
Type 2 Diabetes Related Mitochondrial Defects in Peripheral Mononucleated Blood Cells from Overweight Postmenopausal Women

 , , ,
, , ,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participant’s Enrollment, Inclusion Criteria and Health Data Collection
2.2. Experimental Design
2.3. Anthropometry and Body Composition
2.4. Handgrip Strength Test
2.5. Leg-Press Test
2.6. VO2max
2.7. Blood Sampling
2.8. Hematological Testing
2.9. Clinical Chemistry and Immunochemistry Test
2.10. PBMCs Purification
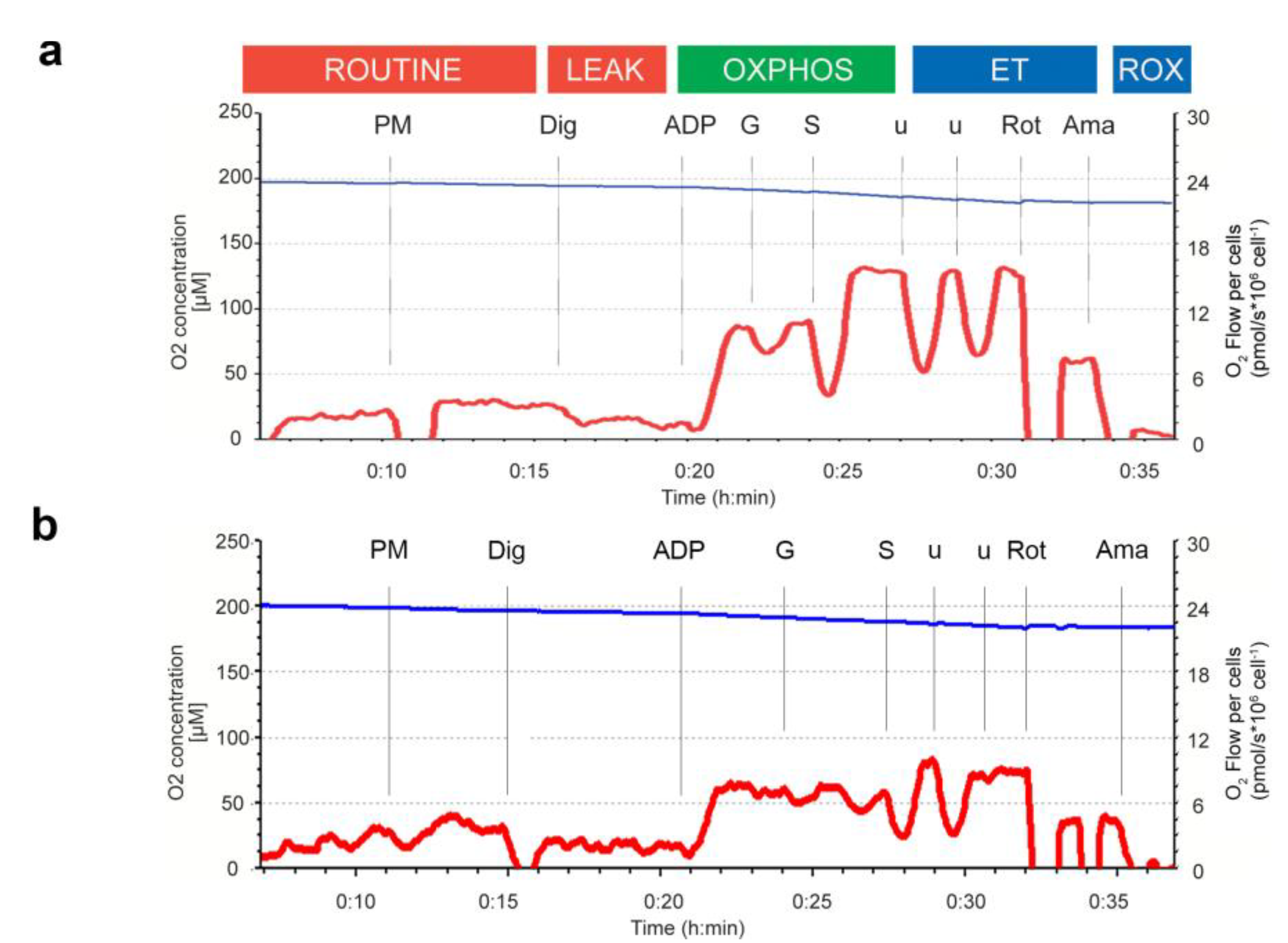
2.11. High Resolution Respirometry
2.12. Data Analysis and Statistics
3. Results
3.1. Anthropometric Characteristics and Body Composition
3.2. Blood Biomarkers
3.3. Cardio-Respiratory Function and Fitness
3.4. Muscle Strength
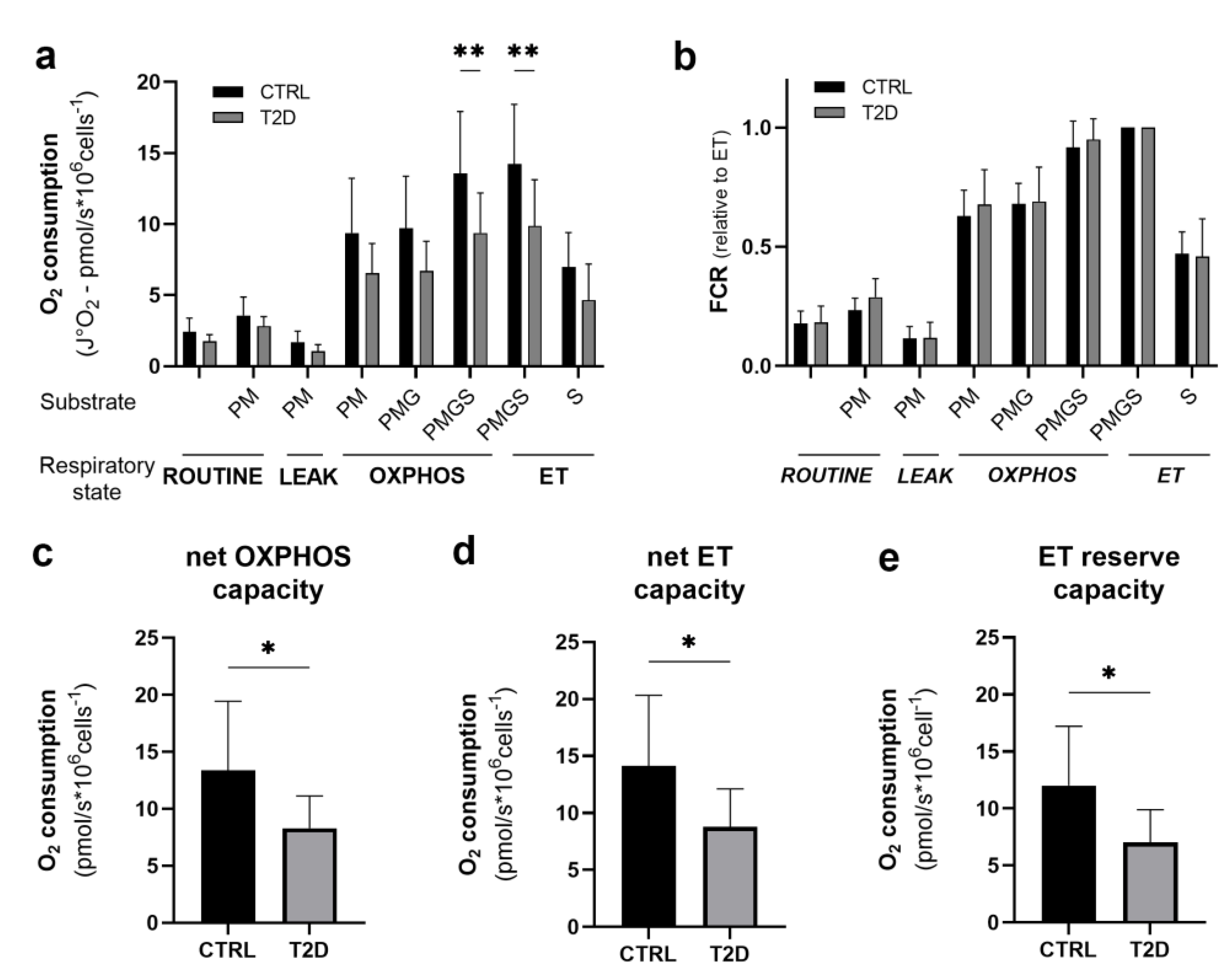
3.5. Mitochondrial Function
3.6. Glutathione Antioxidant Pool
3.7. Correlation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Unnikrishnan, R.; Pradeepa, R.; Joshi, S.R.; Mohan, V. Type 2 Diabetes: Demystifying the Global Epidemic. Diabetes 2017, 66, 1432–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krako Jakovljevic, N.; Pavlovic, K.; Jotic, A.; Lalic, K.; Stoiljkovic, M.; Lukic, L.; Milicic, T.; Macesic, M.; Stanarcic Gajovic, J.; Lalic, N.M. Targeting Mitochondria in Diabetes. Int. J. Mol. Sci. 2021, 22, 6642. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. S1), S81–S90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiner, G.G.; Emmert-Fees, K.M.F.; Becker, J.; Rathmann, W.; Thorand, B.; Peters, A.; Quante, A.S.; Schwettmann, L.; Laxy, M. Toward targeted prevention: Risk factors for prediabetes defined by impaired fasting glucose, impaired glucose tolerance and increased HbA1c in the population-based KORA study from Germany. Acta Diabetol. 2020, 57, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gregg, E.W.; Williamson, D.F.; Barker, L.E.; Thomas, W.; Bullard, K.M.; Imperatore, G.; Williams, D.E.; Albright, A.L. A1C level and future risk of diabetes: A systematic review. Diabetes Care 2010, 33, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.C.K. The diabesity epidemic in the light of evolution: Insights from the capacity-load model. Diabetologia 2019, 62, 1740–1750. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Parhofer, K.G. Diabetic dyslipidemia. Metabolism 2014, 63, 1469–1479. [Google Scholar] [CrossRef]
- Verges, B. Pathophysiology of diabetic dyslipidaemia: Where are we? Diabetologia 2015, 58, 886–899. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.L.; Koh, W.P.; Talaei, M.; Yuan, J.M.; Pan, A. Association between the ratio of triglyceride to high-density lipoprotein cholesterol and incident type 2 diabetes in Singapore Chinese men and women. J. Diabetes 2017, 9, 689–698. [Google Scholar] [CrossRef]
