
IGF2: A Role in Metastasis and Tumor Evasion from Immune Surveillance?
 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
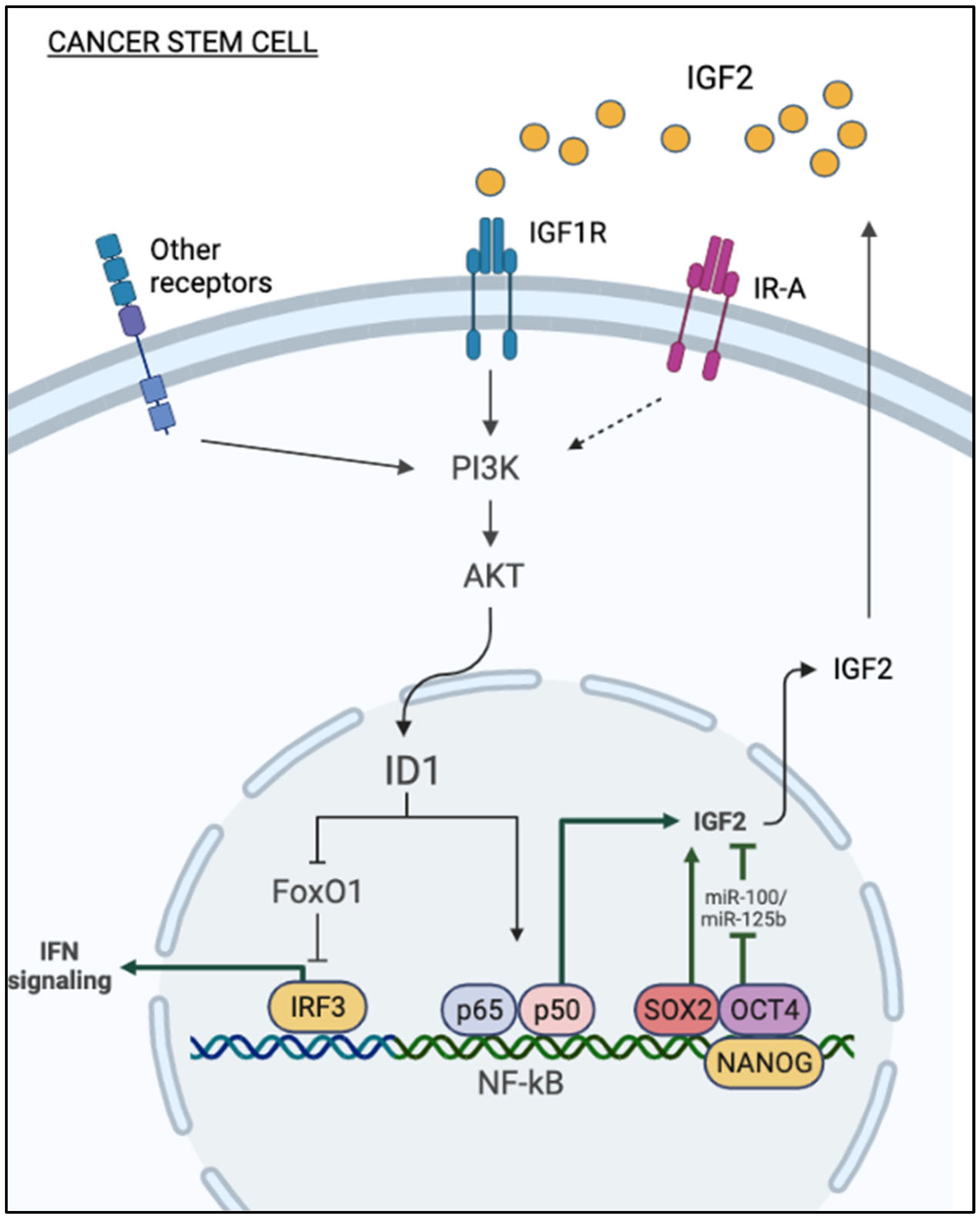
2. IGF2: A Role in Stem-Like Phenotype of Cancer Cells?
2.1. Cancer Cells with Stem-Like Phenotype
2.2. Autocrine IGF2 and CSCs
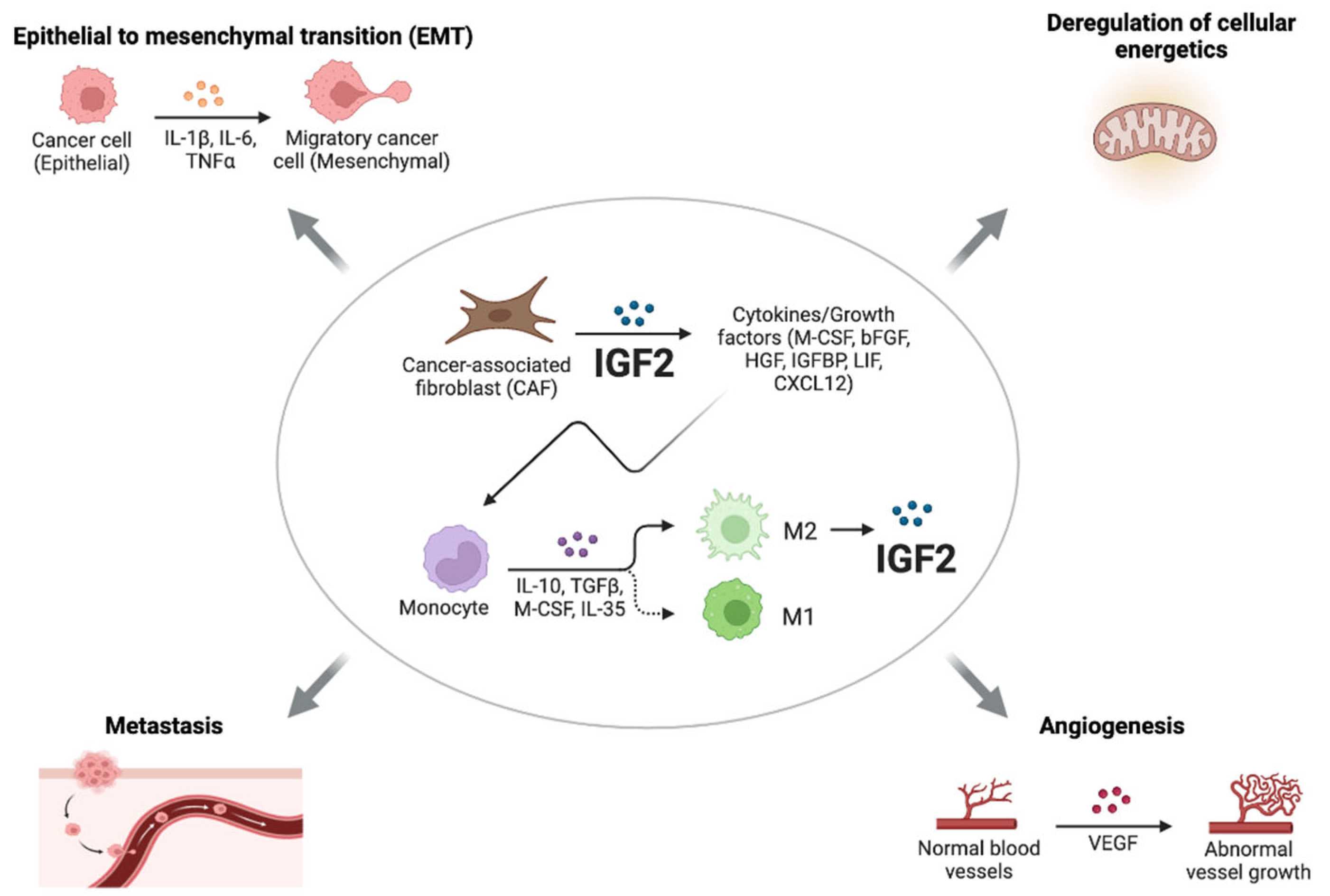
2.3. Paracrine IGF2 and CSCs
3. IGF2 Role in the Pre-Metastatic Niche and Metastasis
4. IGF2 and Immunosuppression
4.1. IGF2 and Dendritic Cells (DCs)
4.2. IGF2 and Myeloid-Derived Suppressor Cells
4.3. IGF2 and T Cells
4.4. IGF2 and Macrophages
4.5. IGF2 and Immune Checkpoint Molecules
5. Therapeutical Implications
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of Deaths from Cancer Caused by Metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of Insulin/IGF Hybrid Receptors: Contributions of the Insulin Receptor L2 and Fn1 Domains and the Alternatively Spliced Exon 11 Sequence to Ligand Binding and Receptor Activation. Biochem. J. 2007, 403, 603–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-like Growth Factor II Receptor in Fetal and Cancer Cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Flier, J.S.; Benecke, H.; Ransil, B.J.; Moller, D.E. Ligand-Binding Properties of the Two Isoforms of the Human Insulin Receptor. Endocrinology 1993, 132, 1132–1138. [Google Scholar] [CrossRef]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/Insulin-like Growth Factor I Hybrid Receptors Have Different Biological Characteristics Depending on the Insulin Receptor Isoform Involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [Green Version]
- Denley, A.; Bonython, E.R.; Booker, G.W.; Cosgrove, L.J.; Forbes, B.E.; Ward, C.W.; Wallace, J.C. Structural Determinants for High-Affinity Binding of Insulin-like Growth Factor II to Insulin Receptor (IR)-A, the Exon 11 Minus Isoform of the IR. Mol. Endocrinol. 2004, 18, 2502–2512. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin Resistance Causes Inflammation in Adipose Tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Ackerman, S.E.; Blackburn, O.A.; Marchildon, F.; Cohen, P. Insights into the Link Between Obesity and Cancer. Curr. Obes. Rep. 2017, 6, 195–203. [Google Scholar] [CrossRef]
- Steele, C.B.; Thomas, C.C.; Henley, S.J.; Massetti, G.M.; Galuska, D.A.; Agurs-Collins, T.; Puckett, M.; Richardson, L.C. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity—United States, 2005-2014. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Pearson-Stuttard, J.; Zhou, B.; Kontis, V.; Bentham, J.; Gunter, M.J.; Ezzati, M. Worldwide Burden of Cancer Attributable to Diabetes and High Body-Mass Index: A Comparative Risk Assessment. Lancet Diabetes Endocrinol. 2018, 6, e6–e15. [Google Scholar] [CrossRef]
- LeRoith, D.; Holly, J.M.P.; Forbes, B.E. Insulin-like Growth Factors: Ligands, Binding Proteins, and Receptors. Mol. Metab. 2021, 52, 101245. [Google Scholar] [CrossRef]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [Green Version]
- Vella, V.; Nicolosi, M.L.; Giuliano, M.; Morrione, A.; Malaguarnera, R.; Belfiore, A. Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation. Cells 2019, 8, 1017. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, L.; Costantino, A.; Pandini, G.; Mineo, R.; Frasca, F.; Scalia, P.; Sbraccia, P.; Goldfine, I.D.; Vigneri, R.; Belfiore, A. Insulin Receptor Activation by IGF-II in Breast Cancers: Evidence for a New Autocrine/Paracrine Mechanism. Oncogene 1999, 18, 2471–2479. [Google Scholar] [CrossRef]
- Vella, V.; Pandini, G.; Sciacca, L.; Mineo, R.; Vigneri, R.; Pezzino, V.; Belfiore, A. A Novel Autocrine Loop Involving IGF-II and the Insulin Receptor Isoform-A Stimulates Growth of Thyroid Cancer. J. Clin. Endocrinol. Metab. 2002, 87, 245–254. [Google Scholar] [CrossRef]
