

Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics


Abstract
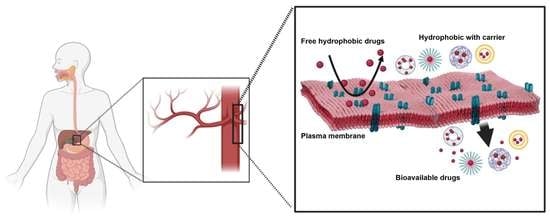
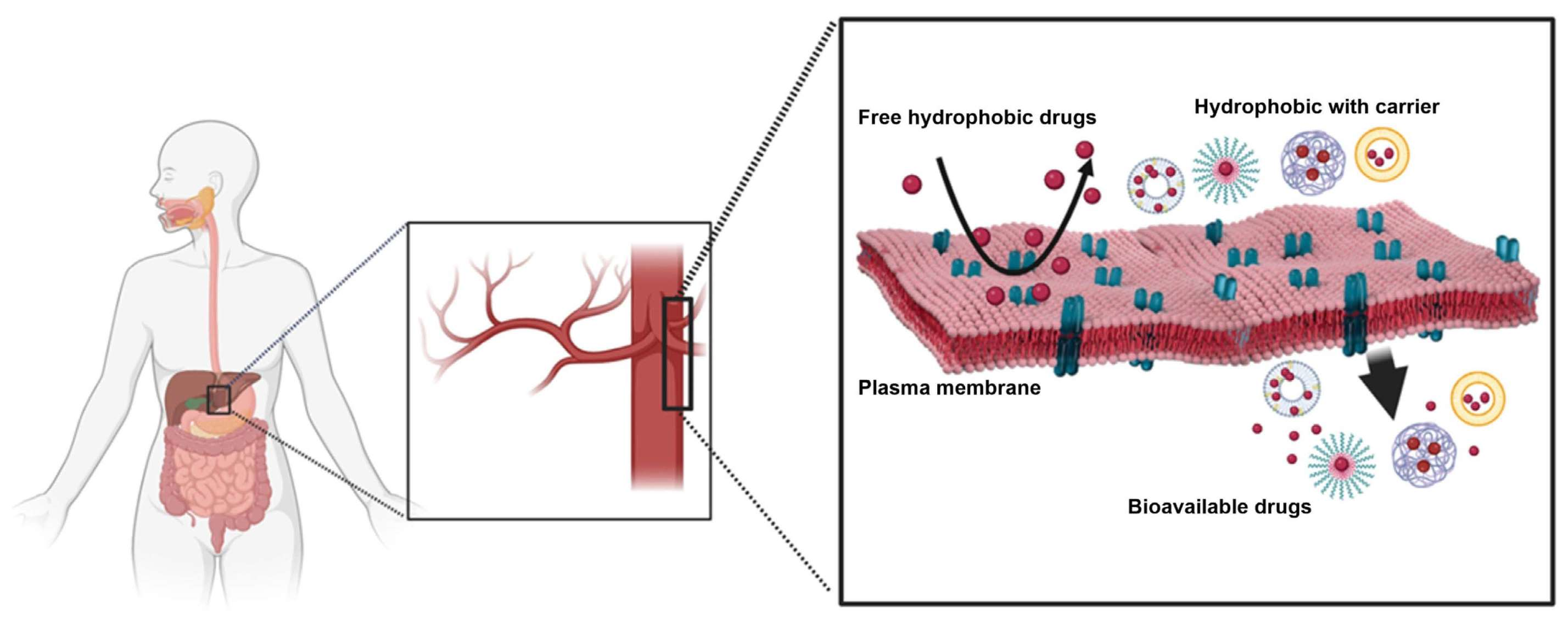
:
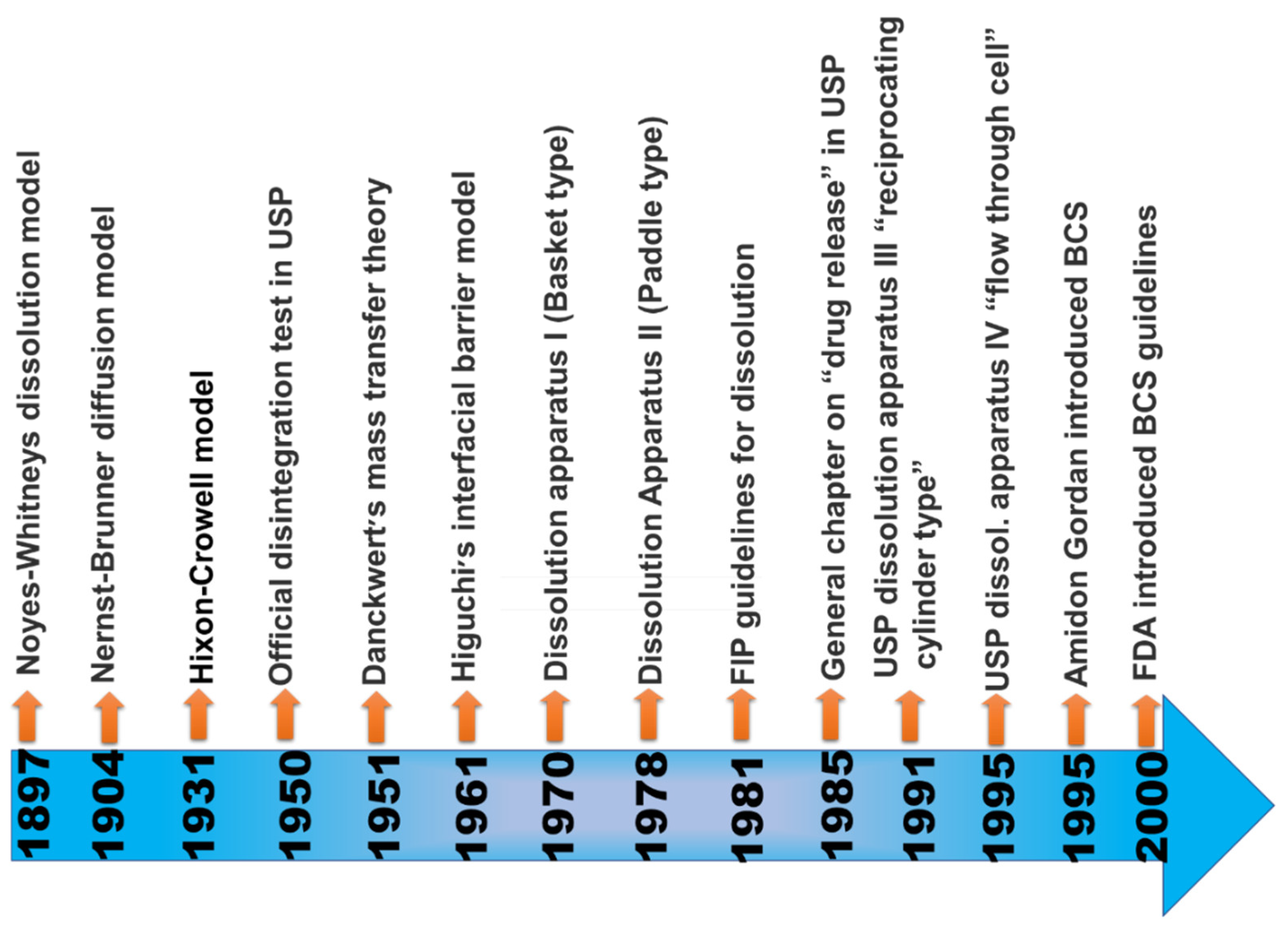
1. Introduction
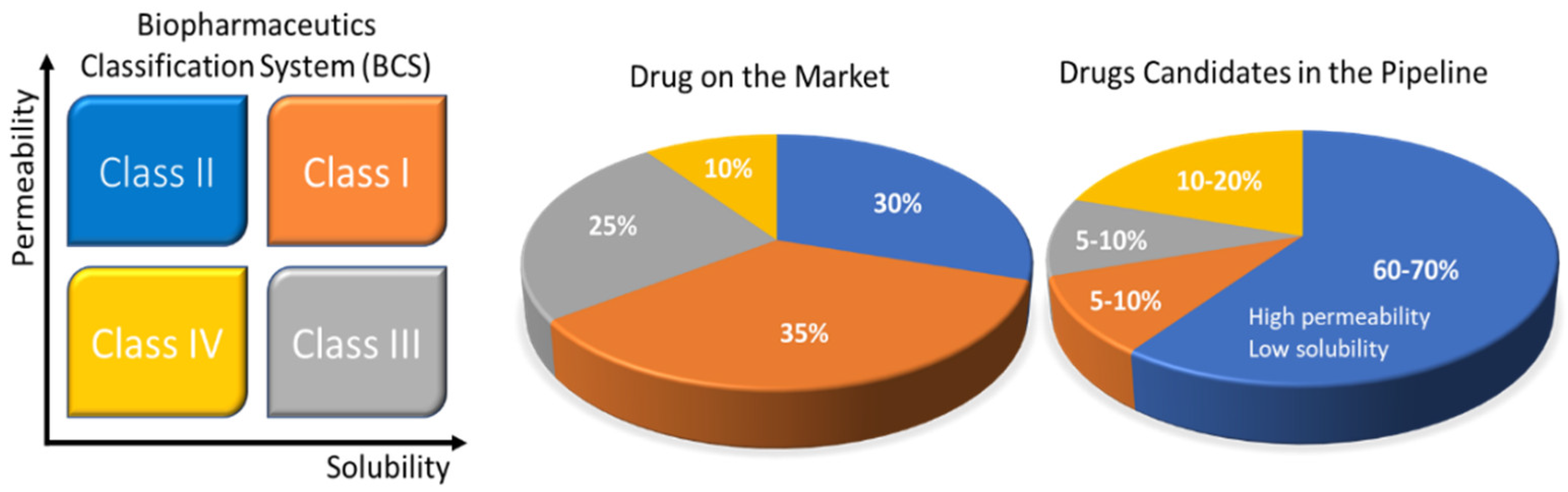
1.1. Biopharmaceutical Classification System
1.2. Importance of Solubility Enhancement
2. Techniques for Solubility Enhancement
2.1. Physical Modifications
2.1.1. Particle Size Reduction
- Advantages of nanosuspension [60]
- Enhancement of drug solubility and its bioavailability
- Higher drug loading
- Suitable for hydrophobic drugs
- Passive drug targeting
- Reduction in dosage
- Increase in drug’s physical and chemical stability.
Milling Techniques
- (a)
- Media milling
- (b) Dry grinding
Lipid Emulsion/Microemulsion Template
Microprecipitation—High-Pressure Homogenization (Nano Edge)
Nanojet Technology
2.1.2. Modification of the Crystal Habit
2.1.3. Drug Dispersion in Carriers
Eutectic Mixtures
Solid Dispersion
Solid Solutions
2.1.4. Solubilization by Surfactants
Microemulsion
Components of microemulsion
Classification of microemulsion
- Oil-in-water microemulsions (o/w)
- Water-in-oil microemulsions (w/o)
- Bi-continuous microemulsions
- (i)
- Phase titration method
- (ii)
- Phase inversion method.
