
Alterations of the Composition and Neurometabolic Profile of Human Gut Microbiota in Major Depressive Disorder

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of the Cohorts of Patients with Depression and Healthy Volunteers
2.2. Preparation and Sequencing of Metagenomic Samples
2.3. Quality Control and Trimming
2.4. Taxonomic Analysis
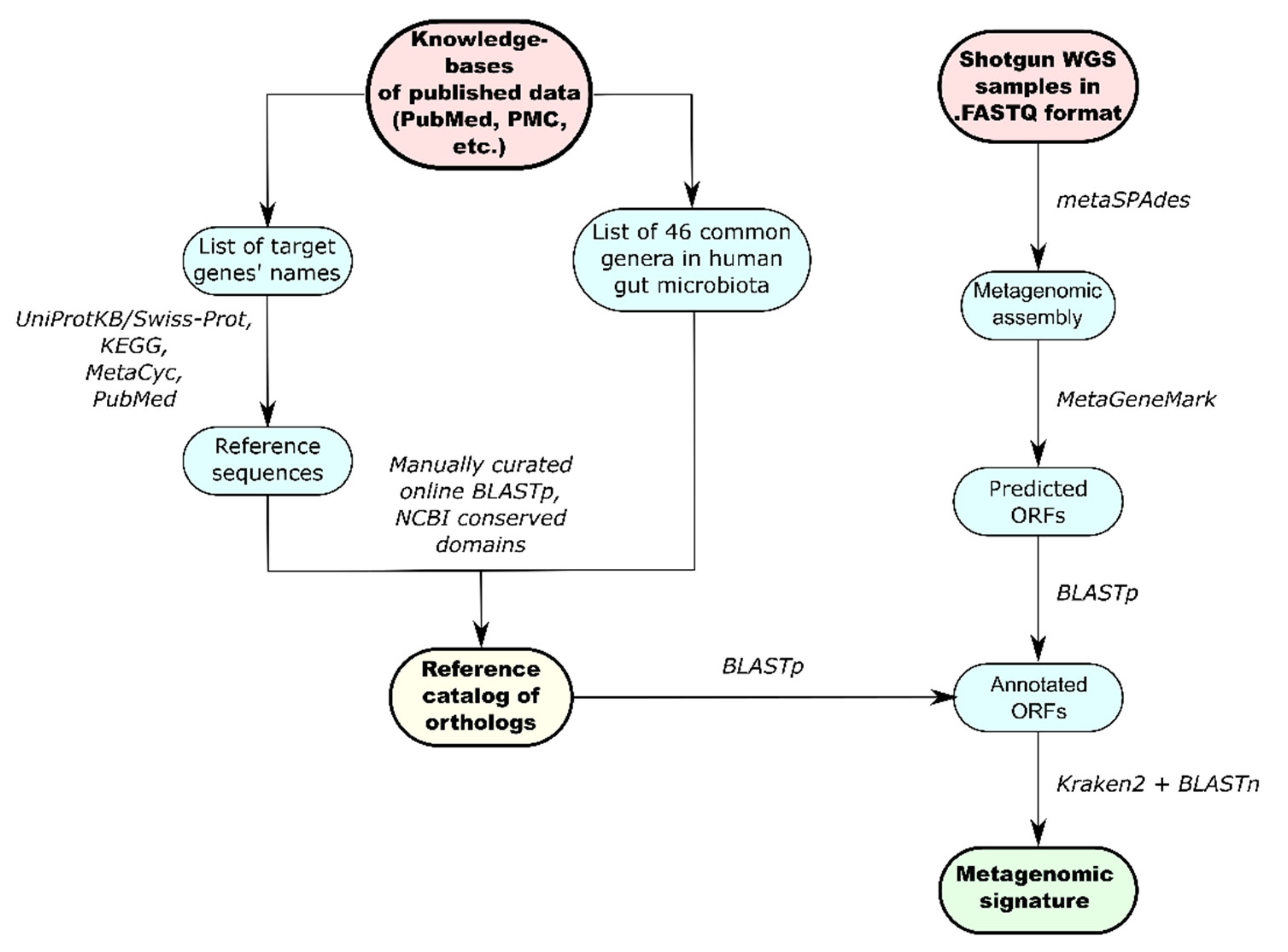
2.5. Development of the Reference Catalog
2.6. Functional Metagenomic Analysis
3. Results
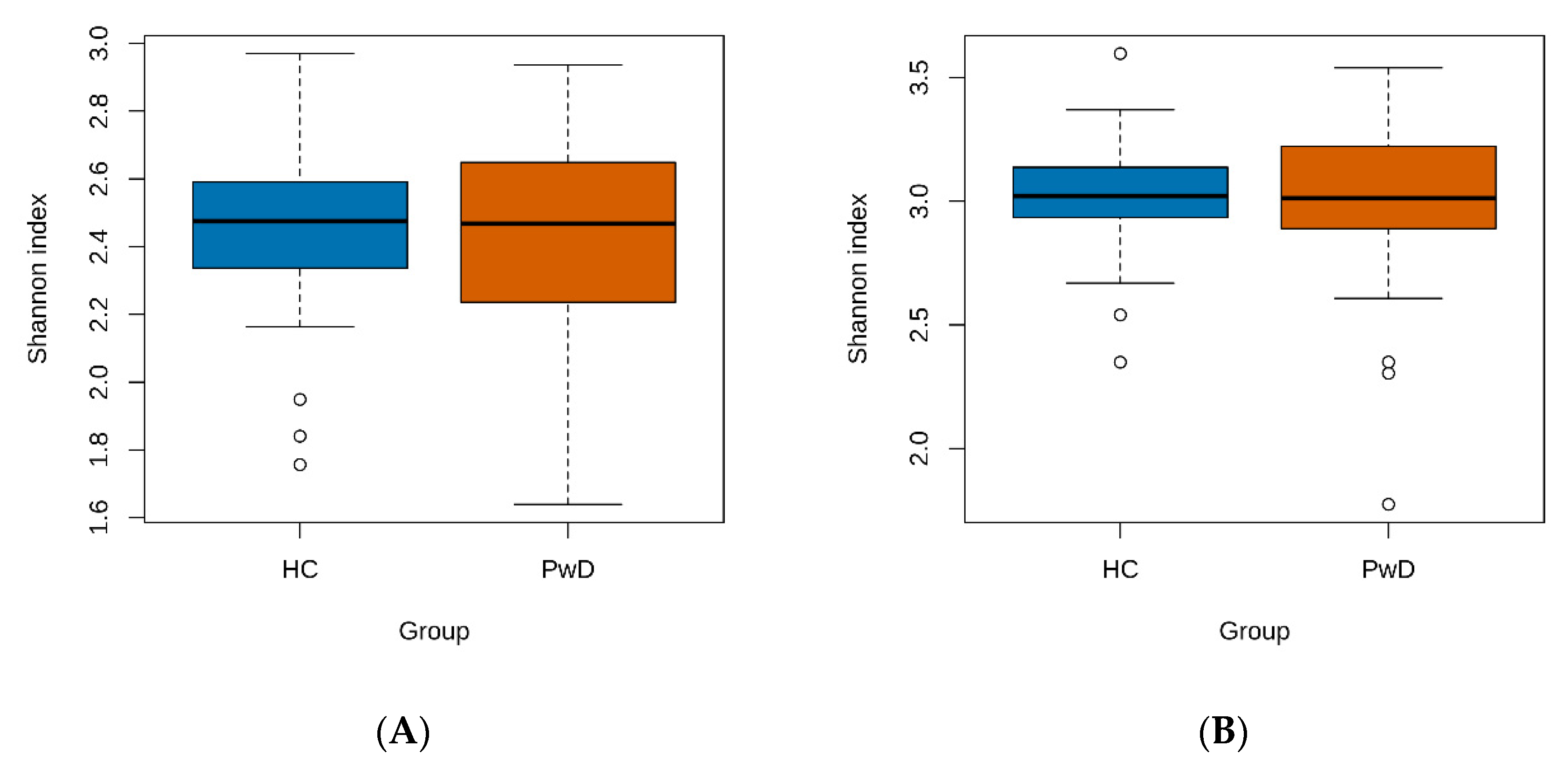
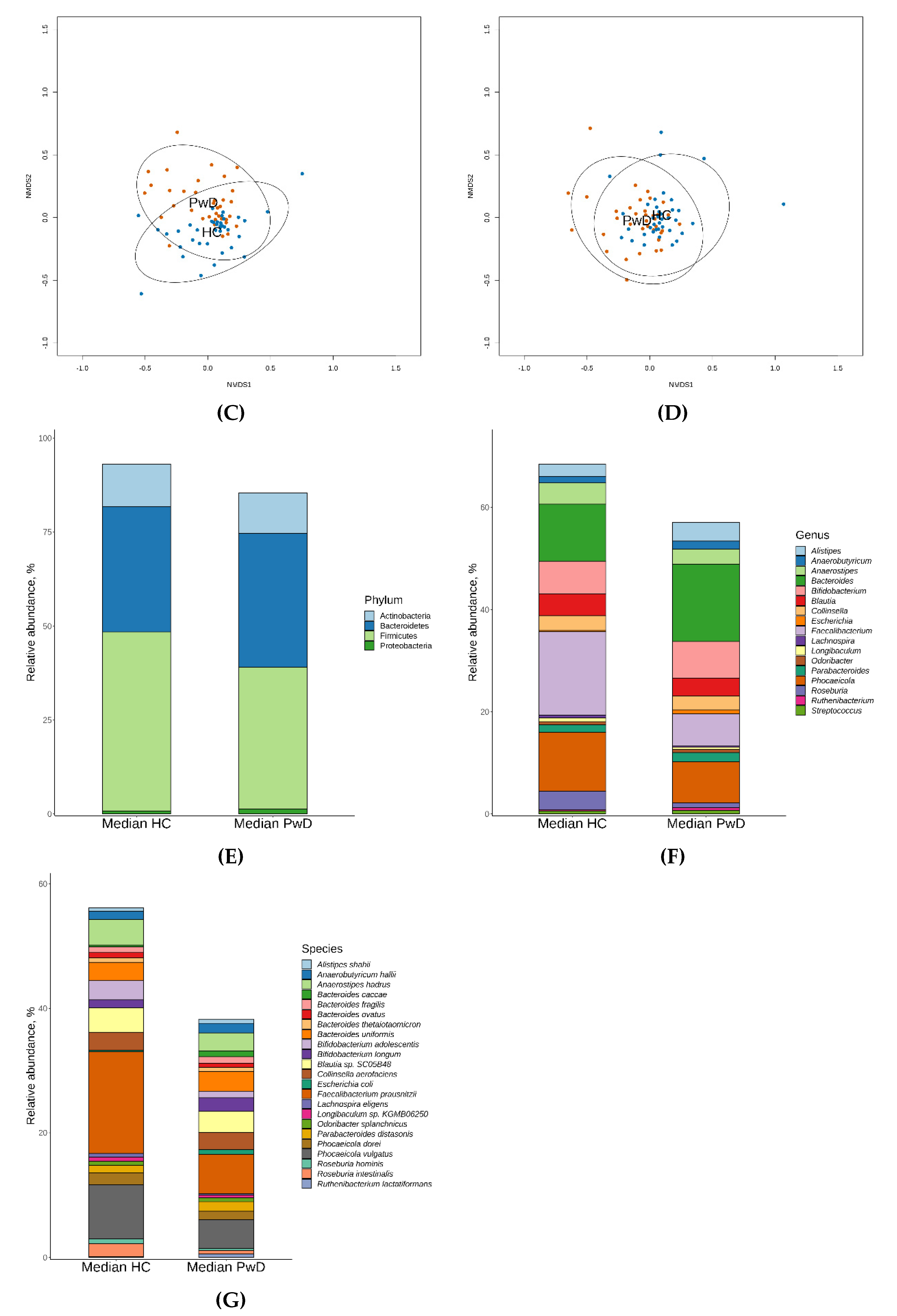
3.1. Taxonomic Analysis of GM of Patients with Depression
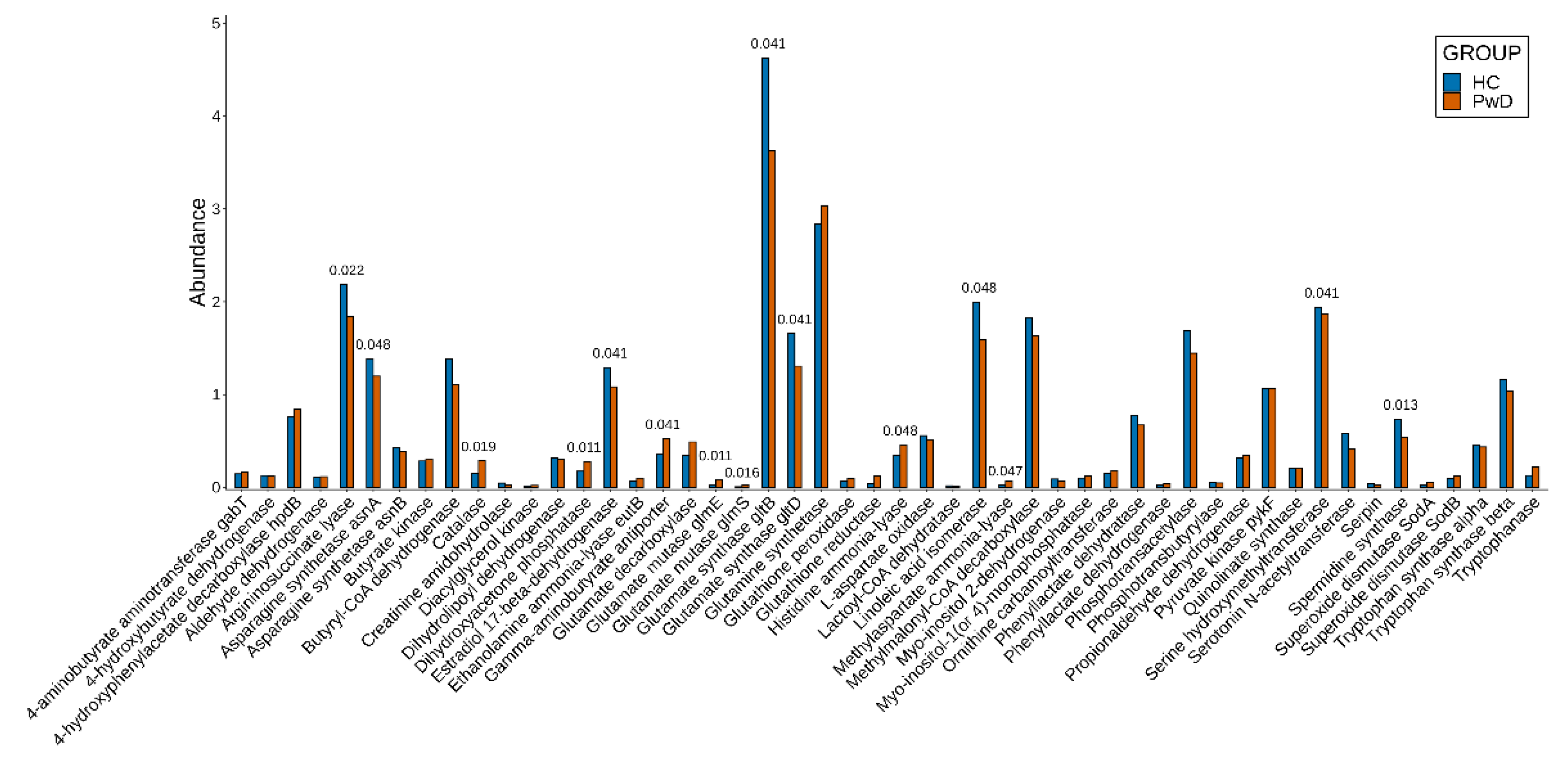
3.2. Genes
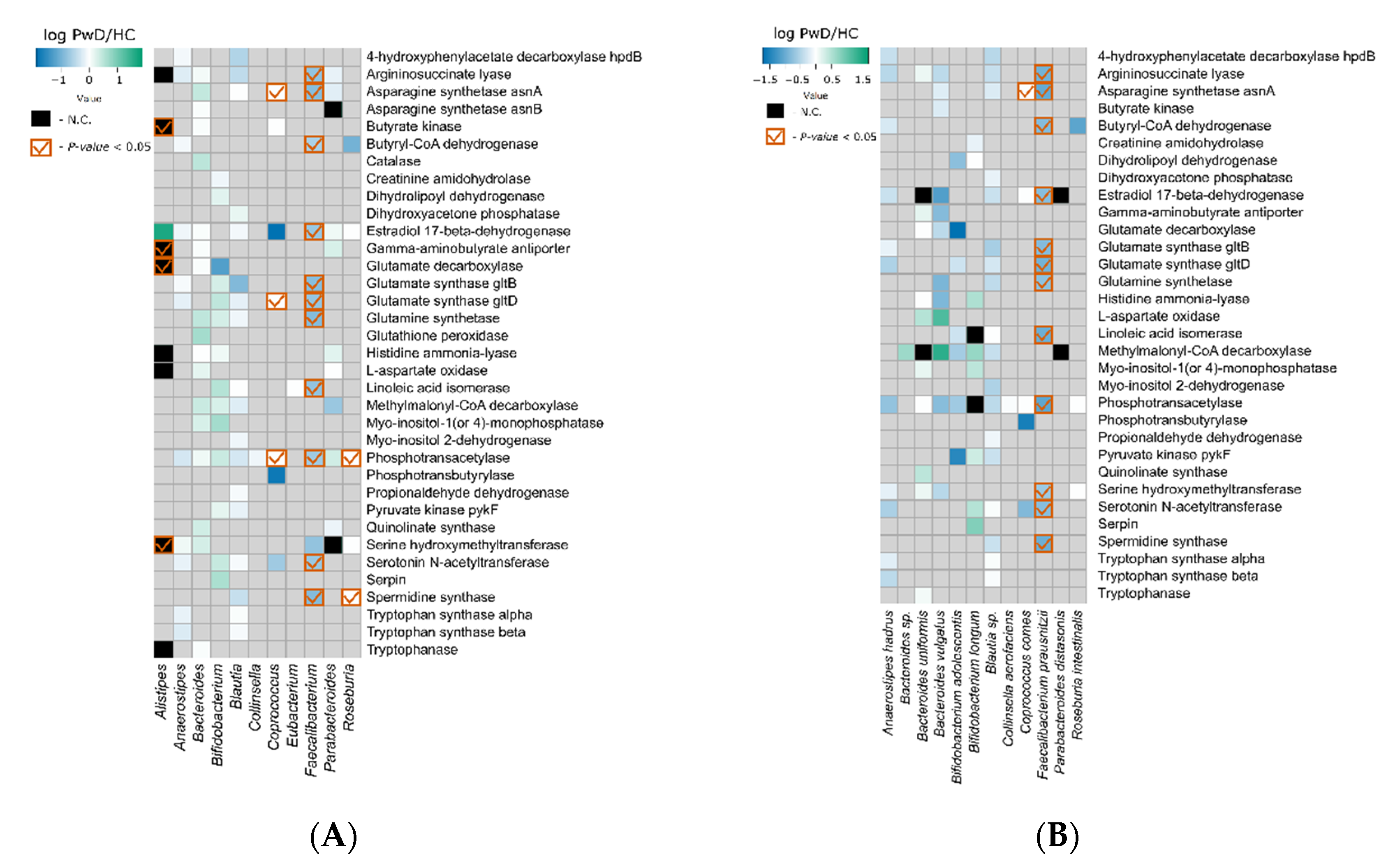
3.3. Metagenomic Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- The Lancet Global Health. Mental health matters. Lancet Glob. Health 2020, 8, e1352. [Google Scholar] [CrossRef]
- Thakur, V.; Jain, A. COVID 2019-suicides: A global psychological pandemic. Brain Behav. Immun. 2020, 88, 952–953. [Google Scholar] [CrossRef] [PubMed]
- Santomauro, D.F.; Mantilla Herrera, A.M.; Shadid, J.; Zheng, P.; Ashbaugh, C.; Pigott, D.M.; Abbafati, C.; Adolph, C.; Amlag, J.O.; Aravkin, A.Y.; et al. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet 2021, 398, 1700–1712. [Google Scholar] [CrossRef]
- Burke, H.M.; Davis, M.C.; Otte, C.; Mohr, D.C. Depression and cortisol responses to psychological stress: A meta-analysis. Psychoneuroendocrinology 2005, 30, 846–856. [Google Scholar] [CrossRef]