- Hu, S.; Gu, Y.; Lu, Z.; Zhang, Q.; Liu, L.; Meng, G.; Yao, Z.; Wu, H.; Bao, X.; Chi, V.T.Q.; et al. Relationship Between Grip Strength and Prediabetes in a Large-Scale Adult Population. Am. J. Prev. Med. 2019, 56, 844–851. [Google Scholar] [CrossRef]
- Qin, H.; Chen, Z.; Zhang, Y.; Wang, L.; Ouyang, P.; Cheng, L.; Zhang, Y. Triglyceride to high-density lipoprotein cholesterol ratio is associated with incident diabetes in men: A retrospective study of Chinese individuals. J. Diabetes Investig. 2020, 11, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Reusch, J.E.; Bridenstine, M.; Regensteiner, J.G. Type 2 diabetes mellitus and exercise impairment. Rev Endocr Metab Disord 2013, 14, 77–86. [Google Scholar] [CrossRef]
- Regensteiner, J.G.; Bauer, T.A.; Reusch, J.E.; Quaife, R.A.; Chen, M.Y.; Smith, S.C.; Miller, T.M.; Groves, B.M.; Wolfel, E.E. Cardiac dysfunction during exercise in uncomplicated type 2 diabetes. Med. Sci. Sports Exerc. 2009, 41, 977–984. [Google Scholar] [CrossRef] [Green Version]
- Gulsin, G.S.; Henson, J.; Brady, E.M.; Sargeant, J.A.; Wilmot, E.G.; Athithan, L.; Htike, Z.Z.; Marsh, A.M.; Biglands, J.D.; Kellman, P.; et al. Cardiovascular Determinants of Aerobic Exercise Capacity in Adults With Type 2 Diabetes. Diabetes Care 2020, 43, 2248–2256. [Google Scholar] [CrossRef]
- Laukkanen, J.A.; Kurl, S.; Salonen, R.; Rauramaa, R.; Salonen, J.T. The predictive value of cardiorespiratory fitness for cardiovascular events in men with various risk profiles: A prospective population-based cohort study. Eur. Heart J. 2004, 25, 1428–1437. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.J.; Echouffo-Tcheugui, J.B.; Golden, S.H.; Chen, H.; Jenny, N.S.; Carnethon, M.R.; Jacobs, D., Jr.; Burke, G.L.; Vaidya, D.; Ouyang, P.; et al. Physical activity, sedentary behaviors and the incidence of type 2 diabetes mellitus: The Multi-Ethnic Study of Atherosclerosis (MESA). BMJ Open Diabetes Res. Care 2016, 4, e000185. [Google Scholar] [CrossRef] [Green Version]
- Wahid, A.; Manek, N.; Nichols, M.; Kelly, P.; Foster, C.; Webster, P.; Kaur, A.; Friedemann Smith, C.; Wilkins, E.; Rayner, M.; et al. Quantifying the Association Between Physical Activity and Cardiovascular Disease and Diabetes: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2016, 5, e002495. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, A.; Stoelwinder, J.; Shortreed, S.; Wolfe, R.; Stevenson, C.; Walls, H.; de Courten, M.; Peeters, A. The duration of obesity and the risk of type 2 diabetes. Public Health Nutr. 2011, 14, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Logue, J.; Walker, J.J.; Colhoun, H.M.; Leese, G.P.; Lindsay, R.S.; McKnight, J.A.; Morris, A.D.; Pearson, D.W.; Petrie, J.R.; Philip, S.; et al. Do men develop type 2 diabetes at lower body mass indices than women? Diabetologia 2011, 54, 3003–3006. [Google Scholar] [CrossRef] [Green Version]
