
Neuroprotective Effects of Licochalcone D in Oxidative-Stress-Induced Primitive Neural Stem Cells from Parkinson’s Disease Patient-Derived iPSCs
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Flow Cytometry Analysis
2.4. Western Blot Analysis
2.5. Immunofluorescence
2.6. Quantitative Real-Time PCR
2.7. In Vitro Pull-Down Assay
2.8. Statistical Analysis
3. Results
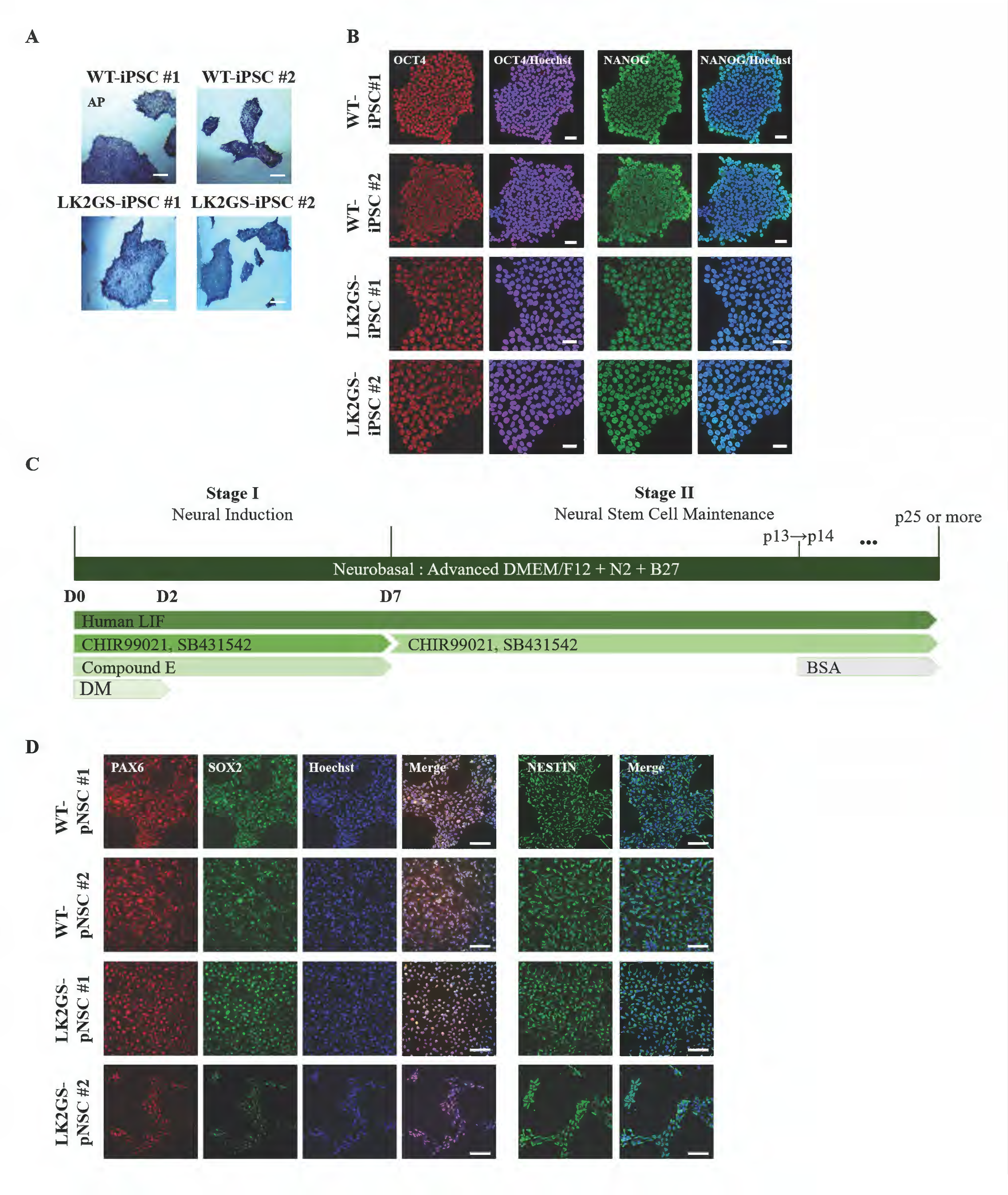
3.1. Generation and Characterization of pNSCs from LK2GS Patients
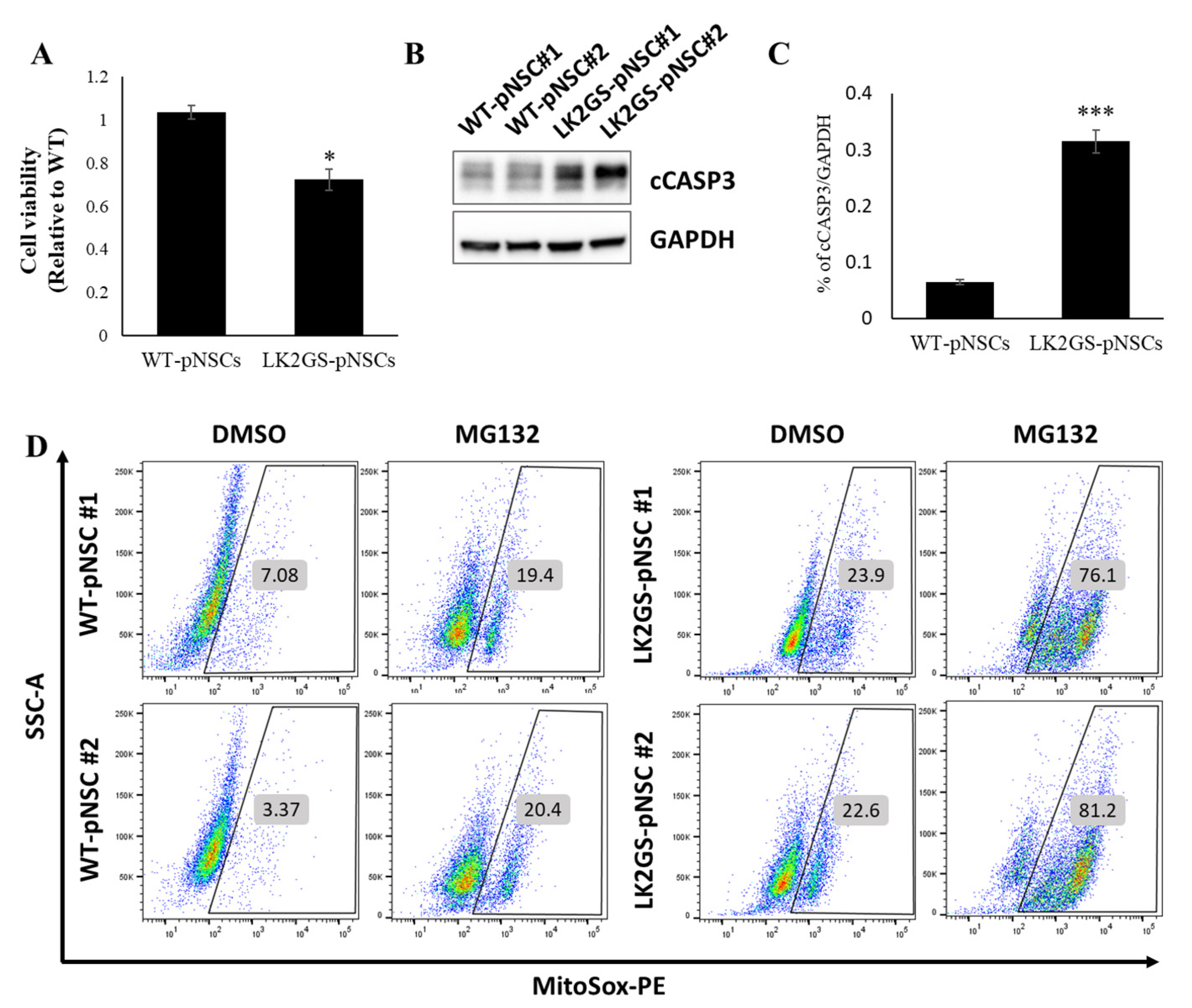
3.2. LK2GS-pNSCs as an In Vitro Model for PD
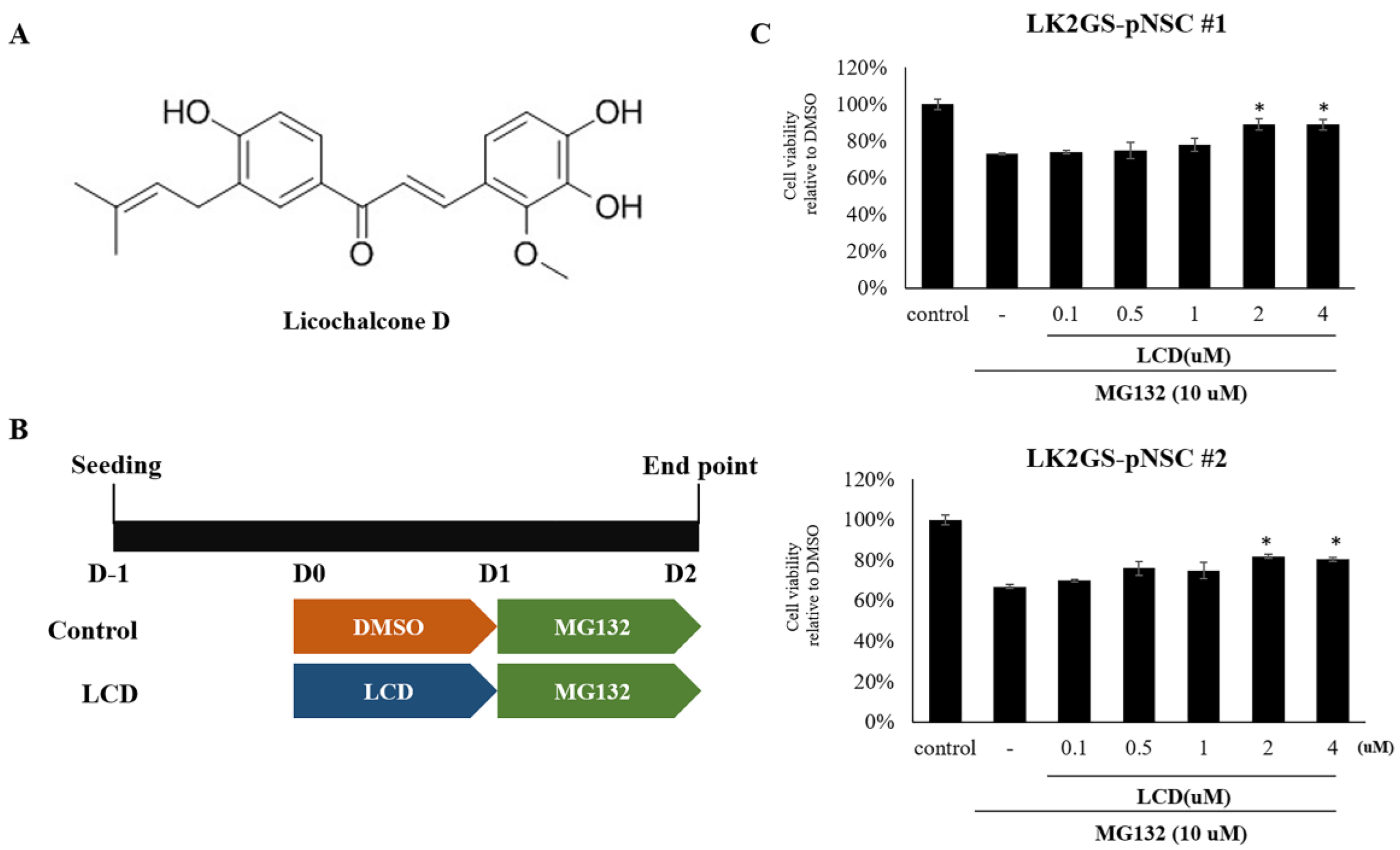
3.3. LCD Attenuated MG132-Induced Cell Death of the PD Model
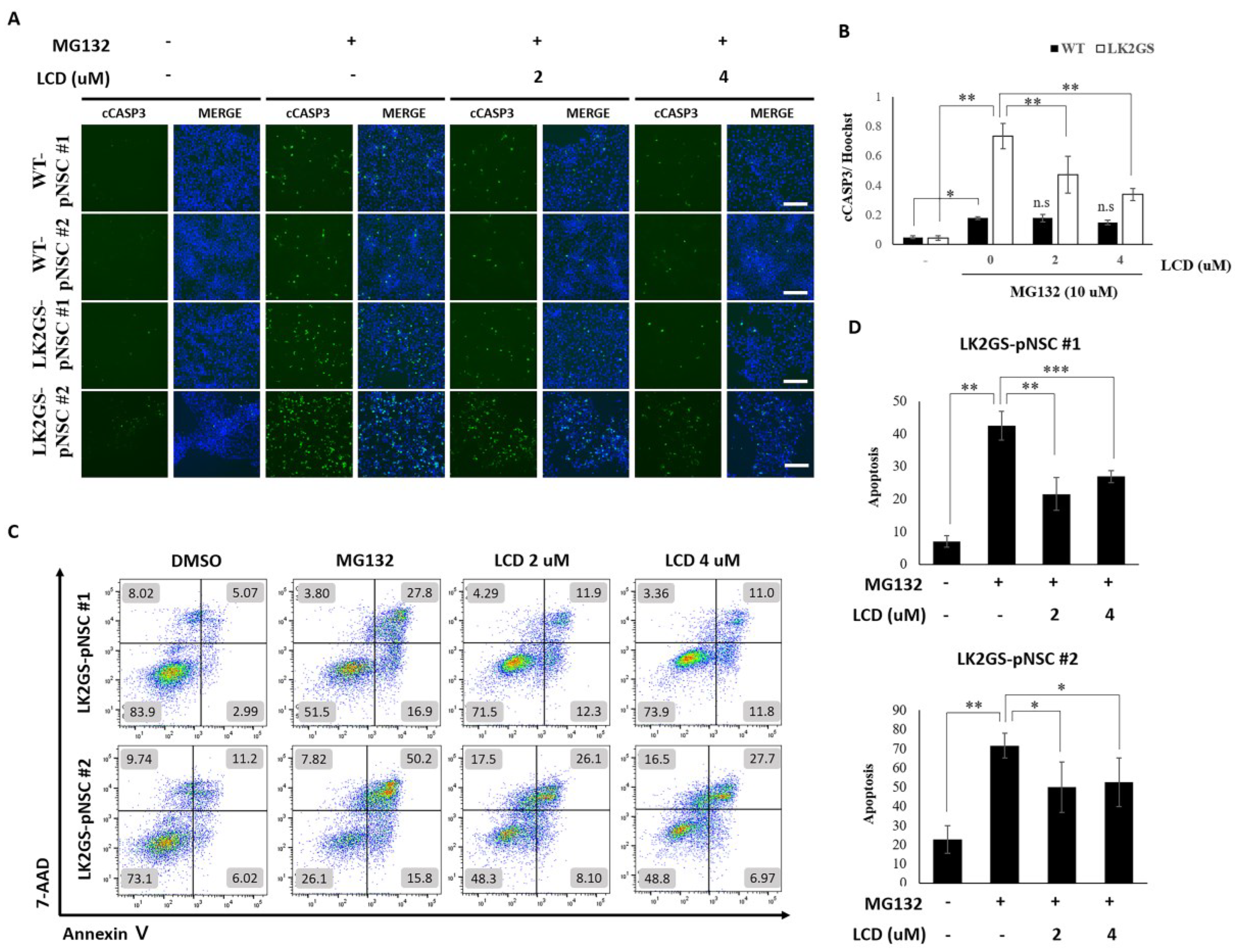
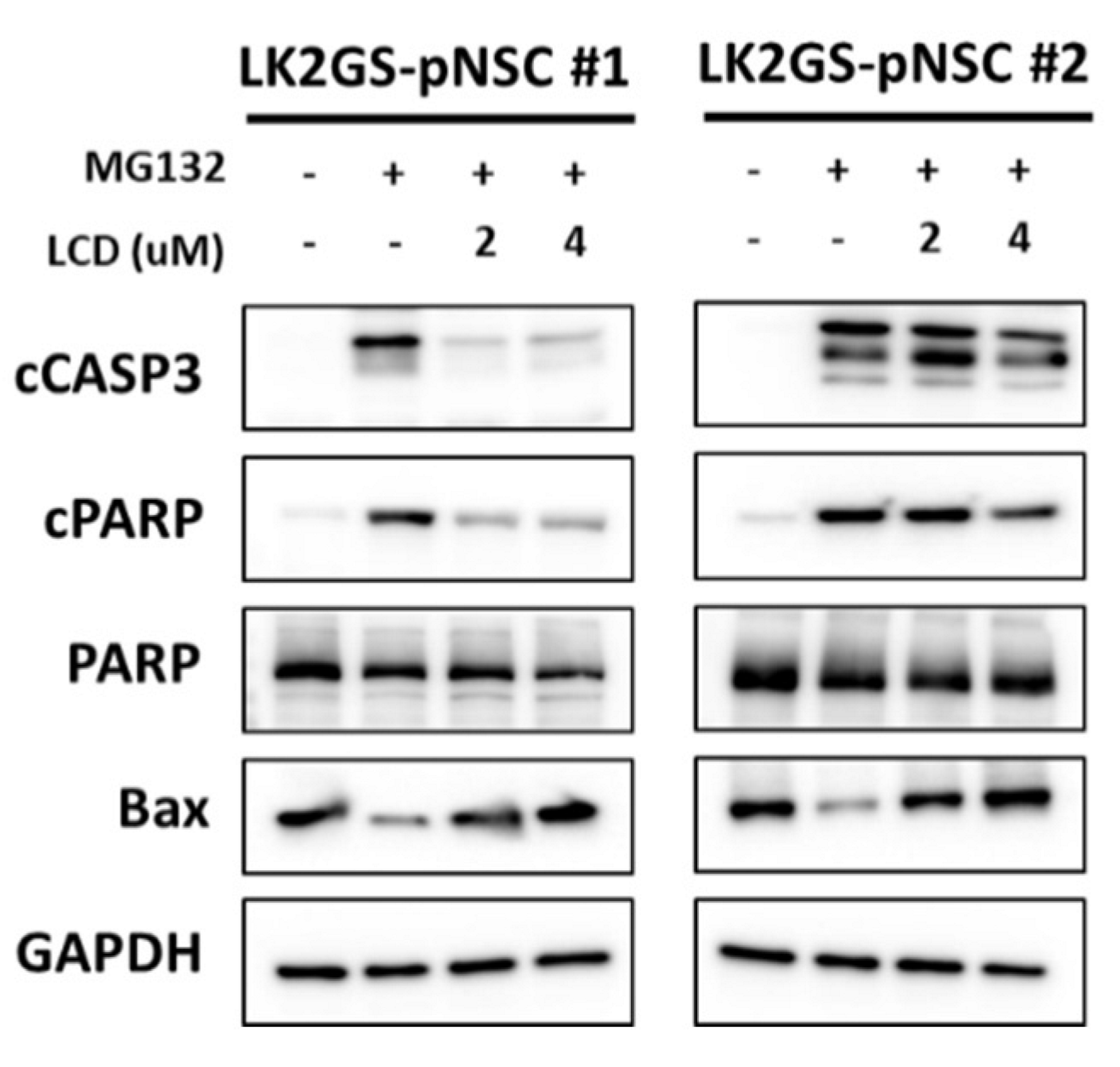
3.4. LCD Protects against MG132-Induced Apoptosis in the PD Model
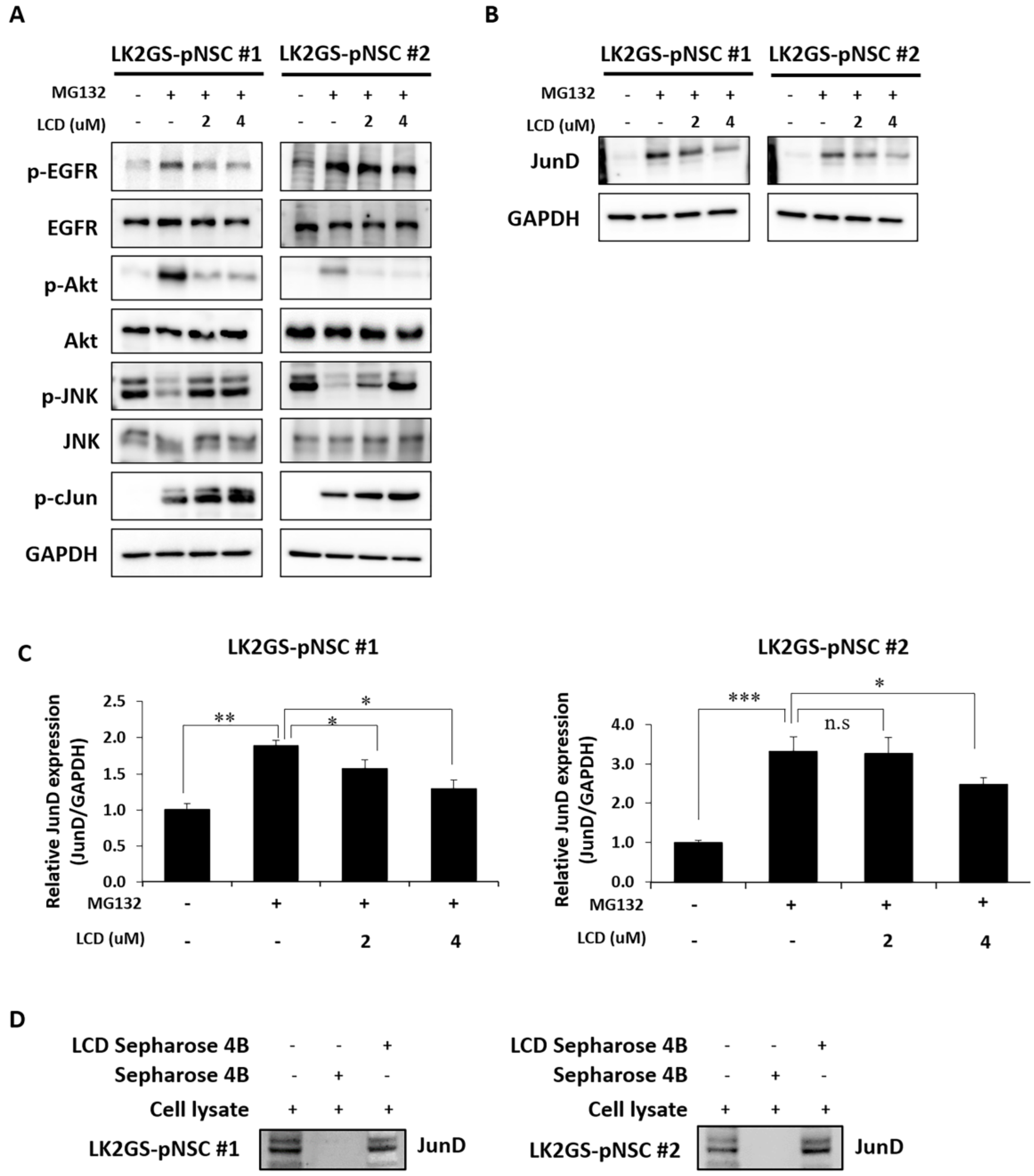
3.5. LCD Administration Modulated Phosphorylation of EGFR/AKT and JNK in the MG132-Treated PD Model
3.6. LCD Regulated the Expression of Apoptosis-Related Molecules in the PD Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| SNpc | substantia nigra pars compacta |
| PFC | prefrontal cortex |
| LCD | Licochalcone D |
| iPSCs | induced pluripotent stem cells |
| pNSCs | primitive neural stem cells |
| LRRK2 | leucine-rich repeat kinase 2 |
| Lk2GS | LRRK2 G2019S |
| ROS | reactive oxygen species |
| PAX6 | paired box 6 |
| SOX2 | SRY-box transcription factor 2 |
| NES | Nestin |
| cCASP3 | cleaved caspase-3 |
| MAPK | mitogen-activated protein kinase |
| EGFR | epidermal growth factor receptor |
| AKT | serine/thereonine protein kinase B |
| JNK | c-Jun NH(2)-terminal kinases |
| PARP | poly (ADP-ribose) polymerase |
| PI3K | phosphoinositide 3-kinase |
