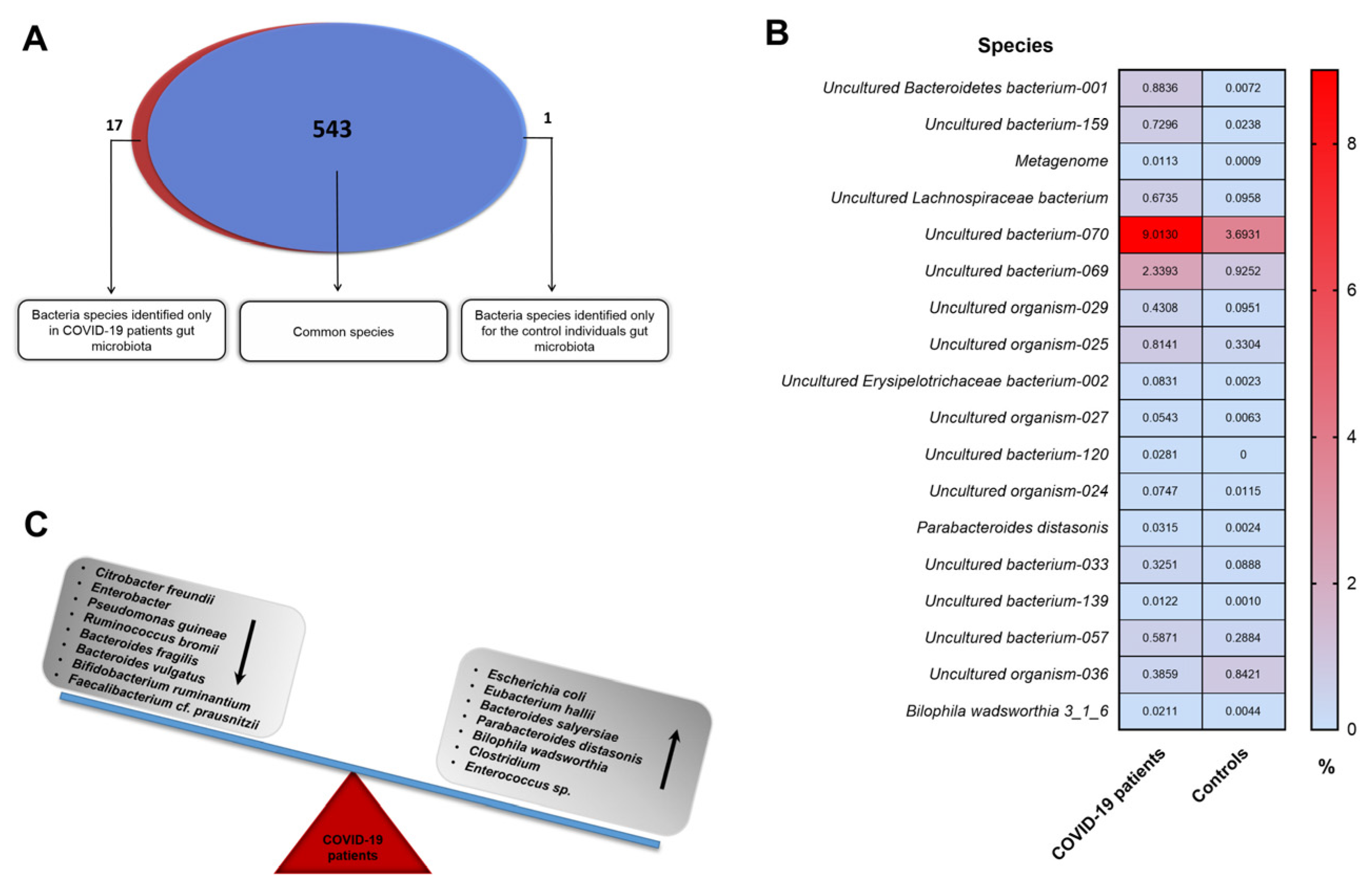
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-COVID-19---11-march-2020 (accessed on 16 December 2021).