Self-Emulsifying Drug Delivery Systems (SEDDS)
- Self-nano emulsifying drug delivery system (SNEDDS)
- Self-micro emulsifying drug delivery system (SMEDDS)
- Oils
- Surfactants
- Co-surfactants
- Viscosity enhancers
- Polymers
- Antioxidant agents
2.1.5. Complexation
Stanching Complexation
Inclusion Complexation
- Kneading method
- Microwave irradiation method
- Co-precipitate method
- Lyophilization/freeze-drying technique
- Spray drying
Peptide Complexation
2.1.6. Cryogenic Techniques
2.2. Chemical Modifications
2.2.1. pH Adjustment
2.2.2. Hydrotrophy
2.2.3. Co-Crystallization
2.2.4. Co-Solvency
2.2.5. Salt Formation
2.2.6. Nanotechnology in Pharmaceuticals
2.3. Miscellaneous Methods
2.3.1. Supercritical Fluid Technology
2.3.2. Micellar Solubilization
- Direct capsule filling
- Electrospinning method
- Dropping method solution
Cyclodextrins
Solid-Lipid Nanoparticles
Polymeric Micellar Carriers
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| NCEs | New Chemical entities |
| TBPOH | Tetrabutylphophonium hydroxide |
| USP | United States Pharmacopoeia |
| HPC | Hydroxypropopyl celluolose |
| HPMC | Hydroxypropyl methylcellulose |
| PVP | Polyvinylpyrrolidone |
| PEG | Poly (ethylene glycol) |
| HPMCAS | Hydroxypropyl methyl cellulose acetate succinate |
| PVA-VA | Polyvinylpyrrolidone vinyl acetate |
| FDA | Food and Drug Administration |
| PDMAm | Poly(N, N dimethylacrylamide) |
| PHEAm | Poly(N-hydroxyethylacrylamide) |
| CMC | Critical Micellar Concentration |
| SLN | Solid-Lipid nanoparticles |
| ATRP | Atom transfer radical polymerization |
| RAFT | Reversible addition fragmentation chain transfer |
References
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Krishnaiah, Y.S. Pharmaceutical technologies for enhancing oral bioavailability of poorly soluble drugs. J. Bioequiv. Availab. 2010, 2, 28–36. [Google Scholar] [CrossRef]
- Nidhi, K.; Indrajeet, S.; Khushboo, M.; Gauri, K.; Sen, D.D.J. Hydrotropy: A promising tool for solubility enhancement: A review. Int. J. Drug Dev. Res. 2011, 3, 26–33. [Google Scholar] [CrossRef]
- Martin, A.; Bustamanate, P.; Chun, A.H.C. Physical Pharmacy; BI Wavely Pvt. Ltd.: New Delhi, India, 1994; Volume 4, p. 223. [Google Scholar]
- Gennaro, A.R. Remington’s Pharmaceutical Sciences, 17th ed.; Mack Publishing Co.: Easton, PA, USA, 1985. [Google Scholar]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef]
- Hu, J.; Johnston, K.P.; Williams, R.O., 3rd. Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Dev. Ind. Pharm. 2004, 30, 233–245. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Curry, S.H. Applied biopharmaceutics and pharmacokinetics, Leon Shargel and Andrew BC Yu, Appleton-Century-Crofts, New York, 1980. Biopharm. Drug Dispos. 1982, 3, 287–290. [Google Scholar] [CrossRef]
- Sinko, P.J. Martin’s Physical Pharmacy and Pharmaceutical Sciences; Lippincott Williams & Wilkins: Philadelphia, PA, USA; London, UK, 2005. [Google Scholar]
- Carstensen, J.T. Pharmaceutical Preformulation; Technomic Publishing Company: Lancaster, PA, USA, 1998; Volume 14, p. 47. [Google Scholar]
- Brahmankar, D.M.; Jaiswal, S.B. Biopharmaceutics and Pharmacokinetics: A Treatise; Vallabh Prakashan: Delhi, India, 2005. [Google Scholar]
- Ting, J.M.; Porter III, W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in polymer design for enhancing oral drug solubility and delivery. Bioconjug. Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef]