| mTOR | mechanistic target of rapamycin |
| BBB | blood–brain barrier |
References
- Corona, J.C. Natural Compounds for the Management of Parkinson’s Disease and Attention-Deficit/Hyperactivity Disorder. BioMed Res. Int. 2018, 2018, 4067597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.D.; De Palma, G.; Ahmadi, A.; Scott, N.W.; Prescott, G.J.; Bennett, J.; Semple, S.; Dick, S.; Counsell, C.; Mozzoni, P.; et al. Environmental risk factors for Parkinson’s disease and parkinsonism: The Geoparkinson study. Occup. Environ. Med. 2007, 64, 666–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the human ventromedial prefrontal cortex support fear learning, fear extinction or both? A commentary on subregional contributions. Mol. Psychiatry 2022, 27, 784–786. [Google Scholar] [CrossRef]
- Battaglia, S.; Cardellicchio, P.; Di Fazio, C.; Nazzi, C.; Fracasso, A.; Borgomaneri, S. Stopping in (e)motion: Reactive action inhibition when facing valence-independent emotional stimuli. Front. Behav. Neurosci. 2022, 16, 998714. [Google Scholar] [CrossRef]
- Battaglia, S.; Cardellicchio, P.; Di Fazio, C.; Nazzi, C.; Fracasso, A.; Borgomaneri, S. The Influence of Vicarious Fear-Learning in "Infecting" Reactive Action Inhibition. Front. Behav. Neurosci. 2022, 16, 946263. [Google Scholar] [CrossRef]
- Battaglia, S.; Thayer, J.F. Functional interplay between central and autonomic nervous systems in human fear conditioning. Trends Neurosci. 2022, 45, 504–506. [Google Scholar] [CrossRef]