- Sharma, A.; Bhatt, N.S.; St Martin, A.; Abid, M.B.; Bloomquist, J.; Chemaly, R.F.; Dandoy, C.; Gauthier, J.; Gowda, L.; Perales, M.-A.; et al. Clinical Characteristics and Outcomes of COVID-19 in Haematopoietic Stem-Cell Transplantation Recipients: An Observational Cohort Study. Lancet Haematol. 2021, 8, e185–e193. [Google Scholar] [CrossRef] [PubMed]
- Jordan, R.E.; Adab, P.; Cheng, K.K. Covid-19: Risk Factors for Severe Disease and Death. BMJ 2020, 368, m1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.D.; Harris, C.; Cain, J.K.; Hummer, C.; Goyal, H.; Perisetti, A. Pulmonary and Extra-Pulmonary Clinical Manifestations of COVID-19. Front. Med. 2020, 7, 526. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Mao, R.; Qiu, Y.; He, J.-S.; Tan, J.-Y.; Li, X.-H.; Liang, J.; Shen, J.; Zhu, L.-R.; Chen, Y.; Iacucci, M.; et al. Manifestations and Prognosis of Gastrointestinal and Liver Involvement in Patients with COVID-19: A Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 667–678. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The Pathogenesis and Treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Abid, M.A.; Nunley, L.; Abid, M.B. Could Coronavirus Disease 2019 (COVID-19) Render Natural Immunity to Re-Infections? A Spotlight on the Therapeutic Pipeline. Front. Immunol. 2020, 11, 1294. [Google Scholar] [CrossRef]
- Viner, R.M.; Whittaker, E. Kawasaki-like Disease: Emerging Complication during the COVID-19 Pandemic. Lancet 2020, 395, 1741–1743. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Yang, Z.; Wang, Y.; Zhou, F.; Li, S.; Li, C.; Li, L.; Zhang, W.; Li, X. Recent Advance of ACE2 and Microbiota Dysfunction in COVID-19 Pathogenesis. Heliyon 2021, 7, e07548. [Google Scholar] [CrossRef] [PubMed]
- Vuille-dit-Bille, R.N.; Camargo, S.M.; Emmenegger, L.; Sasse, T.; Kummer, E.; Jando, J.; Hamie, Q.M.; Meier, C.F.; Hunziker, S.; Forras-Kaufmann, Z.; et al. Human Intestine Luminal ACE2 and Amino Acid Transporter Expression Increased by ACE-Inhibitors. Amino Acids 2015, 47, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, X.; Zhu, B.; Liang, H.; Fang, C.; Gong, Y.; Guo, Q.; Sun, X.; Zhao, D.; Shen, J.; et al. Characteristics of Pediatric SARS-CoV-2 Infection and Potential Evidence for Persistent Fecal Viral Shedding. Nat. Med. 2020, 26, 502–505. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Du, R.-H.; Li, B.; Zheng, X.-S.; Yang, X.-L.; Hu, B.; Wang, Y.-Y.; Xiao, G.-F.; Yan, B.; Shi, Z.-L.; et al. Molecular and Serological Investigation of 2019-NCoV Infected Patients: Implication of Multiple Shedding Routes. Emerg. Microbes Infect. 2020, 9, 386–389. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.; Tong, Z.; Wang, H.; Dai, Y.; Li, K.; Liu, J.; Wu, W.; Yuan, C.; Yu, M.; Li, P.; et al. Detection of Novel Coronavirus by RT-PCR in Stool Specimen from Asymptomatic Child, China. Emerg. Infect. Dis. 2020, 26, 1337–1339. [Google Scholar] [CrossRef]
- Noce, A.; Marrone, G.; Di Daniele, F.; Ottaviani, E.; Wilson Jones, G.; Bernini, R.; Romani, A.; Rovella, V. Impact of Gut Microbiota Composition on Onset and Progression of Chronic Non-Communicable Diseases. Nutrients 2019, 11, 1073. [Google Scholar] [CrossRef] [Green Version]
- Zakerska-Banaszak, O.; Tomczak, H.; Gabryel, M.; Baturo, A.; Wolko, L.; Michalak, M.; Malinska, N.; Mankowska-Wierzbicka, D.; Eder, P.; Dobrowolska, A.; et al. Dysbiosis of Gut Microbiota in Polish Patients with Ulcerative Colitis: A Pilot Study. Sci. Rep. 2021, 11, 2166. [Google Scholar] [CrossRef] [PubMed]