- Ratajczak, M.Z. Igf2-H19, an Imprinted Tandem Gene, Is an Important Regulator of Embryonic Development, a Guardian of Proliferation of Adult Pluripotent Stem Cells, a Regulator of Longevity, and a “passkey” to Cancerogenesis. Folia Histochem. Cytobiol. 2012, 50, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Alipoor, B.; Parvar, S.N.; Sabati, Z.; Ghaedi, H.; Ghasemi, H. An Updated Review of the H19 LncRNA in Human Cancer: Molecular Mechanism and Diagnostic and Therapeutic Importance. Mol. Biol. Rep. 2020, 47, 6357–6374. [Google Scholar] [CrossRef]
- Venkatraman, A.; He, X.C.; Thorvaldsen, J.L.; Sugimura, R.; Perry, J.M.; Tao, F.; Zhao, M.; Christenson, M.K.; Sanchez, R.; Yu, J.Y.; et al. Maternal Imprinting at the H19-Igf2 Locus Maintains Adult Haematopoietic Stem Cell Quiescence. Nature 2013, 500, 345–349. [Google Scholar] [CrossRef]
- Rotwein, P. The Complex Genetics of Human Insulin-like Growth Factor 2 Are Not Reflected in Public Databases. J. Biol. Chem. 2018, 293, 4324–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, T.H.; Hoffman, A.R. Promoter-Specific Imprinting of the Human Insulin-like Growth Factor-II Gene. Nature 1994, 371, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Blyth, A.J.; Kirk, N.S.; Forbes, B.E. Understanding IGF-II Action through Insights into Receptor Binding and Activation. Cells 2020, 9, 2276. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C. IGF2 and Cancer. Endocr. Relat. Cancer 2013, 20, R321–R339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, T.; Nishimoto, I.; Murayama, Y.; Ohkuni, Y.; Ogata, E. Insulin-like Growth Factor-II/Mannose 6-Phosphate Receptor Is Incapable of Activating GTP-Binding Proteins in Response to Mannose 6-Phosphate, but Capable in Response to Insulin-like Growth Factor-II. Biochem. Biophys. Res. Commun. 1990, 168, 1201–1210. [Google Scholar] [CrossRef]
- Okamoto, T.; Katada, T.; Murayama, Y.; Ui, M.; Ogata, E.; Nishimoto, I. A Simple Structure Encodes G Protein-Activating Function of the IGF-II/Mannose 6-Phosphate Receptor. Cell 1990, 62, 709–717. [Google Scholar] [CrossRef]
- Maeng, Y.-S.; Choi, H.-J.; Kwon, J.-Y.; Park, Y.-W.; Choi, K.-S.; Min, J.-K.; Kim, Y.-H.; Suh, P.-G.; Kang, K.-S.; Won, M.-H.; et al. Endothelial Progenitor Cell Homing: Prominent Role of the IGF2-IGF2R-PLCbeta2 Axis. Blood 2009, 113, 233–243. [Google Scholar] [CrossRef]
- Harris, L.K.; Westwood, M. Biology and Significance of Signalling Pathways Activated by IGF-II. Growth Factors 2012, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- DeChiara, T.M.; Robertson, E.J.; Efstratiadis, A. Parental Imprinting of the Mouse Insulin-like Growth Factor II Gene. Cell 1991, 64, 849–859. [Google Scholar] [CrossRef]
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of Insulin-like Growth Factors in Embryonic and Postnatal Growth. Cell 1993, 75, 73–82. [Google Scholar] [CrossRef]
- Burns, J.L.; Hassan, A.B. Cell Survival and Proliferation Are Modified by Insulin-like Growth Factor 2 between Days 9 and 10 of Mouse Gestation. Dev. 2001, 128, 3819–3830. [Google Scholar] [CrossRef]
- Clemmons, D.R. Insulin-like Growth Factor Binding Proteins and Their Role in Controlling IGF Actions. Cytokine Growth Factor Rev. 1997, 8, 45–62. [Google Scholar] [CrossRef] [Green Version]
- Morcavallo, A.; Gaspari, M.; Pandini, G.; Palummo, A.; Cuda, G.; Larsen, M.R.; Vigneri, R.; Belfiore, A. Research Resource: New and Diverse Substrates for the Insulin Receptor Isoform A Revealed by Quantitative Proteomics after Stimulation with IGF-II or Insulin. Mol. Endocrinol. 2011, 25, 1456–1468. [Google Scholar] [CrossRef] [Green Version]
- Pandini, G.; Conte, E.; Medico, E.; Sciacca, L.; Vigneri, R.; Belfiore, A. IGF-II Binding to Insulin Receptor Isoform A Induces a Partially Different Gene Expression Profile from Insulin Binding. Ann. N. Y. Acad. Sci. 2004, 1028, 450–456. [Google Scholar] [CrossRef]