- Battaglia, S.; Orsolini, S.; Borgomaneri, S.; Barbieri, R.; Diciotti, S.; di Pellegrino, G. Characterizing cardiac autonomic dynamics of fear learning in humans. Psychophysiology 2022, 59, e14122. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabo, A.; Vecsei, L. Integrating Armchair, Bench, and Bedside Research for Behavioral Neurology and Neuropsychiatry: Editorial. Biomedicines 2022, 10, 2999. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Fabius, J.H.; Moravkova, K.; Fracasso, A.; Borgomaneri, S. The Neurobiological Correlates of Gaze Perception in Healthy Individuals and Neurologic Patients. Biomedicines 2022, 10, 627. [Google Scholar] [CrossRef]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-dependent extinction of threat memories: Influences of healthy aging. Sci. Rep. 2018, 8, 12592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasser, T. Mendelian forms of Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Toulouse, A.; Sullivan, A.M. Progress in Parkinson’s disease-where do we stand? Prog. Neurobiol. 2008, 85, 376–392. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [Green Version]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Senkevich, K.; Rudakou, U.; Gan-Or, Z. New therapeutic approaches to Parkinson’s disease targeting GBA, LRRK2 and Parkin. Neuropharmacology 2022, 202, 108822. [Google Scholar] [CrossRef]
- Berwick, D.C.; Heaton, G.R.; Azeggagh, S.; Harvey, K. LRRK2 Biology from structure to dysfunction: Research progresses, but the themes remain the same. Mol. Neurodegener. 2019, 14, 49. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Schapira, A.H.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: Future therapeutic perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Chesselet, M.F.; Fleming, S.; Mortazavi, F.; Meurers, B. Strengths and limitations of genetic mouse models of Parkinson’s disease. Park. Relat. Disord. 2008, 14 (Suppl. 2), S84–S87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef]