- Sirichoat, A.; Sankuntaw, N.; Engchanil, C.; Buppasiri, P.; Faksri, K.; Namwat, W.; Chantratita, W.; Lulitanond, V. Comparison of Different Hypervariable Regions of 16S RRNA for Taxonomic Profiling of Vaginal Microbiota Using Next-Generation Sequencing. Arch. Microbiol. 2021, 203, 1159–1166. [Google Scholar] [CrossRef]
- Gao, F.; Guo, R.; Ma, Q.; Li, Y.; Wang, W.; Fan, Y.; Ju, Y.; Zhao, B.; Gao, Y.; Qian, L.; et al. Stressful Events Induce Long-Term Gut Microbiota Dysbiosis and Associated Post-Traumatic Stress Symptoms in Healthcare Workers Fighting against COVID-19. J. Affect. Disord. 2022, 303, 187–195. [Google Scholar] [CrossRef]
- Mazzarelli, A.; Giancola, M.L.; Farina, A.; Marchioni, L.; Rueca, M.; Gruber, C.E.M.; Bartolini, B.; Ascoli Bartoli, T.; Maffongelli, G.; Capobianchi, M.R.; et al. 16S RRNA Gene Sequencing of Rectal Swab in Patients Affected by COVID-19. PLoS ONE 2021, 16, e0247041. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, C.; Gentile, I.; Setaro, M.; Di Domenico, C.; Pinchera, B.; Buonomo, A.R.; Zappulo, E.; Scotto, R.; Scaglione, G.L.; Castaldo, G.; et al. Nasopharyngeal Microbiome Signature in COVID-19 Positive Patients: Can We Definitively Get a Role to Fusobacterium Periodonticum? Front. Cell Infect. Microbiol. 2021, 11, 625581. [Google Scholar] [CrossRef] [PubMed]
- Uehara, O.; Abiko, Y.; Nagasawa, T.; Morikawa, T.; Hiraki, D.; Harada, F.; Kawano, Y.; Toraya, S.; Matsuoka, H.; Paudel, D.; et al. Alterations in the Oral Microbiome of Individuals with a Healthy Oral Environment Following COVID-19 Vaccination. BMC Oral Health 2022, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Matijašić, M.; Meštrović, T.; Čipčić Paljetak, H.; Perić, M.; Barešić, A.; Verbanac, D. Gut Microbiota beyond Bacteria—Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. IJMS 2020, 21, 2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.B.; Tolonen, A.C.; Xavier, R.J. Human Genetic Variation and the Gut Microbiome in Disease. Nat. Rev. Genet. 2017, 18, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.B.; Mazmanian, S.K. The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 2017, 46, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.A.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging Pathogenic Links between Microbiota and the Gut–Lung Axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef] [PubMed]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef]
- Cardinale, V.; Capurso, G.; Ianiro, G.; Gasbarrini, A.; Arcidiacono, P.G.; Alvaro, D. Intestinal Permeability Changes with Bacterial Translocation as Key Events Modulating Systemic Host Immune Response to SARS-CoV-2: A Working Hypothesis. Dig. Liver Dis. 2020, 52, 1383–1389. [Google Scholar] [CrossRef]
- Dickson, R.P.; Singer, B.H.; Newstead, M.W.; Falkowski, N.R.; Erb-Downward, J.R.; Standiford, T.J.; Huffnagle, G.B. Enrichment of the Lung Microbiome with Gut Bacteria in Sepsis and the Acute Respiratory Distress Syndrome. Nat. Microbiol. 2016, 1, 16113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burcelin, R.; Garidou, L.; Pomié, C. Immuno-Microbiota Cross and Talk: The New Paradigm of Metabolic Diseases. Semin. Immunol. 2012, 24, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Lian, J.-S.; Hu, J.-H.; Gao, J.; Zheng, L.; Zhang, Y.-M.; Hao, S.-R.; Jia, H.-Y.; Cai, H.; Zhang, X.-L.; et al. Epidemiological, Clinical and Virological Characteristics of 74 Cases of Coronavirus-Infected Disease 2019 (COVID-19) with Gastrointestinal Symptoms. Gut 2020, 69, 1002–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e8. [Google Scholar] [CrossRef]