- Kakran, M.; Li, L.; Müller, R.H. Overcoming the challenge of poor drug solubility. Pharm. Eng. 2012, 32, 82–89. [Google Scholar]
- Janssens, S.; Van den Mooter, G. Review: Physical chemistry of solid dispersions. J. Pharm. Pharm. 2009, 61, 1571–1586. [Google Scholar] [CrossRef]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Patel, D.; Wang, G. Use of Spray-Dried Dispersions in Early Pharmaceutical Development: Theoretical and Practical Challenges. AAPS J. 2017, 19, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.K.; Dhote, V.; Bhargava, A.; Jain, D.K.; Mishra, P.K. Amorphous solid dispersion technique for improved drug delivery: Basics to clinical applications. Drug Deliv. Transl. Res. 2015, 5, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Li, Z.; LaBelle, A.J.; Bates, F.S.; Lodge, T.P.; Hillmyer, M.A. Impact of Polymer Excipient Molar Mass and End Groups on Hydrophobic Drug Solubility Enhancement. Macromolecules 2017, 50, 1102–1112. [Google Scholar] [CrossRef]
- Johnson, L.M.; Hillmyer, M.A. Critical Excipient Properties for the Dissolution Enhancement of Phenytoin. ACS Omega 2019, 4, 19116–19127. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Yang, H.-W.; Chi, S.-M.; Yue, L.-L.; Ruan, Q.; Lei, Z.; Zhu, H.-Y.; Zhao, Y. Solubility and biological activity enhancement of docetaxel via formation of inclusion complexes with three alkylenediamine-modified β-cyclodextrins. RSC Adv. 2021, 11, 6292–6303. [Google Scholar] [CrossRef]
- Alshehri, S.; Imam, S.S.; Hussain, A.; Altamimi, M.A.; Alruwaili, N.K.; Alotaibi, F.; Alanazi, A.; Shakeel, F. Potential of solid dispersions to enhance solubility, bioavailability, and therapeutic efficacy of poorly water-soluble drugs: Newer formulation techniques, current marketed scenario and patents. Drug Deliv. 2020, 27, 1625–1643. [Google Scholar] [CrossRef]
- Papich, M.G.; Martinez, M.N. Applying Biopharmaceutical Classification System (BCS) Criteria to Predict Oral Absorption of Drugs in Dogs: Challenges and Pitfalls. AAPS J. 2015, 17, 948–964. [Google Scholar] [CrossRef]
- Bai, G.; Armenante, P.M.; Plank, R.V.; Gentzler, M.; Ford, K.; Harmon, P. Hydrodynamic Investigation of USP Dissolution Test Apparatus II. J. Pharm. Sci. 2007, 96, 2327–2349. [Google Scholar] [CrossRef]
- Shekhawat, P.B.; Pokharkar, V.B. Understanding peroral absorption: Regulatory aspects and contemporary approaches to tackling solubility and permeability hurdles. Acta Pharm. Sin. B 2017, 7, 260–280. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. Int. Sch. Res. Not. ChemInform 2012, 42, 195727. [Google Scholar] [CrossRef] [PubMed]
- Varandal, A.B.; Magar, D.; Saudagar, R. Different approaches toward the enhancement of drug solubility: A review. J. Adv. Pharm. Educ. Res. 2013, 3, 415–426. [Google Scholar]
- Di, L.; Kerns, E. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Sharma, D.; Soni, M.; Kumar, S.; Gupta, G. Solubility enhancement—Eminent role in poorly soluble drugs. Res. J. Pharm. Technol. 2009, 2, 220–224. [Google Scholar]
- Kumar, A.; Sahoo, S.K.; Padhee, K.; Kochar, P.S.; Sathapathy, A.; Pathak, N. Review on solubility enhancement techniques for hydrophobic drugs. Pharm. Glob. 2011, 3, 1–7. [Google Scholar]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef]
- Varshosaz, J.; Ghassami, E.; Ahmadipour, S. Crystal engineering for enhanced solubility and bioavailability of poorly soluble drugs. Curr. Pharm. Des. 2018, 24, 2473–2496. [Google Scholar] [CrossRef]