- Beal, M.F. Experimental models of Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 325–334. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.C. Neural subtype specification from embryonic stem cells. Brain Pathol. 2006, 16, 132–142. [Google Scholar] [CrossRef]
- Sim, H.; Seo, J.H.; Kim, J.; Oh, M.; Lee, J.E.; Baek, A.; Lee, S.Y.; Chung, S.K.; Son, M.Y.; Chae, J.I.; et al. Quantitative Proteomic Analysis of Primitive Neural Stem Cells from LRRK2 G2019S-Associated Parkinson’s Disease Patient-Derived iPSCs. Life 2020, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Maharajan, N.; Ganesan, C.D.; Moon, C.; Jang, C.H.; Oh, W.K.; Cho, G.W. Licochalcone D Ameliorates Oxidative Stress-Induced Senescence via AMPK Activation. Int. J. Mol. Sci. 2021, 22, 7324. [Google Scholar] [CrossRef]
- Seo, J.H.; Choi, H.W.; Oh, H.N.; Lee, M.H.; Kim, E.; Yoon, G.; Cho, S.S.; Park, S.M.; Cho, Y.S.; Chae, J.I.; et al. Licochalcone D directly targets JAK2 to induced apoptosis in human oral squamous cell carcinoma. J. Cell. Physiol. 2019, 234, 1780–1793. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Niu, H.T.; Wang, P.L.; Lu, J.; Zhao, H.; Liu, S.H.; Zheng, Q.S.; Li, C.G. Cardioprotective Effect of Licochalcone D against Myocardial Ischemia/Reperfusion Injury in Langendorff-Perfused Rat Hearts. PLoS ONE 2015, 10, e0128375. [Google Scholar] [CrossRef]
- Ke, M.; Chong, C.M.; Su, H. Using induced pluripotent stem cells for modeling Parkinson’s disease. World J. Stem Cells 2019, 11, 634–649. [Google Scholar] [CrossRef] [PubMed]
- Son, M.Y.; Sim, H.; Son, Y.S.; Jung, K.B.; Lee, M.O.; Oh, J.H.; Chung, S.K.; Jung, C.R.; Kim, J. Distinctive genomic signature of neural and intestinal organoids from familial Parkinson’s disease patient-derived induced pluripotent stem cells. Neuropathol. Appl. Neurobiol. 2017, 43, 584–603. [Google Scholar] [CrossRef]