- Köhler, C.; Freitas, T.; Maes, M.D.; De Andrade, N.; Liu, C.; Fernandes, B.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.S.; Allen, J.J. Vagal tone as an indicator of treatment response in major depression. Psychophysiology 2002, 39, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Hare, B.D.; Duman, R.S. Prefrontal cortex GABAergic deficits and circuit dysfunction in the pathophysiology and treatment of chronic stress and depression. Curr. Opin. Beha. Sci. 2017, 14, 1–8. [Google Scholar] [CrossRef]
- Kim, I.-B.; Lee, J.-H.; Park, S.-C. The relationship between stress, inflammation, and depression. Biomedicines 2022, 10, 1929. [Google Scholar] [CrossRef]
- Moncrieff, J.; Cooper, R.E.; Stockmann, T.; Amendola, S.; Hengartner, M.P.; Horowitz, M.A. The serotonin theory of depression: A systematic umbrella review of the evidence. Mol. Psychiatry 2022. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Salbaum, J.M.; Berthoud, H.-R. Harnessing gut microbes for mental health: Getting from here to there. Biol. Psychiatry 2018, 83, 214–223. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial impairment: A common motif in neuropsychiatric presentation? The link to the tryptophan-kynurenine metabolic system. Cells 2022, 11, 2607. [Google Scholar] [CrossRef]
- Gheshlagh, R.G.; Parizad, N.; Sayehmiri, K. The relationship between depression and metabolic syndrome: Systematic review and meta-analysis study. Iran. Red Crescent Med. J. 2016, 18, e26523. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, Ó.; García-Montero, C.; Alvarez-Mon, M.A.; Lahera, G.; Monserrat, J.; Llavero-Valero, M.; Mora, F.; Rodríguez-Jiménez, R.; Fernandez-Rojo, S.; et al. Nutrition, epigenetics, and major depressive disorder: Understanding the connection. Front. Nutr. 2022, 9, 867150. [Google Scholar] [CrossRef]