- Kannel, W.B. The Framingham Study: Historical insight on the impact of cardiovascular risk factors in men versus women. J. Gend Specif. Med. 2002, 5, 27–37. [Google Scholar]
- Kenchaiah, S.; Vasan, R.S. Heart Failure in Women--Insights from the Framingham Heart Study. Cardiovasc Drugs Ther. 2015, 29, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Wexler, D.J.; Grant, R.W.; Meigs, J.B.; Nathan, D.M.; Cagliero, E. Sex disparities in treatment of cardiac risk factors in patients with type 2 diabetes. Diabetes Care 2005, 28, 514–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.C.; Cristofaro, M.R.; Gentile, S.; Lucisano, G.; Manicardi, V.; Mulas, M.F.; Napoli, A.; Nicolucci, A.; Pellegrini, F.; Suraci, C.; et al. Sex disparities in the quality of diabetes care: Biological and cultural factors may play a different role for different outcomes: A cross-sectional observational study from the AMD Annals initiative. Diabetes Care 2013, 36, 3162–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedisinghe, L.; Perera, M. Diabetes and the menopause. Maturitas 2009, 63, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Franquesa, A.; Patti, M.E. Insulin Resistance and Mitochondrial Dysfunction. Adv. Exp. Med. Biol. 2017, 982, 465–520. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Invest. 2018, 128, 3671–3681. [Google Scholar] [CrossRef] [Green Version]
- Pacheu-Grau, D.; Rucktaschel, R.; Deckers, M. Mitochondrial dysfunction and its role in tissue-specific cellular stress. Cell Stress 2018, 2, 184–199. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Kosiol, M.; Mayr, M.; Schulz, R.; Rohrbach, S. Mitochondria and ageing: Role in heart, skeletal muscle and adipose tissue. J. Cachexia Sarcopenia Muscle 2017, 8, 349–369. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, M.; Sahlin, K.; Fernstrom, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Hojlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Porcu, S.; Lapolla, A.; Biasutto, L.; Zoratti, M.; Piarulli, F.; Eliana, G.; Basso, D.; Roverso, M.; Seraglia, R. A preliminary fastview of mitochondrial protein profile from healthy and type 2 diabetic subjects. Eur. J. Mass Spectrom 2014, 20, 307–315. [Google Scholar] [CrossRef]
- Sreekumar, R.; Halvatsiotis, P.; Schimke, J.C.; Nair, K.S. Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 2002, 51, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta. Mol. Basis. Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Karabatsiakis, A.; Bock, C.; Salinas-Manrique, J.; Kolassa, S.; Calzia, E.; Dietrich, D.E.; Kolassa, I.T. Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl. Psychiatry 2014, 4, e397. [Google Scholar] [CrossRef] [Green Version]
- Tyrrell, D.J.; Bharadwaj, M.S.; Jorgensen, M.J.; Register, T.C.; Shively, C.; Andrews, R.N.; Neth, B.; Keene, C.D.; Mintz, A.; Craft, S.; et al. Blood-Based Bioenergetic Profiling Reflects Differences in Brain Bioenergetics and Metabolism. Oxid. Med. Cell Longev 2017, 2017, 7317251. [Google Scholar] [CrossRef] [Green Version]