- Morcavallo, A.; Genua, M.; Palummo, A.; Kletvikova, E.; Jiracek, J.; Brzozowski, A.M.; Iozzo, R.V.; Belfiore, A.; Morrione, A. Insulin and Insulin-like Growth Factor II Differentially Regulate Endocytic Sorting and Stability of Insulin Receptor Isoform A. J. Biol. Chem. 2012, 287, 11422–11436. [Google Scholar] [CrossRef] [Green Version]
- Brouwer-Visser, J.; Huang, G.S. IGF2 Signaling and Regulation in Cancer. Cytokine Growth Factor Rev. 2015, 26, 371–377. [Google Scholar] [CrossRef]
- Ward, A. Beck-Wiedemann Syndrome and Wilms’ Tumour. Mol. Hum. Reprod. 1997, 3, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Kashanchi, F.; Zhan, Q.; Zhan, S.; Brady, J.N.; Fornace, A.J.; Seth, P.; Helman, L.J. Regulation of Insulin-like Growth Factor II P3 Promotor by P53: A Potential Mechanism for Tumorigenesis. Cancer Res. 1996, 56, 1367–1373. [Google Scholar]
- Bates, P.; Fisher, R.; Ward, A.; Richardson, L.; Hill, D.J.; Graham, C.F. Mammary Cancer in Transgenic Mice Expressing Insulin-like Growth Factor II (IGF-II). Br. J. Cancer 1995, 72, 1189–1193. [Google Scholar] [CrossRef] [Green Version]
- Moorehead, R.A.; Sanchez, O.H.; Baldwin, R.M.; Khokha, R. Transgenic Overexpression of IGF-II Induces Spontaneous Lung Tumors: A Model for Human Lung Adenocarcinoma. Oncogene 2003, 22, 853–857. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-Dependent PD-L1 Accumulation on Cancer Stem Cells Promotes Immune Evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Fillmore, C.M.; Koyama, S.; Wu, H.; Zhao, Y.; Chen, Z.; Herter-Sprie, G.S.; Akbay, E.A.; Tchaicha, J.H.; Altabef, A.; et al. Loss of Lkb1 and Pten Leads to Lung Squamous Cell Carcinoma with Elevated PD-L1 Expression. Cancer Cell 2014, 25, 590–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhood, B.; Najafi, M.; Salehi, E.; Hashemi Goradel, N.; Nashtaei, M.S.; Khanlarkhani, N.; Mortezaee, K. Disruption of the Redox Balance with Either Oxidative or Anti-Oxidative Overloading as a Promising Target for Cancer Therapy. J. Cell. Biochem. 2019, 120, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Salvadori, G.; Zanardi, F.; Iannelli, F.; Lobefaro, R.; Vernieri, C.; Longo, V.D. Fasting-Mimicking Diet Blocks Triple-Negative Breast Cancer and Cancer Stem Cell Escape. Cell Metab. 2021, 33, 2247–2259.e6. [Google Scholar] [CrossRef]
- Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. [Google Scholar] [CrossRef] [Green Version]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting Signalling Pathways and the Immune Microenvironment of Cancer Stem Cells—A Clinical Update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Kreso, A.; Ryan, P.; Hermans, K.G.; Gibson, L.; Wang, Y.; Tsatsanis, A.; Gallinger, S.; Dick, J.E. ID1 and ID3 Regulate the Self-Renewal Capacity of Human Colon Cancer-Initiating Cells through P21. Cancer Cell 2012, 21, 777–792. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, K.; Shimamura, T.; Kimura, N.; Murayama, T.; Matsubara, D.; Kanauchi, H.; Niida, A.; Shimizu, S.; Nishioka, K.; Tsuji, E.-I.; et al. Addiction to the IGF2-ID1-IGF2 Circuit for Maintenance of the Breast Cancer Stem-like Cells. Oncogene 2017, 36, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Tsao, S.W.; Chan, K.W.; Ludwig, D.L.; Novosyadlyy, R.; Li, Y.Y.; He, Q.Y.; Cheung, A.L.M. Id1-Induced IGF-II and Its Autocrine/Endocrine Promotion of Esophageal Cancer Progression and Chemoresistance--Implications for IGF-II and IGF-IR-Targeted Therapy. Clin. Cancer Res. 2014, 20, 2651–2662. [Google Scholar] [CrossRef]
- Xu, W.W.; Li, B.; Zhao, J.F.; Yang, J.G.; Li, J.Q.; Tsao, S.W.; He, Q.-Y.; Cheung, A.L.M. IGF2 Induces CD133 Expression in Esophageal Cancer Cells to Promote Cancer Stemness. Cancer Lett. 2018, 425, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Murayama, T.; Nakaoku, T.; Enari, M.; Nishimura, T.; Tominaga, K.; Nakata, A.; Tojo, A.; Sugano, S.; Kohno, T.; Gotoh, N. Oncogenic Fusion Gene CD74-NRG1 Confers Cancer Stem Cell-like Properties in Lung Cancer through a IGF2 Autocrine/Paracrine Circuit. Cancer Res. 2016, 76, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.; Chen, H.; Liu, X.; Wang, Y.; Fan, A.; Qi, L.; Pan, L.; Bai, W.; Zhang, Y.; Sun, Y. ID1 Inhibits Foot-and-Mouth Disease Virus Replication via Targeting of Interferon Pathways. FEBS J. 2021, 288, 4364–4381. [Google Scholar] [CrossRef]