- Lee, M.; Ha, J.; Son, Y.S.; Ahn, H.; Jung, K.B.; Son, M.Y.; Kim, J. Efficient exogenous DNA-free reprogramming with suicide gene vectors. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Chung, S.K. Generation of gene-corrected iPSC line, KIOMi002-A, from Parkinson’s disease patient iPSC with LRRK2 G2019S mutation using BAC-based homologous recombination. Stem Cell Res. 2019, 41, 101649. [Google Scholar] [CrossRef]
- Chae, J.I.; Jeon, Y.J.; Shim, J.H. Downregulation of Sp1 is involved in honokiol-induced cell cycle arrest and apoptosis in human malignant pleural mesothelioma cells. Oncol. Rep. 2013, 29, 2318–2324. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.H.; Choi, H.S.; Pugliese, A.; Lee, S.Y.; Chae, J.I.; Choi, B.Y.; Bode, A.M.; Dong, Z. (-)-Epigallocatechin gallate regulates CD3-mediated T cell receptor signaling in leukemia through the inhibition of ZAP-70 kinase. J. Biol. Chem. 2008, 283, 28370–28379. [Google Scholar] [CrossRef]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentea, E.; Verbruggen, L.; Massie, A. The Proteasome Inhibition Model of Parkinson’s Disease. J. Park. Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Tarjanyi, O.; Haerer, J.; Vecsernyes, M.; Berta, G.; Stayer-Harci, A.; Balogh, B.; Farkas, K.; Boldizsar, F.; Szeberenyi, J.; Setalo, G., Jr. Prolonged treatment with the proteasome inhibitor MG-132 induces apoptosis in PC12 rat pheochromocytoma cells. Sci. Rep. 2022, 12, 5808. [Google Scholar] [CrossRef]