- Kramer, P.A.; Ravi, S.; Chacko, B.; Johnson, M.S.; Darley-Usmar, V.M. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biol. 2014, 2, 206–210. [Google Scholar] [CrossRef] [Green Version]
- Thon, J.N.; Italiano, J.E. Platelets: Production, morphology and ultrastructure. Handb. Exp. Pharmacol. 2012, 210, 3–22. [Google Scholar] [CrossRef]
- Hoppel, F.; Garcia-Souza, L.F.; Kantner-Rumplmair, W.; Burtscher, M.; Gnaiger, E.; Pesta, D.; Calabria, E. Human Platelet Mitochondrial Function Reflects Systemic Mitochondrial Alterations: A Protocol for Application in Field Studies. Cells 2021, 10, 2088. [Google Scholar] [CrossRef]
- Mossberg, K.; Olausson, J.; Fryk, E.; Jern, S.; Jansson, P.A.; Brogren, H. The role of the platelet pool of Plasminogen Activator Inhibitor-1 in well-controlled type 2 diabetes patients. PLoS ONE 2022, 17, e0267833. [Google Scholar] [CrossRef] [PubMed]
- Tschoepe, D.; Roesen, P.; Esser, J.; Schwippert, B.; Nieuwenhuis, H.K.; Kehrel, B.; Gries, F.A. Large platelets circulate in an activated state in diabetes mellitus. Semin. Thromb. Hemost 1991, 17, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Sumbalova, Z.D.S.; Hiller, E.; Chang, S.; Garcia-Souza, L.; Calabria, E.; Volani, C.; Krumschnabel, G.; Gneiger, E. O2k-Protocols: Isolation of peripheral blood mononuclear cells and platelets from human blood for HRR. MiPNet 2018, 21, 1–16. [Google Scholar]
- Braganza, A.; Annarapu, G.K.; Shiva, S. Blood-based bioenergetics: An emerging translational and clinical tool. Mol. Aspects Med. 2020, 71, 100835. [Google Scholar] [CrossRef] [PubMed]
- Widlansky, M.E.; Wang, J.; Shenouda, S.M.; Hagen, T.M.; Smith, A.R.; Kizhakekuttu, T.J.; Kluge, M.A.; Weihrauch, D.; Gutterman, D.D.; Vita, J.A. Altered mitochondrial membrane potential, mass, and morphology in the mononuclear cells of humans with type 2 diabetes. Transl. Res. 2010, 156, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.L.; Shirihai, O.S.; Holbrook, M.; Xu, G.; Kocherla, M.; Shah, A.; Fetterman, J.L.; Kluge, M.A.; Frame, A.A.; Hamburg, N.M.; et al. Relation of mitochondrial oxygen consumption in peripheral blood mononuclear cells to vascular function in type 2 diabetes mellitus. Vasc. Med. 2014, 19, 67–74. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes, A. (2) Classification and diagnosis of diabetes. Diabetes Care 2015, 38, S8–S16. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.C.; Cole, T.J.; Steam, A.S. Adjustment of fat-free mass and fat mass for height in children aged 8 y. Int. J. Obes. Relat. Metab. Disord 2002, 26, 947–952. [Google Scholar] [CrossRef] [Green Version]
- Gunther, C.M.; Burger, A.; Rickert, M.; Crispin, A.; Schulz, C.U. Grip strength in healthy caucasian adults: Reference values. J. Hand Surg. Am. 2008, 33, 558–565. [Google Scholar] [CrossRef]