- Vella, V.; De Francesco, E.M.; Bonavita, E.; Lappano, R.; Belfiore, A. IFN-I Signaling in Cancer: The Connection with Dysregulated Insulin/IGF Axis. Trends Endocrinol. Metab. 2022, 33, 569–586. [Google Scholar] [CrossRef]
- Chiu, Y.-F.; Wu, C.-C.; Kuo, M.-H.; Miao, C.-C.; Zheng, M.-Y.; Chen, P.-Y.; Lin, S.-C.; Chang, J.-L.; Wang, Y.-H.; Chou, Y.-T. Critical Role of SOX2-IGF2 Signaling in Aggressiveness of Bladder Cancer. Sci. Rep. 2020, 10, 8261. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Shi, L.; Zhang, L.; Li, R.; Liang, J.; Yu, W.; Sun, L.; Yang, X.; Wang, Y.; Zhang, Y.; et al. The Molecular Mechanism Governing the Oncogenic Potential of SOX2 in Breast Cancer. J. Biol. Chem. 2008, 283, 17969–17978. [Google Scholar] [CrossRef] [Green Version]
- Bass, A.J.; Watanabe, H.; Mermel, C.H.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 Is an Amplified Lineage-Survival Oncogene in Lung and Esophageal Squamous Cell Carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef]
- Gebeshuber, C.A.; Martinez, J. MiR-100 Suppresses IGF2 and Inhibits Breast Tumorigenesis by Interfering with Proliferation and Survival Signaling. Oncogene 2013, 32, 3306–3310. [Google Scholar] [CrossRef]
- Ge, Y.; Sun, Y.; Chen, J. IGF-II Is Regulated by MicroRNA-125b in Skeletal Myogenesis. J. Cell Biol. 2011, 192, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Seol, H.S.; Akiyama, Y.; Lee, S.-E.; Shimada, S.; Jang, S.J. Loss of MiR-100 and MiR-125b Results in Cancer Stem Cell Properties through IGF2 Upregulation in Hepatocellular Carcinoma. Sci. Rep. 2020, 10, 21412. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Lobe, C.; Tahraoui, S.; Clapéron, A.; Mergey, M.; Merabtene, F.; Wendum, D.; Coulouarn, C.; Housset, C.; Desbois-Mouthon, C.; et al. The IGF2/IR/IGF1R Pathway in Tumor Cells and Myofibroblasts Mediates Resistance to EGFR Inhibition in Cholangiocarcinoma. Clin. Cancer Res. 2018, 24, 4282–4296. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Liu, C.; Chen, F.-K.; Feng, Z.-P.; Jia, L.; Liu, P.-J.; Yang, Z.-X.; Hou, F.; Deng, Z.-Y. M2-like Tumour-associated Macrophage-secreted IGF Promotes Thyroid Cancer Stemness and Metastasis by Activating the PI3K/AKT/MTOR Pathway. Mol. Med. Rep. 2021, 24, 604. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Singer, C.; Rasmussen, A.; Smith, H.S.; Lippman, M.E.; Lynch, H.T.; Cullen, K.J. Malignant Breast Epithelium Selects for Insulin-like Growth Factor II Expression in Breast Stroma: Evidence for Paracrine Function. Cancer Res. 1995, 55, 2448–2454. [Google Scholar]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S.; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-β Signaling in a Subpopulation of Human Stromal Cells Promotes Prostatic Carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-J.; Ho, C.-C.; Chang, Y.-L.; Chen, H.-Y.; Lin, C.-A.; Ling, T.-Y.; Yu, S.-L.; Yuan, S.-S.; Chen, Y.-J.L.; Lin, C.-Y.; et al. Cancer-Associated Fibroblasts Regulate the Plasticity of Lung Cancer Stemness via Paracrine Signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef] [Green Version]
- Richards, M.; Fong, C.-Y.; Chan, W.-K.; Wong, P.-C.; Bongso, A. Human Feeders Support Prolonged Undifferentiated Growth of Human Inner Cell Masses and Embryonic Stem Cells. Nat. Biotechnol. 2002, 20, 933–936. [Google Scholar] [CrossRef]