- Brasso, C.; Bellino, S.; Blua, C.; Bozzatello, P.; Rocca, P. The impact of SARS-CoV-2 infection on youth mental health: A narrative review. Biomedicines 2022, 10, 772. [Google Scholar] [CrossRef]
- Slavich, G.M.; Sacher, J. Stress, sex hormones, inflammation, and major depressive disorder: Extending Social Signal Transduction Theory of Depression to account for sex differences in mood disorders. Psychopharmacology 2019, 236, 3063–3079. [Google Scholar] [CrossRef]
- Kucukkarapinar, M.; Yay-Pence, A.; Yildiz, Y.; Buyukkoruk, M.; Yaz-Aydin, G.; Deveci-Bulut, T.S.; Gulbahar, O.; Senol, E.; Candansaya, S. Psychological outcomes of COVID 19 survivors at sixth months after diagnose: The role of kynurenine pathway metabolites in depression, anxiety, and stress. J. Neural Transm. 2022, 129, 1077–1089. [Google Scholar] [CrossRef]
- Holtzheimer III, P.E.; Nemeroff, C.B. Future prospects in depression research. Dialogues Clin. Neurosci. 2006, 8, 175–189. [Google Scholar] [CrossRef]
- Friedrich, M.J. Depression is the leading cause of disability around the world. JAMA 2017, 317, 1517. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Meyyappan, A.C.; Forth, E.; Wallace, C.J.K.; Milev, R. Effect of fecal microbiota transplant on symptoms of psychiatric disorders: A systematic review. BMC Psychiatry 2020, 20, 299. [Google Scholar] [CrossRef]
- Caspani, G.; Swann, J. Small talk: Microbial metabolites involved in the signaling from microbiota to brain. Curr. Opin. Pharmacol. 2019, 48, 99–106. [Google Scholar] [CrossRef]