- Melikova, M.S.; Kondratov, K.A.; Kornilova, E.S. Two different stages of epidermal growth factor (EGF) receptor endocytosis are sensitive to free ubiquitin depletion produced by proteasome inhibitor MG132. Cell. Biol. Int. 2006, 30, 31–43. [Google Scholar] [CrossRef]
- Hernandez, J.M.; Floyd, D.H.; Weilbaecher, K.N.; Green, P.L.; Boris-Lawrie, K. Multiple facets of junD gene expression are atypical among AP-1 family members. Oncogene 2008, 27, 4757–4767. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Haraguchi, H.; Ishikawa, H.; Mizutani, K.; Tamura, Y.; Kinoshita, T. Antioxidative and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflata. Bioorg. Med. Chem. 1998, 6, 339–347. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Tavassoly, O.; Sato, T.; Tavassoly, I. Inhibition of Brain Epidermal Growth Factor Receptor Activation: A Novel Target in Neurodegenerative Diseases and Brain Injuries. Mol. Pharmacol. 2020, 98, 13–22. [Google Scholar] [CrossRef]
- Liu, B.; Neufeld, A.H. Activation of epidermal growth factor receptor causes astrocytes to form cribriform structures. Glia 2004, 46, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Neufeld, A.H. Activation of the epidermal growth factor receptor in optic nerve astrocytes leads to early and transient induction of cyclooxygenase-2. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2035–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Chen, H.; Johns, T.G.; Neufeld, A.H. Epidermal growth factor receptor activation: An upstream signal for transition of quiescent astrocytes into reactive astrocytes after neural injury. J. Neurosci. 