- Unger, C.; Kramer, N.; Unterleuthner, D.; Scherzer, M.; Burian, A.; Rudisch, A.; Stadler, M.; Schlederer, M.; Lenhardt, D.; Riedl, A.; et al. Stromal-Derived IGF2 Promotes Colon Cancer Progression via Paracrine and Autocrine Mechanisms. Oncogene 2017, 36, 5341–5355. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Krebs, M.G.; Hou, J.-M.; Ward, T.H.; Blackhall, F.H.; Dive, C. Circulating Tumour Cells: Their Utility in Cancer Management and Predicting Outcomes. Ther. Adv. Med. Oncol. 2010, 2, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.W.; Li, B.; Guan, X.Y.; Chung, S.K.; Wang, Y.; Yip, Y.L.; Law, S.Y.K.; Chan, K.T.; Lee, N.P.Y.; Chan, K.W.; et al. Cancer Cell-Secreted IGF2 Instigates Fibroblasts and Bone Marrow-Derived Vascular Progenitor Cells to Promote Cancer Progression. Nat. Commun. 2017, 8, 14399. [Google Scholar] [CrossRef] [Green Version]
- Gui, Y.; Aguilar-Mahecha, A.; Krzemien, U.; Hosein, A.; Buchanan, M.; Lafleur, J.; Pollak, M.; Ferrario, C.; Basik, M. Metastatic Breast Carcinoma-Associated Fibroblasts Have Enhanced Protumorigenic Properties Related to Increased IGF2 Expression. Clin. Cancer Res. 2019, 25, 7229–7242. [Google Scholar] [CrossRef] [Green Version]
- Molnár, K.; Mészáros, Á.; Fazakas, C.; Kozma, M.; Győri, F.; Reisz, Z.; Tiszlavicz, L.; Farkas, A.E.; Nyúl-Tóth, Á.; Haskó, J.; et al. Pericyte-Secreted IGF2 Promotes Breast Cancer Brain Metastasis Formation. Mol. Oncol. 2020, 14, 2040–2057. [Google Scholar] [CrossRef]
- Salminen, A. Activation of Immunosuppressive Network in the Aging Process. Ageing Res. Rev. 2020, 57, 100998. [Google Scholar] [CrossRef]
- Liu, E.; Law, H.K.W.; Lau, Y.-L. Insulin-like Growth Factor I Promotes Maturation and Inhibits Apoptosis of Immature Cord Blood Monocyte-Derived Dendritic Cells through MEK and PI 3-Kinase Pathways. Pediatr. Res. 2003, 54, 919–925. [Google Scholar] [CrossRef] [Green Version]
- Matte, I.; Lane, D.; Laplante, C.; Rancourt, C.; Piché, A. Profiling of Cytokines in Human Epithelial Ovarian Cancer Ascites. Am. J. Cancer Res. 2012, 2, 566–580. [Google Scholar]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 Expression Identifies a Novel Suppressive Macrophage Population in Human Ovarian Carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef]
- Giuntoli, R.L.; Webb, T.J.; Zoso, A.; Rogers, O.; Diaz-Montes, T.P.; Bristow, R.E.; Oelke, M. Ovarian Cancer-Associated Ascites Demonstrates Altered Immune Environment: Implications for Antitumor Immunity. Anticancer Res. 2009, 29, 2875–2884. [Google Scholar] [PubMed]
- Somri-Gannam, L.; Meisel-Sharon, S.; Hantisteanu, S.; Groisman, G.; Limonad, O.; Hallak, M.; Bruchim, I. IGF1R Axis Inhibition Restores Dendritic Cell Antitumor Response in Ovarian Cancer. Transl. Oncol. 2020, 13, 100790. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, S.; Gennaro, M.; Vaccari, M.; Ferraris, C.; Nicola, S.; Riva, A.; Clerici, M.; Greco, M.; Villa, M.L. Altered Maturation of Peripheral Blood Dendritic Cells in Patients with Breast Cancer. Br. J. Cancer 2003, 89, 1463–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaspyridonos, M.; Matei, I.; Huang, Y.; do Rosario Andre, M.; Brazier-Mitouart, H.; Waite, J.C.; Chan, A.S.; Kalter, J.; Ramos, I.; Wu, Q.; et al. Id1 Suppresses Anti-Tumour Immune Responses and Promotes Tumour Progression by Impairing Myeloid Cell Maturation. Nat. Commun. 2015, 6, 6840. [Google Scholar] [CrossRef] [Green Version]
- Collins, N.B.; Al Abosy, R.; Miller, B.C.; Bi, K.; Zhao, Q.; Quigley, M.; Ishizuka, J.J.; Yates, K.B.; Pope, H.W.; Manguso, R.T.; et al. PI3K Activation Allows Immune Evasion by Promoting an Inhibitory Myeloid Tumor Microenvironment. J. Immunother. Cancer 2022, 10, e003402. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.A.; Drake, V.; Huang, H.-S.; Chiu, S.; Zheng, L. Reprogramming the Tumor Microenvironment: Tumor-Induced Immunosuppressive Factors Paralyze T Cells. Oncoimmunology 2015, 4, e1016700. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Z.; Roden, D.L.; Wang, C.; Holliday, H.; Harvey, K.; Cazet, A.S.; Murphy, K.J.; Pereira, B.; Al-Eryani, G.; Bartonicek, N.; et al. Stromal Cell Diversity Associated with Immune Evasion in Human Triple-Negative Breast Cancer. EMBO J. 2020, 39, e104063. [Google Scholar] [CrossRef]