- Margolis, K.G.; Cryan, J.F.; Mayer, E.A. The microbiota-gut-brain axis: From motility to mood. Gastroenterology 2021, 160, 1486–1501. [Google Scholar] [CrossRef]
- Generoso, J.S.; Giridharan, V.V.; Lee, J.; Macedo, D.; Barichello, T. The role of the microbiota-gut-brain axis in neuropsychiatric disorders. Rev. Bras. Psiquiatr. 2021, 43, 293–305. [Google Scholar] [CrossRef]
- Averina, O.V.; Zorkina, Y.A.; Yunes, R.A.; Kovtun, A.S.; Ushakova, V.M.; Morozova, A.Y.; Kostyuk, G.P.; Danilenko, V.N.; Chekhonin, V.P. Bacterial metabolites of human gut microbiota correlating with depression. Int. J. Mol. Sci. 2020, 21, 9234. [Google Scholar] [CrossRef]
- Grenham, S.; Clarke, G.; Cryan, J.F.; Dinan, T.G. Brain–gut–microbe communication in health and disease. Front. Physiol. 2011, 2, 94. [Google Scholar] [CrossRef]
- Rogers, G.B.; Keating, D.J.; Young, R.L.; Wong, M.-L.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef]
- Harsch, I.A.; Konturek, P.C. The role of gut microbiota in obesity and type 2 and type 1 diabetes mellitus: New insights into “old” diseases. Med. Sci. 2018, 6, 32. [Google Scholar] [CrossRef]
- Rodriguez, J.; Hiel, S.; Delzenne, N.M. Metformin: Old friend, new ways of action-implication of the gut microbiome? Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 294–301. [Google Scholar] [CrossRef]
- Du, Y.; Gao, X.-R.; Peng, L.; Ge, J.-F. Crosstalk between the microbiota-gut-brain axis and depression. Heliyon 2020, 6, e04097. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Caprio, G.G.; Ormando, V.M.; Loguercio, C. Gut microbiota and the liver. Minerva Gastroenterol. Dietol. 2017, 63, 385–398. [Google Scholar] [CrossRef]
- Depken, D.E. Gut microbiota; impacts on human health and wellbeing. In Good Health and Well-Being; Filho, W.L., Wall, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–9. ISBN 978-3-319-95680-0. [Google Scholar]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Rajilic-Stojanovic, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Bistoletti, M.; Bosi, A.; Banfi, D.; Cristina, G.; Baj, A. The microbiota-gut-brain axis: Focus on the fundamental communication pathways. In Progress in Molecular Biology and Translational Science; Kasselman, L.J., Elsevier, B.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 176, pp. 43–110. ISBN 978-0-12-820797-0. [Google Scholar]
- Knudsen, J.K.; Bundgaard-Nielsen, C.; Hjerrild, S.; Nielsen, R.E.; Leutscher, P.; Sørensen, S. Gut microbiota variations in patients diagnosed with major depressive disorder—A systematic review. Brain Behav. 2021, 11, e02177. [Google Scholar] [CrossRef]
- The International Human Microbiome Standards (IHMS) Project. Available online: http://www.microbiome-standards.org/ (accessed on 1 August 2020).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 13 September 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Lu, J.; Breitwieser, F.B.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, G.F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. Available online: https://CRAN.R-project.org/package=vegan (accessed on 7 January 2022).