- Xu, R.; Lu, R.; Zhang, T.; Wu, Q.; Cai, W.; Han, X.; Wan, Z.; Jin, X.; Zhang, Z.; Zhang, C. Temporal Association between Human Upper Respiratory and Gut Bacterial Microbiomes during the Course of COVID-19 in Adults. Commun. Biol. 2021, 4, 240. [Google Scholar] [CrossRef]
- Liu, T.F.D.; Philippou, E.; Kolokotroni, O.; Siakallis, G.; Rahima, K.; Constantinou, C. Gut and Airway Microbiota and Their Role in COVID-19 Infection and Pathogenesis: A Scoping Review. Infection 2021, 50, 815–847. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.-Y.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut Microbiota Composition Reflects Disease Severity and Dysfunctional Immune Responses in Patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef]
- Zuo, T.; Liu, Q.; Zhang, F.; Lui, G.C.-Y.; Tso, E.Y.; Yeoh, Y.K.; Chen, Z.; Boon, S.S.; Chan, F.K.; Chan, P.K.; et al. Depicting SARS-CoV-2 Faecal Viral Activity in Association with Gut Microbiota Composition in Patients with COVID-19. Gut 2020, 70, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling Serum Cytokines in COVID-19 Patients Reveals IL-6 and IL-10 Are Disease Severity Predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Diversity, Metabolism and Microbial Ecology of Butyrate-Producing Bacteria from the Human Large Intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia Muciniphila Gen. Nov., Sp. Nov., a Human Intestinal Mucin-Degrading Bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, Y.; Uchiyama, K.; Takagi, T. A Next-Generation Beneficial Microbe: Akkermansia muciniphila. J. Clin. Biochem. Nutr. 2018, 63, 33–35. [Google Scholar] [CrossRef] [Green Version]
- Mańkowska-Wierzbicka, D.; Stelmach-Mardas, M.; Gabryel, M.; Tomczak, H.; Skrzypczak-Zielińska, M.; Zakerska-Banaszak, O.; Sowińska, A.; Mahadea, D.; Baturo, A.; Wolko, Ł.; et al. The Effectiveness of Multi-Session FMT Treatment in Active Ulcerative Colitis Patients: A Pilot Study. Biomedicines 2020, 8, 268. [Google Scholar] [CrossRef]
- Pascoal, L.B.; Rodrigues, P.B.; Genaro, L.M.; dos Santos Pereira Gomes, A.B.; Toledo-Teixeira, D.A.; Parise, P.L.; Bispo-Dos-Santos, K.; Simeoni, C.L.; Guimarães, P.V.; Buscaratti, L.I.; et al. Microbiota-Derived Short-Chain Fatty Acids Do Not Interfere with SARS-CoV-2 Infection of Human Colonic Samples. Gut Microbes 2021, 13, 1–9. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; Bayless, A.J.; Kominsky, D.J.; et al. HIF-Dependent Regulation of Claudin-1 Is Central to Intestinal Epithelial Tight Junction Integrity. Mol. Biol. Cell 2015, 26, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, A.; Licata, G.; Calabrese, G.; De Rosa, A.; Pagliuca, F.; Alfano, R.; Argenziano, G. CMV Infection: A Clinical Challenge in Biological Therapy? The Case of Asymptomatic Patients with Persistent Positive Immunoglobulin M Anti-CMV Treated with Secukinumab. Psoriasis Targets Ther. 2020, 10, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Liévin-Le Moal, V.; Servin, A.L. The Front Line of Enteric Host Defense against Unwelcome Intrusion of Harmful Microorganisms: Mucins, Antimicrobial Peptides, and Microbiota. Clin. Microbiol. Rev. 2006, 19, 315–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlot, T.; Penninger, J.M. ACE2—From the Renin–Angiotensin System to Gut Microbiota and Malnutrition. Microbes Infect. 2013, 15, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Heine, W.; Radke, M.; Wutzke, K.-D. The Significance of Tryptophan in Human Nutrition. Amino Acids 1995, 9, 91–205. [Google Scholar] [CrossRef]