- Lal, G.; Zhang, N.; van der Touw, W.; Ding, Y.; Ju, W.; Bottinger, E.P.; Reid, S.P.; Levy, D.E.; Bromberg, J.S. Epigenetic Regulation of Foxp3 Expression in Regulatory T Cells by DNA Methylation. J. Immunol. 2009, 182, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.-C.; Li, C.-H.; Chu, L.-H.; Huang, P.-S.; Sheu, B.-C.; Huang, S.-C. Regulatory T Cells Suppress Natural Killer Cell Immunity in Patients With Human Cervical Carcinoma. Int. J. Gynecol. Cancer 2016, 26, 156–162. [Google Scholar] [CrossRef]
- Pallandre, J.-R.; Brillard, E.; Créhange, G.; Radlovic, A.; Remy-Martin, J.-P.; Saas, P.; Rohrlich, P.-S.; Pivot, X.; Ling, X.; Tiberghien, P.; et al. Role of STAT3 in CD4+CD25+FOXP3+ Regulatory Lymphocyte Generation: Implications in Graft-versus-Host Disease and Antitumor Immunity. J. Immunol. 2007, 179, 7593–7604. [Google Scholar] [CrossRef]
- Bilbao, D.; Luciani, L.; Johannesson, B.; Piszczek, A.; Rosenthal, N. Insulin-like Growth Factor-1 Stimulates Regulatory T Cells and Suppresses Autoimmune Disease. EMBO Mol. Med. 2014, 6, 1423–1435. [Google Scholar] [CrossRef]
- Yang, G.; Geng, X.-R.; Song, J.-P.; Wu, Y.; Yan, H.; Zhan, Z.; Yang, L.; He, W.; Liu, Z.-Q.; Qiu, S.; et al. Insulin-like Growth Factor 2 Enhances Regulatory T-Cell Functions and Suppresses Food Allergy in an Experimental Model. J. Allergy Clin. Immunol. 2014, 133, 1702–1708.e5. [Google Scholar] [CrossRef]
- Miyagawa, I.; Nakayamada, S.; Nakano, K.; Yamagata, K.; Sakata, K.; Yamaoka, K.; Tanaka, Y. Induction of Regulatory T Cells and Its Regulation with Insulin-like Growth Factor/Insulin-like Growth Factor Binding Protein-4 by Human Mesenchymal Stem Cells. J. Immunol. 2017, 199, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Johannesson, B.; Sattler, S.; Semenova, E.; Pastore, S.; Kennedy-Lydon, T.M.; Sampson, R.D.; Schneider, M.D.; Rosenthal, N.; Bilbao, D. Insulin-like Growth Factor-1 Induces Regulatory T Cell-Mediated Suppression of Allergic Contact Dermatitis in Mice. Dis. Model. Mech. 2014, 7, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism Governs Dendritic Cell and Macrophage Function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.-C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the MTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Lin, L.; Li, Q.; Liu, K.; Huang, Y.; Wang, X.; Cao, K.; Chen, X.; Cao, W.; Li, F.; et al. IGF-2 Preprograms Maturing Macrophages to Acquire Oxidative Phosphorylation-Dependent Anti-Inflammatory Properties. Cell Metab. 2019, 29, 1363–1375.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, L.; Lan, B.; Wang, Y.; Du, L.; Chen, X.; Li, Q.; Liu, K.; Hu, M.; Xue, Y.; et al. IGF2R-Initiated Proton Rechanneling Dictates an Anti-Inflammatory Property in Macrophages. Sci. Adv. 2020, 6, eabb7389. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.C.C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e9. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-C.; Hu, Y.-W.; Sha, Y.-H.; Gao, J.-J.; Ma, X.; Li, S.-F.; Zhao, J.-Y.; Qiu, Y.-R.; Lu, J.-B.; Huang, C.; et al. Ox-LDL Upregulates IL-6 Expression by Enhancing NF-ΚB in an IGF2-Dependent Manner in THP-1 Macrophages. Inflammation 2015, 38, 2116–2123. [Google Scholar] [CrossRef]
- Ieronymaki, E.; Theodorakis, E.M.; Lyroni, K.; Vergadi, E.; Lagoudaki, E.; Al-Qahtani, A.; Aznaourova, M.; Neofotistou-Themeli, E.; Eliopoulos, A.G.; Vaporidi, K.; et al. Insulin Resistance in Macrophages Alters Their Metabolism and Promotes an M2-Like Phenotype. J. Immunol. 2019, 202, 1786–1797. [Google Scholar] [CrossRef] [Green Version]