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Riley, M.; Paley, S.M.; Pellegrini-Toole, A. The MetaCyc Database. Nucleic Acids Res. 2002, 30, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: How to use the entry view. Methods Mol. Biol. 2016, 1374, 23–54. [Google Scholar] [CrossRef]
- Madden, T.L.; Tatusov, R.L.; Zhang, J. Applications of network BLAST server. Methods Enzymol. 1996, 266, 131–141. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Averina, O.V.; Kovtun, A.S.; Polyakova, S.I.; Savilova, A.M.; Rebrikov, D.V.; Danilenko, V.N. The bacterial neurometabolic signature of the gut microbiota of young children with autism spectrum disorders. J. Med. Microbiol. 2020, 69, 558–571. [Google Scholar] [CrossRef]
- Kovtun, A.S.; Averina, O.V.; Zakharevich, N.V.; Kasianov, A.S.; Danilenko, V.N. In silico identification of metagenomic signature describing neurometabolic potential of normal human gut microbiota. Russ. J. Gen. 2018, 54, 1101–1110. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef]
- Chahwan, B.; Kwan, S.; Isik, A.; van Hemert, S.; Burke, C.; Roberts, L. Gut feelings: A randomised, triple-blind, placebo-controlled trial of probiotics for depressive symptoms. J. Affect. Disord. 2019, 253, 317–326. [Google Scholar] [CrossRef]
- Vinberg, M.; Ottesen, N.M.; Meluken, I.; Sørensen, N.; Pedersen, O.; Kessing, L.V.; Miskowiak, K.W. Remitted affective disorders and high familial risk of affective disorders associate with aberrant intestinal microbiota. Acta Psychiat. Scand. 2019, 139, 174–184. [Google Scholar] [CrossRef]
- Mason, B.L.; Li, Q.; Minhajuddin, A.; Czysz, A.H.; Coughlin, L.A.; Hussain, S.K.; Koh, A.Y.; Trivedi, M.H. Reduced anti-inflammatory gut microbiota are associated with depression and anhedonia. J. Affect. Disord. 2020, 266, 394–401. [Google Scholar] [CrossRef]
- Lai, W.; Deng, W.-F.; Xu, S.-X.; Zhao, J.; Xu, D.; Liu, Y.-H.; Guo, Y.-Y.; Wang, M.-B.; He, F.-S.; Ye, S.-W.; et al. Shotgun metagenomics reveals both taxonomic and tryptophan pathway differences of gut microbiota in major depressive disorder patients. Psychol. Med. 2021, 51, 90–101. [Google Scholar] [CrossRef]
- Sanada, K.; Nakajima, S.; Kurokawa, S.; Barcelo-Soler, A.; Ikuse, D.; Hirata, A.; Yoshizawa, A.; Tomizawa, Y.; Salas-Valero, M.; Noda, Y.; et al. Gut microbiota and major depressive disorder: A systematic review and meta-analysis. J. Affect. Disord. 2020, 266, 1–13. [Google Scholar] [CrossRef]
- Huang, Y.; Shi, X.; Li, Z.; Shen, Y.; Shi, X.; Wang, L.; Li, G.; Yuan, Y.; Wang, J.; Zhang, Y.; et al. Possible association of Firmicutes in the gut microbiota of patients with major depressive disorder. Neuropsychiatr. Dis. Treat. 2018, 14, 3329–3337. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.-T.; Lai, J.-B.; Du, Y.-L.; Xu, Y.; Ruan, L.-M.; Hu, S.-H. Current understanding of gut microbiota in mood disorders: An update of human studies. Front. Genet. 2019, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Barandouzi, Z.A.; Starkweather, A.R.; Henderson, W.A.; Gyamfi, A.; Cong, X.S. Altered composition of gut microbiota in depression: A systematic review. Front. Psychiatry 2020, 11, 541. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermudez-Humaran, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.E.; Bäumler, A.J. Dysbiosis in the inflamed intestine: Chance favors the prepared microbe. Gut Microbes 2014, 5, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200. [Google Scholar] [CrossRef]
- Wong, M.-L.; Inserra, A.; Lewis, M.D.; Mastronardi, C.A.; Leong, L.; Choo, J.; Kentish, S.; Xie, P.; Morrison, M.; Wesselingh, S.L.; et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol. Psychiatry 2016, 21, 797–805. [Google Scholar] [CrossRef]
- Löwe, B.; Andresen, V.; Fraedrich, K.; Gappmayer, K.; Wegscheider, K.; Treszl, A.; Riegel, B.; Rose, M.; Lohse, A.W.; Broicher, W. Psychological outcome, fatigue, and quality of life after infection with shiga toxin-producing Escherichia coli O104. Clin. Gastroenterol. Hepatol. 2014, 12, 1848–1855. [Google Scholar] [CrossRef]