- Ninomiya, S.; Nakamura, N.; Nakamura, H.; Mizutani, T.; Kaneda, Y.; Yamaguchi, K.; Matsumoto, T.; Kitagawa, J.; Kanemura, N.; Shiraki, M.; et al. Low Levels of Serum Tryptophan Underlie Skeletal Muscle Atrophy. Nutrients 2020, 12, 978. [Google Scholar] [CrossRef] [Green Version]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut Microbiota: Role in Pathogen Colonization, Immune Responses, and Inflammatory Disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Di Pierro, F. Gut Microbiota Parameters Potentially Useful in Clinical Perspective. Microorganisms 2021, 9, 2402. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Total n = 22 | COVID-19 Patients n = 8 | Healthy Controls n = 14 | p-Value |
|---|---|---|---|---|
| Gender | 0.0541 | |||
| Female, n (%) | 8 (36.36) | 5 (62.5) | 3 (21.43) | |
| Male, n (%) | 14 (63.64) | 3 (37.5) | 11 (78.57) | |
| Age, (years) mean ± SD (min.–max.) | 36.23 ± 12.51 (20–59) | 34.88 ± 11.21 (20–59) | 37 ± 110.91 (22–57) | 0.6680 |
| BMI (kg/m2), mean ± SD (min.–max.) | 25,64 ± 3.525 (20.90–32.87) | 24.86 ± 3.784 (20.90–32.27) | 26,09 ± 3.431 (20.94–32.87) | 0.4468 |
| Smoker, n (%) | 0 (0.00) | 0 (0.00) | 0 (0.00) | NA |
| Probiotics taking, n (%) | 0 (0.00) | 0 (0.00) | 0 (0.00) | NA |
| Supplements taking, n (%) | 7 (31.82) | 4 (50.00) | 3 (21.43) | 0.1663 |
| Antibiotics, last 6 months, n (%) | 0 (0.00) | 0 (0.00) | 0 (0.00) | NA |
| COVID-19 symptoms | ||||
| Cough, n (%) | 3 (13.64) | 3 (37.50) | - | NA |
| Fever, n (%) | 5 (22.73) | 5 (62.50) | - | NA |
| Dyspnea, n (%) | 1 (4.55) | 1 (12.50) | - | NA |
| Headache, n (%) | 3 (13.64) | 3 (37.50) | - | NA |
| Sinus pain | 2 (9.09) | 2 (25.00) | - | NA |
| Myalgia, n (%) | 3 (13.64) | 3 (37.50) | - | NA |
| Chest pain, n (%) | 2 (9.09) | 2 (25.00) | - | NA |
| Fatigue, n (%) | 6 (27.27) | 6 (75.00) | - | NA |
| Taste loss, n (%) | 5 (22.73) | 5 (62.50) | - | NA |
| Sense of smell loss, n (%) | 4 (18.18) | 4 (50.00) | - | NA |
| Symptom duration (days), mean ± SD (min.–max.) | - | 7.125 ± 3.40 (4–14) | - | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mańkowska-Wierzbicka, D.; Zuraszek, J.; Wierzbicka, A.; Gabryel, M.; Mahadea, D.; Baturo, A.; Zakerska-Banaszak, O.; Slomski, R.; Skrzypczak-Zielinska, M.; Dobrowolska, A. Alterations in Gut Microbiota Composition in Patients with COVID-19: A Pilot Study of Whole Hypervariable 16S rRNA Gene Sequencing. Biomedicines 2023, 11, 367. https://doi.org/10.3390/biomedicines11020367
Mańkowska-Wierzbicka D, Zuraszek J, Wierzbicka A, Gabryel M, Mahadea D, Baturo A, Zakerska-Banaszak O, Slomski R, Skrzypczak-Zielinska M, Dobrowolska A. Alterations in Gut Microbiota Composition in Patients with COVID-19: A Pilot Study of Whole Hypervariable 16S rRNA Gene Sequencing. Biomedicines. 2023; 11(2):367. https://doi.org/10.3390/biomedicines11020367
Chicago/Turabian StyleMańkowska-Wierzbicka, Dorota, Joanna Zuraszek, Adrianna Wierzbicka, Marcin Gabryel, Dagmara Mahadea, Alina Baturo, Oliwia Zakerska-Banaszak, Ryszard Slomski, Marzena Skrzypczak-Zielinska, and Agnieszka Dobrowolska. 2023. "Alterations in Gut Microbiota Composition in Patients with COVID-19: A Pilot Study of Whole Hypervariable 16S rRNA Gene Sequencing" Biomedicines 11, no. 2: 367. https://doi.org/10.3390/biomedicines11020367