- Ieronymaki, E.; Daskalaki, M.G.; Lyroni, K.; Tsatsanis, C. Insulin Signaling and Insulin Resistance Facilitate Trained Immunity in Macrophages Through Metabolic and Epigenetic Changes. Front. Immunol. 2019, 10, 1330. [Google Scholar] [CrossRef]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Schworer, S.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The Kinase Akt1 Controls Macrophage Response to Lipopolysaccharide by Regulating MicroRNAs. Immunity 2009, 31, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Lee, S.-J.; Jang, B.-C.; Lee, S.-W.; Yang, Y.-I.; Suh, S.-I.; Park, Y.-M.; Oh, S.; Shin, J.-G.; Yao, S.; Chen, L.; et al. Interferon Regulatory Factor-1 Is Prerequisite to the Constitutive Expression and IFN-Gamma-Induced Upregulation of B7-H1 (CD274). FEBS Lett. 2006, 580, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic Kinase NPM/ALK Induces through STAT3 Expression of Immunosuppressive Protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [Green Version]
- Crane, C.A.; Panner, A.; Murray, J.C.; Wilson, S.P.; Xu, H.; Chen, L.; Simko, J.P.; Waldman, F.M.; Pieper, R.O.; Parsa, A.T. PI(3) Kinase Is Associated with a Mechanism of Immunoresistance in Breast and Prostate Cancer. Oncogene 2009, 28, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, M.; Wu, P.; Chen, C.; Xu, Z.P.; Gu, W. Increased PD-L1 Expression in Breast and Colon Cancer Stem Cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 602–604. [Google Scholar] [CrossRef]
- Chen, M.; Sharma, A.; Lin, Y.; Wu, Y.; He, Q.; Gu, Y.; Xu, Z.P.; Monteiro, M.; Gu, W. Insluin and Epithelial Growth Factor (EGF) Promote Programmed Death Ligand 1(PD-L1) Production and Transport in Colon Cancer Stem Cells. BMC Cancer 2019, 19, 153. [Google Scholar] [CrossRef]
- Heckl, S.M.; Mau, F.; Senftleben, A.; Daunke, T.; Beckinger, S.; Abdullazade, S.; Schreiber, S.; Röcken, C.; Sebens, S.; Schäfer, H. Programmed Death-Ligand 1 (PD-L1) Expression Is Induced by Insulin in Pancreatic Ductal Adenocarcinoma Cells Pointing to Its Role in Immune Checkpoint Control. Med. Sci. 2021, 9, 48. [Google Scholar] [CrossRef]
- Chen, C.; Li, S.; Xue, J.; Qi, M.; Liu, X.; Huang, Y.; Hu, J.; Dong, H.; Ling, K. PD-L1 Tumor-Intrinsic Signaling and Its Therapeutic Implication in Triple-Negative Breast Cancer. JCI Insight 2021, 6, e131458. [Google Scholar] [CrossRef]
- Alsuliman, A.; Colak, D.; Al-Harazi, O.; Fitwi, H.; Tulbah, A.; Al-Tweigeri, T.; Al-Alwan, M.; Ghebeh, H. Bidirectional Crosstalk between PD-L1 Expression and Epithelial to Mesenchymal Transition: Significance in Claudin-Low Breast Cancer Cells. Mol. Cancer 2015, 14, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The Immune Checkpoint Ligand PD-L1 Is Upregulated in EMT-Activated Human Breast Cancer Cells by a Mechanism Involving ZEB-1 and MiR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Carpino, G.; Nicolazzo, C.; Gradilone, A.; Gianni, W.; Gelibter, A.; Gaudio, E.; Cortesi, E.; Gazzaniga, P. PD-L1 and Epithelial-Mesenchymal Transition in Circulating Tumor Cells from Non-Small Cell Lung Cancer Patients: A Molecular Shield to Evade Immune System? Oncoimmunology 2017, 6, e1315488. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jang, J.-Y.; Koh, J.; Kwon, D.; Kim, Y.A.; Paeng, J.C.; Ock, C.-Y.; Keam, B.; Kim, M.; Kim, T.M.; et al. Programmed Cell Death Ligand-1-Mediated Enhancement of Hexokinase 2 Expression Is Inversely Related to T-Cell Effector Gene Expression in Non-Small-Cell Lung Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 462. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liang, Y.; Li, D.; Wang, L.; Liang, Z.; Chen, Y.; Ma, G.; Wu, H.; Jiao, W.; Niu, H. Glucose Metabolism Involved in PD-L1-Mediated Immune Escape in the Malignant Kidney Tumour Microenvironment. Cell Death Discov. 2021, 7, 15. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, H.; Yuan, Y.; He, Q.; Zhou, J.; Li, S.; Sun, Y.; Li, D.Y.; Qiu, H.