- Borgo, F.; Riva, A.; Benetti, A.; Casiraghi, M.C.; Bertelli, S.; Garbossa, S.; Anselmetti, S.; Scarone, S.; Pontiroli, A.E.; Morace, G.; et al. Microbiota in anorexia nervosa: The triangle between bacterial species, metabolites and psychological tests. PLoS ONE 2017, 12, e0179739. [Google Scholar] [CrossRef]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Dinan, T.G.; Stanton, C.; Cryan, J.F. Psychobiotics: A novel class of psychotropic. Biol. Psychiatry 2013, 74, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Lehto, S.M.; Harty, S.; Dinan, T.G.; Cryan, J.F.; Burnet, P.W.J. Psychobiotics and the manipulation of bacteria-gut-brain signals. Trends Neurosci. 2016, 39, 763–781. [Google Scholar] [CrossRef] [PubMed]
- Yunes, R.A.; Poluektova, E.U.; Dyachkova, M.S.; Klimina, K.M.; Kovtun, A.S.; Averina, O.V.; Orlova, V.S.; Danilenko, V.N. GABA production and structure of gadB/gadC genes in Lactobacillus and Bifidobacterium strains from human microbiota. Anaerobe 2016, 42, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Yunes, R.A.; Poluektova, E.U.; Vasileva, E.V.; Odorskaya, M.V.; Marsova, M.V.; Kovalev, G.I.; Danilenko, V.N. A multi-strain potential probiotic formulation of GABA-producing Lactobacillus plantarum 90sk and Bifidobacterium adolescentis 150 with antidepressant effects. Probiotics Antimicrob. Proteins 2020, 12, 973–979. [Google Scholar] [CrossRef]
- Nezametdinova, V.Z.; Yunes, R.A.; Dukhinova, M.S.; Alekseeva, M.G.; Danilenko, V.N. Therole of the PFNA operon of Bifidobacteria in the recognition of host’s immune signals: Prospects for the use of the FN3 protein in the treatment of COVID-19. Int. J. Mol. Sci. 2021, 22, 9219. [Google Scholar] [CrossRef]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X.; et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 2016, 21, 786–796. [Google Scholar] [CrossRef]
- Naseribafrouei, A.; Hestad, K.; Avershina, E.; Sekelja, M.; Linløkken, A.; Wilson, R.; Rudi, K. Correlation between the human fecal microbiota and depression. Neurogastroenterol. Motil. 2014, 26, 1155–1162. [Google Scholar] [CrossRef]
- Lukiw, W.J. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer’s disease. Front. Microbiol. 2016, 7, 1544. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Hill, J.M.; Zhao, Y.; Bond, T.; Taylor, C.M.; Percy, M.E.; Li, W.; Lukiw, W.J. Aluminum-induced generation of lipopolysaccharide (LPS) from the human gastrointestinal (GI)-tract microbiome-resident Bacteroides fragilis. J. Inorg. Biochem. 2020, 203, 110886. [Google Scholar] [CrossRef]
- Ye, X.; Wang, D.; Zhu, H.; Wang, D.; Li, J.; Tang, Y.; Wu, J. Gut microbiota changes in patients with major depressive disorder treated with vortioxetine. Front. Psychiatry 2021, 12, 641491. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Yu, L.; Tong, Y.; Shen, G.; Fu, A.; Lai, Y.; Zhou, X.; Yuan, Y.; Wang, Y.; Pan, Y.; Yu, Z.; et al. Immunodepletion with hypoxemia: A potential high risk subtype of coronavirus disease 2019. medRxiv 2020. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology ofbutyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Wang, W.; Guo, R.; Liu, H. Faecalibacterium prausnitzii (ATCC 27766) has preventive and therapeutic effects on chronic unpredictable mild stress-induced depression-like and anxiety-like behavior in rats. Psychoneuroendocrinology 2019, 104, 132–142. [Google Scholar] [CrossRef]
- Duncan, S.H.; Holtrop, G.; Lobley, G.E.; Calder, A.G.; Stewart, C.S.; Flint, H.J. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 2004, 91, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; He, M.; Sun, Q.; Wang, Y.; Zhang, J.; Fang, Y.; Liu, S.; Duan, L. Roseburia hominis increases intestinal melatonin level by activating p-CREB-AANAT pathway. Nutrients 2022, 14, 117. [Google Scholar] [CrossRef]
- Lu, Y.-R.; Fu, X.-Y.; Shi, L.-G.; Jiang, Y.; Wu, J.-L.; Weng, X.-J.; Wang, Z.-P.; Wu, X.-Y.; Lin, Z.; Liu, W.-B. Decreased plasma neuroactive amino acids and increased nitric oxide levels in melancholic major depressive disorder. BMC Psychiatry 2014, 14, 123. [Google Scholar] [CrossRef]
- Hess, S.; Baker, G.; Gyenes, G.; Tsuyuki, R.; Newman, S.; Le Melledo, J.-M. Decreased serum L-arginine and L-citrulline levels in major depression. Psychopharmacology 2017, 234, 3241–3247. [Google Scholar] [CrossRef]
- Stilling, R.M.; Dinan, T.G.; Cryan, J.F. Microbial genes, brain & behaviour—Epigenetic regulation of the gut–brain axis. Genes Brain Behav. 2014, 13, 69–86. [Google Scholar] [CrossRef]