- Jimenez-Pavon, D.; Ortega, F.B.; Valtuena, J.; Castro-Pinero, J.; Gomez-Martinez, S.; Zaccaria, M.; Gottrand, F.; Molnar, D.; Sjostrom, M.; Gonzalez-Gross, M.; et al. Muscular strength and markers of insulin resistance in European adolescents: The HELENA Study. Eur. J. Appl. Physiol. 2012, 112, 2455–2465. [Google Scholar] [CrossRef]
- Abdul-Hameed, U.; Rangra, P.; Shareef, M.Y.; Hussain, M.E. Reliability of 1-repetition maximum estimation for upper and lower body muscular strength measurement in untrained middle aged type 2 diabetic patients. Asian J. Sports Med. 2012, 3, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Brzycki, M. Strength Testing—Predicting a One-Rep Max from Reps-to-Fatigue. J. Phys. Educ. Recreat. Danc. 1993, 64, 3. [Google Scholar] [CrossRef]
- Bruseghini, P.; Calabria, E.; Tam, E.; Milanese, C.; Oliboni, E.; Pezzato, A.; Pogliaghi, S.; Salvagno, G.L.; Schena, F.; Mucelli, R.P.; et al. Effects of eight weeks of aerobic interval training and of isoinertial resistance training on risk factors of cardiometabolic diseases and exercise capacity in healthy elderly subjects. Oncotarget 2015, 6, 16998–17015. [Google Scholar] [CrossRef] [Green Version]
- Simundic, A.M.; Bolenius, K.; Cadamuro, J.; Church, S.; Cornes, M.P.; van Dongen-Lases, E.C.; Eker, P.; Erdeljanovic, T.; Grankvist, K.; Guimaraes, J.T.; et al. Joint EFLM-COLABIOCLI Recommendation for venous blood sampling. Clin. Chem. Lab. Med. 2018, 56, 2015–2038. [Google Scholar] [CrossRef] [Green Version]
- Venturelli, M.; Ruzzante, F.; Villa, F.; Rudi, D.; Tarperi, C.; Milanese, C.; Cavedon, V.; Fonte, C.; Picelli, A.; Smania, N.; et al. Response: Commentary: Neuromuscular and Muscle Metabolic Functions in MELAS Before and After Resistance Training: A Case Study. Front Physiol. 2020, 11, 337. [Google Scholar] [CrossRef]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Meszaros, A.T.; Gnaiger, E. High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. Methods Mol. Biol. 2018, 1782, 31–70. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal 2010, 12, 537–577. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Nisoli, E.; Carruba, M.O. Nitric oxide and mitochondrial biogenesis. J. Cell Sci. 2006, 119, 2855–2862. [Google Scholar] [CrossRef] [Green Version]
- Goodpaster, B.H. Mitochondrial deficiency is associated with insulin resistance. Diabetes 2013, 62, 1032–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloszy, J.O. "Deficiency" of mitochondria in muscle does not cause insulin resistance. Diabetes 2013, 62, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Befroy, D.E.; Petersen, K.F.; Dufour, S.; Mason, G.F.; de Graaf, R.A.; Rothman, D.L.; Shulman, G.I. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes 2007, 56, 1376–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Ritov, V.B.; Menshikova, E.V.; Azuma, K.; Wood, R.; Toledo, F.G.; Goodpaster, B.H.; Ruderman, N.B.; Kelley, D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E49–E58. [Google Scholar] [CrossRef] [Green Version]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.P.; Hesselink, M.K.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [Green Version]