-B.; Wang, W.; et al. Fatty Acid Oxidation Controls CD8+ Tissue-Resident Memory T-Cell Survival in Gastric Adenocarcinoma. Cancer Immunol. Res. 2020, 8, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Pacher, M.; Seewald, M.J.; Mikula, M.; Oehler, S.; Mogg, M.; Vinatzer, U.; Eger, A.; Schweifer, N.; Varecka, R.; Sommergruber, W.; et al. Impact of Constitutive IGF1/IGF2 Stimulation on the Transcriptional Program of Human Breast Cancer Cells. Carcinogenesis 2007, 28, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Yang, J.; Bai, J.; Ren, J. Reverse of Non-Small Cell Lung Cancer Drug Resistance Induced by Cancer-Associated Fibroblasts via a Paracrine Pathway. Cancer Sci. 2018, 109, 944–955. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Sugihara, E.; Yamaguchi-Iwai, S.; Tamaki, S.; Koyama, Y.; Kamel, W.; Ueki, A.; Ishikawa, T.; Chiyoda, T.; Osuka, S.; et al. IGF2 Preserves Osteosarcoma Cell Survival by Creating an Autophagic State of Dormancy That Protects Cells against Chemotherapeutic Stress. Cancer Res. 2014, 74, 6531–6541. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Zhang, Z.; Qiu, N.; Ling, L.; Jia, X.; Song, Y.; Li, H.; Li, J.; Lyu, H.; Liu, H.; et al. Disruption of FOXO3a-MiRNA Feedback Inhibition of IGF2/IGF-1R/IRS1 Signaling Confers Herceptin Resistance in HER2-Positive Breast Cancer. Nat. Commun. 2021, 12, 2699. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Campbell, F.; Figueiredo, C.; Hammond, D.; Ellies, L.G.; Weyer-Czernilofsky, U.; Bogenrieder, T.; Schmid, M.; Mielgo, A. Blockade of Insulin-like Growth Factors Increases Efficacy of Paclitaxel in Metastatic Breast Cancer. Oncogene 2018, 37, 2022–2036. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Das, M.; Oberg, A.; Sahoo, D.; Wu, P.; Sauceda, C.; Jih, L.; Ellies, L.G.; Langiewicz, M.T.; Sen, S.; et al. Hepatocyte Deletion of IGF2 Prevents DNA Damage and Tumor Formation in Hepatocellular Carcinoma. Adv. Sci. 2022, 9, e2105120. [Google Scholar] [CrossRef]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I.; Wild, R.; et al. Compensatory Insulin Receptor (IR) Activation on Inhibition of Insulin-like Growth Factor-1 Receptor (IGF-1R): Rationale for Cotargeting IGF-1R and IR in Cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin Receptor Functionally Enhances Multistage Tumor Progression and Conveys Intrinsic Resistance to IGF-1R Targeted Therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, C.; Manara, M.C.; Nicoletti, G.; Marino, M.T.; Lollini, P.-L.; Astolfi, A.; Pandini, G.; López-Guerrero, J.A.; Schaefer, K.-L.; Belfiore, A.; et al. Efficacy of and Resistance to Anti-IGF-1R Therapies in Ewing’s Sarcoma Is Dependent on Insulin Receptor Signaling. Oncogene 2011, 30, 2730–2740. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Kang, J.-H.; Boo, H.-J.; Hwang, S.-J.; Hong, S.; Lee, S.-C.; Park, Y.-J.; Chung, T.-M.; Youn, H.; Mi Lee, S.; et al. STAT3-Mediated IGF-2 Secretion in the Tumour Microenvironment Elicits Innate Resistance to Anti-IGF-1R Antibody. Nat. Commun. 2015, 6, 8499. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Konda, J.D.; Perrino, S.; Celia Fernandez, M.; Lowy, A.M.; Brodt, P. Targeting the IGF-Axis Potentiates Immunotherapy for Pancreatic Ductal Adenocarcinoma Liver Metastases by Altering the Immunosuppressive Microenvironment. Mol. Cancer Ther. 2021, 20, 2469–2482. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belfiore, A.; Rapicavoli, R.V.; Le Moli, R.; Lappano, R.; Morrione, A.; De Francesco, E.M.; Vella, V. IGF2: A Role in Metastasis and Tumor Evasion from Immune Surveillance? Biomedicines 2023, 11, 229. https://doi.org/10.3390/biomedicines11010229
Belfiore A, Rapicavoli RV, Le Moli R, Lappano R, Morrione A, De Francesco EM, Vella V. IGF2: A Role in Metastasis and Tumor Evasion from Immune Surveillance? Biomedicines. 2023; 11(1):229. https://doi.org/10.3390/biomedicines11010229
Chicago/Turabian StyleBelfiore, Antonino, Rosaria Valentina Rapicavoli, Rosario Le Moli, Rosamaria Lappano, Andrea Morrione, Ernestina Marianna De Francesco, and Veronica Vella. 2023. "IGF2: A Role in Metastasis and Tumor Evasion from Immune Surveillance?" Biomedicines 11, no. 1: 229. https://doi.org/10.3390/biomedicines11010229