2006, 26, 7532–7540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakatsuki, S.; Furuno, A.; Ohshima, M.; Araki, T. Oxidative stress-dependent phosphorylation activates ZNRF1 to induce neuronal/axonal degeneration. J. Cell. Biol. 2015, 211, 881–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakatsuki, S.; Araki, T. NADPH oxidases promote apoptosis by activating ZNRF1 ubiquitin ligase in neurons treated with an exogenously applied oxidant. Commun. Integr. Biol. 2016, 9, e1143575. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadimitrakopoulou, V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lin, A. Role of JNK activation in apoptosis: A double-edged sword. Cell. Res. 2005, 15, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xie, Y.; Yang, D.; Ren, D. Oxidative stress-induced apoptosis in granulosa cells involves JNK, p53 and Puma. Oncotarget 2017, 8, 25310–25322. [Google Scholar] [CrossRef]
- Suh, Y. Cell signaling in aging and apoptosis. Mech. Ageing Dev. 2002, 123, 881–890. [Google Scholar] [CrossRef] [PubMed]
- White, L.R.; Toft, M.; Kvam, S.N.; Farrer, M.J.; Aasly, J.O. MAPK-pathway activity, Lrrk2 G2019S, and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Weng, Y.H.; Chien, K.Y.; Lin, K.J.; Yeh, T.H.; Cheng, Y.P.; Lu, C.S.; Wang, H.L. (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 2012, 19, 1623–1633. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, N.; Budka, J.A.; Ferris, M.W.; Plotnik, J.P.; Hollenhorst, P.C. Extracellular signal-regulated kinase signaling regulates the opposing roles of JUN family transcription factors at ETS/AP-1 sites and in cell migration. Mol. Cell. Biol. 2015, 35, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, M.; Kolbus, A.; Piu, F.; Szabowski, A.; Mohle-Steinlein, U.; Tian, J.; Karin, M.; Angel, P.; Wagner, E.F. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999, 13, 607–619. [Google Scholar] [CrossRef]