- Müller, C.E. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem. Biodivers. 2009, 6, 2071–2083. [Google Scholar] [CrossRef]
- Sanches, B.M.A.; Ferreira, E.I. Is prodrug design an approach to increase water solubility? Int. J. Pharm. 2019, 568, 118498. [Google Scholar] [CrossRef]
- Cerreia Vioglio, P.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef]
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Mu, R.H. DissoCubes—A novel formulation for poorly soluble and poorly bioavailable drugs. In Modified-Release Drug Delivery Technology; CRC Press: Boca Raton, FL, USA, 2002; pp. 159–174. [Google Scholar]
- Kesisoglou, F.; Panmai, S.; Wu, Y. Nanosizing—Oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Vemula, V.; Lagishetty, V.; Lingala, S. Solubility Enhancement Techniques. Int. J. Pharm. Sci. Rev. Res. 2010, 5, 41–51. [Google Scholar]
- Jouyban, A. Review of the cosolvency models for predicting solubility of drugs in water-cosolvent mixtures. J. Pharm. Pharm. Sci. 2008, 11, 32–58. [Google Scholar] [CrossRef]
- Strickley, R.G. Solubilizing excipients in oral and injectable formulations. Pharm. Res. 2004, 21, 201–230. [Google Scholar] [CrossRef]
- Keskin, D.; Tezcaner, A. Micelles as Delivery System for Cancer Treatment. Curr. Pharm. Des. 2017, 23, 5230–5241. [Google Scholar] [CrossRef]
- Zhang, T.; Luo, J.; Fu, Y.; Li, H.; Ding, R.; Gong, T.; Zhang, Z. Novel oral administrated paclitaxel micelles with enhanced bioavailability and antitumor efficacy for resistant breast cancer. Colloids Surf. B Biointerfaces 2017, 150, 89–97. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.; Liversidge, G.G.; Cooper, E.R. Nanosizing: A formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003, 18, 113–120. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.M.; Liversidge, G.G. Drug nanoparticles: Formulating poorly water-soluble compounds. Toxicol. Pathol. 2008, 36, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Tang, X.; Cui, F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J. Pharm. Pharmacol. 2004, 56, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, F.; Uekama, K. Cyclodextrin-based controlled drug release system. Adv. Drug Deliv. Rev. 1999, 36, 125–141. [Google Scholar] [CrossRef]
- Kim, D.-H.; Lee, S.-e.; Pyo, Y.-C.; Tran, P.; Park, J.-S. Solubility enhancement and application of cyclodextrins in local drug delivery. J. Pharm. Investig. 2019, 50, 17–27. [Google Scholar] [CrossRef]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S93–S98. [Google Scholar] [CrossRef]
- Ahn, H.; Park, J.H. Liposomal delivery systems for intestinal lymphatic drug transport. Biomater. Res. 2016, 20, 36. [Google Scholar] [CrossRef]
- Menzel, C.; Holzeisen, T.; Laffleur, F.; Zaichik, S.; Abdulkarim, M.; Gumbleton, M.; Bernkop-Schnürch, A. In vivo evaluation of an oral self-emulsifying drug delivery system (SEDDS) for exenatide. J. Control. Release 2018, 277, 165–172. [Google Scholar] [CrossRef]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in oral drug delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef]
- Leleux, J.; Williams, R.O., 3rd. Recent advancements in mechanical reduction methods: Particulate systems. Drug Dev. Ind. Pharm. 2014, 40, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Rasenack, N.; Müller, B.W. Micron-size drug particles: Common and novel micronization techniques. Pharm. Dev. Technol. 2004, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liversidge, G.G.; Cundy, K.C. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int. J. Pharm. 1995, 125, 91–97. [Google Scholar] [CrossRef]