- Strandwitz, P.; Kim, K.H.; Terekhova, D.; Liu, J.K.; Sharma, A.; Levering, J.; McDonald, D.; Dietrich, D.; Ramadhar, T.R.; Lekbua, A. GABA-modulating bacteria of the human gut microbiota. Nat. Microbiol. 2019, 4, 396–403. [Google Scholar] [CrossRef]
- Capuco, A.; Urits, I.; Hasoon, J.; Chun, R.; Gerald, B.; Wang, J.K.; Ngo, A.L.; Simopoulos, T.; Kaye, A.D.; Colontonio, M.M.; et al. Gut microbiome dysbiosis and depression: A comprehensive review. Curr. Pain Headache Rep. 2020, 24, 36. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.L.; Bernstein, C.N. Brain-gut interactions in inflammatory bowel disease. Gastroenterology 2013, 144, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Szentirmai, É.; Millican, N.S.; Massie, A.R.; Kapás, L. Butyrate, a metabolite of intestinal bacteria, enhances sleep. Sci. Rep. 2019, 9, 7035. [Google Scholar] [CrossRef]
- Tsuno, N.; Besset, A.; Ritchie, K. Sleep and depression. J. Clin. Psychiatry 2005, 66, 1254–1269. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Sankhe, R.; Mudgal, J.; Arora, D.; Nampoothiri, M. Spermidine, an autophagy inducer, as a therapeutic strategy in neurological disorders. Neuropeptides 2020, 83, 102083. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, S.; Hofer, S.J.; Zimmermann, A.; Pechlaner, R.; Dammbrueck, C.; Pendl, T.; Marcello, G.M.; Pogatschnigg, V.; Bergmann, M.; Müller, M.; et al. Dietary spermidine improves cognitive function. Cell Rep. 2021, 35, 108985. [Google Scholar] [CrossRef]
- Pan, Z.; Park, C.; Brietzke, E.; Zuckerman, H.; Rong, C.; Mansur, R.B.; Fus, D.; Subramaniapillai, M.; Lee, Y.; McIntyre, R.S. Cognitive impairment in major depressive disorder. CNS Spectr. 2019, 24, 22–29. [Google Scholar] [CrossRef]
- Fiori, L.M.; Bureau, A.; Labbe, A.; Croteau, J.; Noël, S.; Mérette, C.; Turecki, G. Global gene expression profiling of the polyamine system in suicide completers. Int. J. Neuropsychopharmacol. 2011, 14, 595–605. [Google Scholar] [CrossRef]
- Gross, J.A.; Turecki, G. Suicide and the Polyamine System. CNS Neurol. Disord. Drug Targets 2013, 12, 980–988. [Google Scholar] [CrossRef] [Green Version]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Averina, O.V.; Poluektova, E.U.; Marsova, M.V.; Danilenko, V.N. Biomarkers and utility of the antioxidant potential of probiotic lactobacilli and bifidobacteria as representatives of the human gut microbiota. Biomedicines 2021, 9, 1340. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Park, Y.H.; Sim, M.; Kim, S.A.; Joung, H.; Shin, D.M. Serum level of sex steroid hormone is associated with diversity and profiles of human gut microbiome. Res. Microbiol. 2019, 170, 192–201. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sex, Male/Female | HC Group | PwD Group |
|---|---|---|
| 19/19 | 19/17 | |
| Age, y.o. (average, (min:max)) | 34, (18:54) | 30, (18:53) |
| BMI, kg/m2 (average, (min:max)) | 24, (18:39) | 22, (16:34) |
| CES-D (average, (min:max)) | 5, (0:17) | 31, (21:52) |
| GAD-7 (average, (min:max)) | 1, (0:10) | 7, (2:10) |
| HAMD-17 (average, (min:max)) | 1, (0:8) | 21, (14:28) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovtun, A.S.; Averina, O.V.; Angelova, I.Y.; Yunes, R.A.; Zorkina, Y.A.; Morozova, A.Y.; Pavlichenko, A.V.; Syunyakov, T.S.; Karpenko, O.A.; Kostyuk, G.P.; et al. Alterations of the Composition and Neurometabolic Profile of Human Gut Microbiota in Major Depressive Disorder. Biomedicines 2022, 10, 2162. https://doi.org/10.3390/biomedicines10092162
Kovtun AS, Averina OV, Angelova IY, Yunes RA, Zorkina YA, Morozova AY, Pavlichenko AV, Syunyakov TS, Karpenko OA, Kostyuk GP, et al. Alterations of the Composition and Neurometabolic Profile of Human Gut Microbiota in Major Depressive Disorder. Biomedicines. 2022; 10(9):2162. https://doi.org/10.3390/biomedicines10092162
Chicago/Turabian StyleKovtun, Alexey S., Olga V. Averina, Irina Y. Angelova, Roman A. Yunes, Yana A. Zorkina, Anna Y. Morozova, Alexey V. Pavlichenko, Timur S. Syunyakov, Olga A. Karpenko, George P. Kostyuk, and et al. 2022. "Alterations of the Composition and Neurometabolic Profile of Human Gut Microbiota in Major Depressive Disorder" Biomedicines 10, no. 9: 2162. https://doi.org/10.3390/biomedicines10092162