- Weitzman, J.B.; Fiette, L.; Matsuo, K.; Yaniv, M. JunD protects cells from p53-dependent senescence and apoptosis. Mol. Cell. 2000, 6, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Mechta-Grigoriou, F.; Gerald, D.; Yaniv, M. The mammalian Jun proteins: Redundancy and specificity. Oncogene 2001, 20, 2378–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Reprogramming Method | Gender | Age | |
|---|---|---|---|---|
| WT-pNSC #1 | ND14317 cor | Episomal vector | Male | 53 |
| WT-pNSC #2 | AG02261 | Sendai virus | Male | 61 |
| LK2GS-pNSC #1 | ND38262 | Episomal vector | Male | 60 |
| LK2GS-pNSC #2 | ND14317 | Episomal vector | Male | 53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, M.; Nam, J.; Baek, A.; Seo, J.-H.; Chae, J.-I.; Lee, S.-Y.; Chung, S.-K.; Park, B.C.; Park, S.G.; Kim, J.; et al. Neuroprotective Effects of Licochalcone D in Oxidative-Stress-Induced Primitive Neural Stem Cells from Parkinson’s Disease Patient-Derived iPSCs. Biomedicines 2023, 11, 228. https://doi.org/10.3390/biomedicines11010228
Oh M, Nam J, Baek A, Seo J-H, Chae J-I, Lee S-Y, Chung S-K, Park BC, Park SG, Kim J, et al. Neuroprotective Effects of Licochalcone D in Oxidative-Stress-Induced Primitive Neural Stem Cells from Parkinson’s Disease Patient-Derived iPSCs. Biomedicines. 2023; 11(1):228. https://doi.org/10.3390/biomedicines11010228
Chicago/Turabian StyleOh, Minyoung, Juhyeon Nam, Areum Baek, Ji-Hye Seo, Jung-Il Chae, Seo-Young Lee, Sun-Ku Chung, Byoung Chul Park, Sung Goo Park, Janghwan Kim, and et al. 2023. "Neuroprotective Effects of Licochalcone D in Oxidative-Stress-Induced Primitive Neural Stem Cells from Parkinson’s Disease Patient-Derived iPSCs" Biomedicines 11, no. 1: 228. https://doi.org/10.3390/biomedicines11010228