- Verma, S.; Gokhale, R.; Burgess, D.J. A comparative study of top-down and bottom-up approaches for the preparation of micro/nanosuspensions. Int. J. Pharm. 2009, 380, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Van den Mooter, G.; Augustijns, P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. Int. J. Pharm. 2008, 364, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Liversidge, G.G.; Conzentino, P. Drug particle size reduction for decreasing gastric irritancy and enhancing absorption of naproxen in rats. Int. J. Pharm. 1995, 125, 309–313. [Google Scholar] [CrossRef]
- Chingunpitak, J. Nanosuspension Technology for Drug Delivery. Walailak J. Sci. Technol. 2006, 4, 139–153. [Google Scholar] [CrossRef]
- Muller, R. Nanosuspension for the Formulation of Poorly Soluble Drugs. In Pharmaceutical Emulsion and Suspension; Neilloud, F., Marti-Mestres, G., Eds.; Routledge: London, UK, 2000. [Google Scholar]
- Clewlow, P. Survival of the smartest. Scrip’s Target World Drug Deliv. News 2004, 35, 316–323. [Google Scholar]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R. Cyclodextrins in drug delivery: An updated review. AAPS PharmSciTech 2005, 6, E329–E357. [Google Scholar] [CrossRef]
- Atul, A.P.; Jorwekar, P.; Chaudhari, P.D. Formulation Development of Aceclofenac Loaded Nanosupension by Three Square (32) Factorial Design. Int. J. Pharm. Sci. Nanotechnol. 2012, 4, 1575–1583. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, J.d.C.; Tenorio Clavijo, J.C.; Alvarez, N.; Ellena, J.; Ayala, A.P. Novel solid solution of the antiretroviral drugs lamivudine and emtricitabine. Cryst. Growth Des. 2018, 18, 3441–3448. [Google Scholar] [CrossRef]
- Ritika, H.S.; Aggarwal, G. Formulation tactics for the delivery of poorly soluble drugs. Int. J. PharmTech Res. 2012, 4, 914–923. [Google Scholar]
- Bhusnure, O.; Kazi, P.; Gholve, S.; Ansari, M.; Kazi, S. Solid Dispersion: An ever green method for solubility enhancement of poorly water soluble drugs. Int. J. Res. Pharm. Chem. 2014, 4, 906–918. [Google Scholar]
- Sareen, S.; Mathew, G.; Joseph, L. Improvement in solubility of poor water-soluble drugs by solid dispersion. Int. J. Pharm. Investig. 2012, 2, 12–17. [Google Scholar] [CrossRef]
- Kumar, S.K.; Sushma, M.; Raju, P.Y. Dissolution enhancement of poorly soluble drugs by using complexation technique-a review. J. Pharm. Sci. Res. 2013, 5, 120. [Google Scholar]
- Nutan, B.; Kumar, A.; Jewrajka, S.K. Library of Derivatizable Multiblock Copolymers by Nucleophilic Substitution Polymerization and Targeting Specific Properties. Biomacromolecules 2020, 21, 5029–5043. [Google Scholar] [CrossRef]
- Nutan, B.; Chandel, A.K.S.; Biswas, A.; Kumar, A.; Yadav, A.; Maiti, P.; Jewrajka, S.K. Gold Nanoparticle Promoted Formation and Biological Properties of Injectable Hydrogels. Biomacromolecules 2020, 21, 3782–3794. [Google Scholar] [CrossRef]
- Moyano, J.; Arias-Blanco, M.; Gines, J.; Giordano, F. Solid-state characterization and dissolution characteristics of gliclazide-β-cyclodextrin inclusion complexes. Int. J. Pharm. 1997, 148, 211–217. [Google Scholar] [CrossRef]