- Chanseaume, E.; Barquissau, V.; Salles, J.; Aucouturier, J.; Patrac, V.; Giraudet, C.; Gryson, C.; Duche, P.; Boirie, Y.; Chardigny, J.M.; et al. Muscle mitochondrial oxidative phosphorylation activity, but not content, is altered with abdominal obesity in sedentary men: Synergism with changes in insulin sensitivity. J. Clin. Endocrinol. Metab. 2010, 95, 2948–2956. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, J.C.; Champagne, C.M.; de Jonge, L.; Xie, H.; Smith, S.R. Increased visceral fat and decreased energy expenditure during the menopausal transition. Int. J. Obes. 2008, 32, 949–958. [Google Scholar] [CrossRef] [Green Version]
- Neeland, I.J.; Turer, A.T.; Ayers, C.R.; Powell-Wiley, T.M.; Vega, G.L.; Farzaneh-Far, R.; Grundy, S.M.; Khera, A.; McGuire, D.K.; de Lemos, J.A. Dysfunctional adiposity and the risk of prediabetes and type 2 diabetes in obese adults. JAMA 2012, 308, 1150–1159. [Google Scholar] [CrossRef] [Green Version]
- Smith, U. Abdominal obesity: A marker of ectopic fat accumulation. J. Clin. Investig. 2015, 125, 1790–1792. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Bharadwaj, M.S.; Van Horn, C.G.; Marsh, A.P.; Nicklas, B.J.; Molina, A.J. Blood-cell bioenergetics are associated with physical function and inflammation in overweight/obese older adults. Exp. Gerontol. 2015, 70, 84–91. [Google Scholar] [CrossRef]
- Kunz, H.E.; Hart, C.R.; Gries, K.J.; Parvizi, M.; Laurenti, M.; Dalla Man, C.; Moore, N.; Zhang, X.; Ryan, Z.; Polley, E.C.; et al. Adipose tissue macrophage populations and inflammation are associated with systemic inflammation and insulin resistance in obesity. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E105–E121. [Google Scholar] [CrossRef]
- Park, S.W.; Goodpaster, B.H.; Strotmeyer, E.S.; Kuller, L.H.; Broudeau, R.; Kammerer, C.; de Rekeneire, N.; Harris, T.B.; Schwartz, A.V.; Tylavsky, F.A.; et al. Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: The health, aging, and body composition study. Diabetes Care 2007, 30, 1507–1512. [Google Scholar] [CrossRef] [Green Version]
- Mesinovic, J.; Zengin, A.; De Courten, B.; Ebeling, P.R.; Scott, D. Sarcopenia and type 2 diabetes mellitus: A bidirectional relationship. Diabetes Metab. Syndr. Obe.s 2019, 12, 1057–1072. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.; de Courten, B.; Ebeling, P.R. Sarcopenia: A potential cause and consequence of type 2 diabetes in Australia’s ageing population? Med. J. Aust. 2016, 205, 329–333. [Google Scholar] [CrossRef]
- Tiainen, K.; Raitanen, J.; Strandberg, T.; Koskinen, S.; Stenholm, S. Type 2 Diabetes as a Predictor of Muscle Strength Decline over 11 years among Men and Women Aged 55 Years and Older. Gerontology 2022, 68, 635–643. [Google Scholar] [CrossRef]
- Murakami, K.; Kondo, T.; Ohtsuka, Y.; Fujiwara, Y.; Shimada, M.; Kawakami, Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism 1989, 38, 753–758. [Google Scholar] [CrossRef]
- Venditti, P.; Di Stefano, L.; Di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef]
- Chen, G.; Chen, Z.; Hu, Y.; Huang, P. Inhibition of mitochondrial respiration and rapid depletion of mitochondrial glutathione by beta-phenethyl isothiocyanate: Mechanisms for anti-leukemia activity. Antioxid. Redox. Signal. 2011, 15, 2911–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, P.G.; Ferrington, D.A.; Kannan, R. Glutathione Metabolism and the Novel Role of Mitochondrial GSH in Retinal Degeneration. Antioxidants 2021, 10, 661. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Description | CTRL (N = 9) | T2D (N = 9) | ∆(T2D vs. CTRL) (%) | p Value (Unpaired Student’s t Test) |
|---|---|---|---|---|
| Age | 54.1 ± 3.8 | 60.9 ± 4.8 | 0.004 | |
| Ethnicity | Caucasian | Caucasian | ||
| Height (cm) | 162 ± 0.05 | 162 ± 0.07 | - | ns |
| BM (kg) | 67.8 ± 16.1 | 78.5 ± 16.8 | +16.2 | ns |
| BMI (Kg/m2) | 25.6 ± 5.2 | 30.0 ± 5.9 | +17.2 | ns |
| LEAN MASS (kg) | 39.7 ± 6.7 | 44.6 ± 7.9 | +13.7 | ns |
| LMI (Kg/m2) | 15.1 ± 1.9 | 17.1 ± 2.6 | +13 | ns |
| FAT MASS (Kg) | 24.9 ± 9.4 | 30.8 ± 9.7 | +23 | ns |
| FMI (Kg/m2) | 9.4 ± 3.2 | 11.8 ± 3.7 | +25 | ns |
| WB FAT(%) | 36.4 ± 5.7 | 38.3 ± 4.4 | +5 | ns |
| ANDROID/GYNOID | 0.45 ± 0.04 | 0.54 ± 0.07 | +20 | 0.005 |
| TRUNK FAT(%) | 33.8 ± 7.2 | 39.8 ± 4.5 | +17 | 0.05 |
| Description | CTRL (N = 9) | T2D (N = 9) | ∆(T2D vs. CTRL) (%) | p Value (Unpaired Student’s t Test) |
|---|---|---|---|---|
| Hb1Ac (mmol/mol) | 34.4 ± 1.7 | 45.2 ± 7.6 | +31.3 | 0.002 |
| Glucose (fasting) | 4.4 ± 0.7 | 6.2 ± 1.5 | +40 | 0.008 |
| CRP (mg/L) | 1 ± 0 | 2.2 ± 2.3 | +120 | ns # |
| CholTOT (mmol/L) | 5.1 ± 0.9 | 4.9 ± 1.2 | −4 | ns |
| CholHDL (mmol/L) | 1.9 ± 0.4 | 1.4 ± 0.3 | −35 | 0.005 |
| CholLDL (mmol/L) | 2.9 ± 0.4 | 3.3 ± 0.7 | +13 | ns |
| TG (mmol/L) | 0.92 ± 0.4 | 1.5 ± 0.7 | +63 | 0.03 |
| TG/CholHDL ratio | 0.54 ± 0.33 | 1.21 ± 0.75 | +125 | 0.03 |
| GSHt (%) | 100 ± 35.7 | 61.8 ± 31.8 | −38 | 0.04 |
| Description | CTRL (N = 9) | T2D (N = 9) | ∆(T2D vs. CTRL) (%) | p Value (Unpaired Student’s t Test) |
|---|---|---|---|---|
| VO2max (mL) | 1857.4 ± 202.5 | 1856.3 ± 256.0 | ns | |
| VO2max/Kg (mL/Kg) | 28.8 ± 7.1 | 24.5 ± 5.6 | −15 | ns |
| PPOmax (Watt) | 167.7 ± 18.5 | 149.1 ± 25.6 | −12 | ns |
| HRmax (bpm) | 167.2 ± 6.7 | 151.3 ± 15.0 | −10 | 0.01 |
| 1RM (Kg) | 168.1 ± 41.1 | 176.7 ± 48.7 | +5 | ns |
| 1RM/BM | 2.53 ± 0.61 | 2.27 ± 0.46 | −11 | ns |
| HG (Kg) | 30.9 ± 4.7 | 29.6 ± 5.8 | −4 | ns |
| HG/BM (Kg/Kg) | 0.47 ± 0.09 | 0.38 ± 0.04 | −23 | 0.02 |
| Factor | r | p Value | |
|---|---|---|---|
| Blood markers | |||
| Hb1Ac | Age | 0.557 | 0.016 |
| CRP | 0.618 | 0.006 | |
| CholHDL | −0.553 | 0.017 | |
| TG | 0.815 | <0.0001 | |
| GSTt | 0.544 | 0.020 | |
| HRmax | −0.651 | 0.003 | |
| Android/Gynoid | 0.570 | 0.013 | |
| Trunk PFAT | 0.503 | 0.034 | |
| LEAKPM | −0.476 | 0.046 | |
| OXPHOSPMGS | −0.502 | 0.034 | |
| ET | −0.529 | 0.024 | |
| ET reserve capacity | −0.501 | 0.034 | |
| Mitochondrial activity | |||
| ROUTINE | TG | −0.490 | 0.039 |
| ROUTINEPM | HDL | 0.487 | 0.041 |
| LEAKPM | Hb1Ac | −0.476 | 0.046 |
| CholHDL | 0.565 | 0.015 | |
| TG | −0.533 | 0.023 | |
| TRUNK_PFAT | −0.556 | 0.017 | |
| OXPHOSPM | VO2max/kg | 0.477 | 0.045 |
| HG/BM | 0.559 | 0.016 | |
| WB PFAT | −0.527 | 0.025 | |
| TRUNK PFAT | −0.606 | 0.008 | |
| OXPHOSPMG | HG/BM | 0.552 | 0.018 |
| WB PFAT | −0.529 | 0.024 | |
| TRUNK PFAT | −0.550 | 0.018 | |
| OXPHOSPMGS | Hb1Ac | −0.502 | 0.034 |
| CholHDL | 0.518 | 0.028 | |
| VO2max/kg | 0.464 | 0.052 | |
| HG/BM | 0.608 | 0.007 | |
| WB PFAT | −0.508 | 0.032 | |
| TRUNK PFAT | −0.602 | 0.008 | |
| ETPMGS | Hb1Ac | −0.529 | 0.024 |
| CholHDL | 0.491 | 0.039 | |
| HG/BM | 0.579 | 0.012 | |
| WB PFAT | −0.481 | 0.043 | |
| TRUNK PFAT | −0.570 | 0.014 | |
| ETS | Hb1Ac | −0.526 | 0.025 |
| netOXPHOS capacity | CholHDL | 0.564 | 0.015 |
| VO2max/kg | 0.504 | 0.033 | |
| HG/BM | 0.615 | 0.007 | |
| TRUNK PFAT | −0.627 | 0.005 | |
| netET capacity | Hb1Ac | −0.467 | 0.051 |
| CholHDL | 0.574 | 0.013 | |
| VO2max/kg | 0.470 | 0.049 | |
| HG/BM | 0.591 | 0.010 | |
| TRUNK PFAT | −0.615 | 0.007 | |
| ET reserve capacity | Hb1Ac | −0.501 | 0.034 |
| CholHDL | 0.597 | 0.009 | |
| TG | −0.460 | 0.055 | |
| VO2max/kg | 0.488 | 0.040 | |
| HG/BM | 0.613 | 0.007 | |
| TRUNK PFAT | −0.649 | 0.004 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabria, E.; Muollo, V.; Cavedon, V.; Capovin, T.; Saccenti, L.; Passarotti, F.; Ghiotto, L.; Milanese, C.; Gelati, M.; Rudi, D.; et al. Type 2 Diabetes Related Mitochondrial Defects in Peripheral Mononucleated Blood Cells from Overweight Postmenopausal Women. Biomedicines 2023, 11, 121. https://doi.org/10.3390/biomedicines11010121
Calabria E, Muollo V, Cavedon V, Capovin T, Saccenti L, Passarotti F, Ghiotto L, Milanese C, Gelati M, Rudi D, et al. Type 2 Diabetes Related Mitochondrial Defects in Peripheral Mononucleated Blood Cells from Overweight Postmenopausal Women. Biomedicines. 2023; 11(1):121. https://doi.org/10.3390/biomedicines11010121
Chicago/Turabian StyleCalabria, Elisa, Valentina Muollo, Valentina Cavedon, Teodora Capovin, Leonardo Saccenti, Francesco Passarotti, Laura Ghiotto, Chiara Milanese, Matteo Gelati, Doriana Rudi, and et al. 2023. "Type 2 Diabetes Related Mitochondrial Defects in Peripheral Mononucleated Blood Cells from Overweight Postmenopausal Women" Biomedicines 11, no. 1: 121. https://doi.org/10.3390/biomedicines11010121