- Kalaiselvan, R.; Mohanta, G.; Manna, P.; Manavalan, R. Studies on mechanism of enhanced dissolution of albendazole solid dispersions with crystalline carriers. Indian J. Pharm. Sci. 2006, 68, 599–607. [Google Scholar] [CrossRef]
- Nokhodchi, A.; Talari, R.; Valizadeh, H.; Jalali, M.B. An investigation on the solid dispersions of chlordiazepoxide. Int. J. Biomed. Sci. IJBS 2007, 3, 211. [Google Scholar]
- Bhise, S.B.; Rajkumar, M. Effect of HPMC on solubility and dissolution of carbamazepine form III in simulated gastrointestinal fluids. Asian J. Pharm. (AJP) 2014, 2, 1. [Google Scholar] [CrossRef]
- Bakatselou, V.; Oppenheim, R.C.; Dressman, J.B. Solubilization and wetting effects of bile salts on the dissolution of steroids. Pharm. Res. 1991, 8, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Betageri, G.V.; Makarla, K.R. Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques. Int. J. Pharm. 1995, 126, 155–160. [Google Scholar] [CrossRef]
- Tsinontides, S.C.; Rajniak, P.; Pham, D.; Hunke, W.A.; Placek, J.; Reynolds, S.D. Freeze drying—Principles and practice for successful scale-up to manufacturing. Int. J. Pharm. 2004, 280, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Vilhelmsen, T.; Eliasen, H.; Schaefer, T. Effect of a melt agglomeration process on agglomerates containing solid dispersions. Int. J. Pharm. 2005, 303, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Mittal, K.L. Handbook of Microemulsion Science and Technology; Marcel Dekker: New York, NY, USA, 1999. [Google Scholar]
- Brouwer, W.; El-Aasser, M.; Vanderhoff, J. The role of water solubility of the oil phase in the formation of miniemulsions. Colloids Surf. 1986, 21, 69–86. [Google Scholar] [CrossRef]
- Dhumal, D.M.; Kothari, P.R.; Kalhapure, R.S.; Akamanchi, K.G. Self-microemulsifying drug delivery system of curcumin with enhanced solubility and bioavailability using a new semi-synthetic bicephalous heterolipid: In vitro and in vivo evaluation. RSC Adv. 2015, 5, 90295–90306. [Google Scholar] [CrossRef]
- Proverbio, Z.E.; Bardavid, S.; Arancibia, E.L.; Schulz, P.C. Hydrophile–lipophile balance and solubility parameter of cationic surfactants. Colloids Surf. A Physicochem. Eng. Asp. 2003, 214, 167–171. [Google Scholar] [CrossRef]
- Ghosh, P.K.; Murthy, R.S. Microemulsions: A potential drug delivery system. Curr. Drug Deliv. 2006, 3, 167–180. [Google Scholar] [CrossRef]
- Glatter, O.; Orthaber, D.; Stradner, A.; Scherf, G.; Fanun, M.; Garti, N.; Clément, V.; Leser, M.E. Sugar-Ester Nonionic Microemulsion: Structural Characterization. J. Colloid Interface Sci. 2001, 241, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Dey, S.K.; Karki, R. Microemulsions—Potential Carrier for Improved Drug Delivery. Asian J. Biomed. Pharm. Sci. 2011, 1, 5–9. [Google Scholar] [CrossRef]
- Rawat, S.; Derle, D.V.; Parve, B.S.; Shinde, P.R. Self emulsifying drug delivery system (sedds): A method for bioavailability enhancement. Int. J. Pharm. Chem. Biol. Sci. 2014, 4, 479–494. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- Vimalson, D.C. Techniques to enhance solubility of hydrophobic drugs: An overview. Asian J. Pharm. (AJP) 2016, 10, 2. [Google Scholar] [CrossRef]
- Hong, S.; Choi, D.W.; Kim, H.N.; Park, C.G.; Lee, W.; Park, H.H. Protein-Based Nanoparticles as Drug Delivery Systems. Pharmaceutics 2020, 12, 604. [Google Scholar] [CrossRef]
- Chang, C.; Meikle, T.G.; Su, Y.; Wang, X.; Dekiwadia, C.; Drummond, C.J.; Conn, C.E.; Yang, Y. Encapsulation in egg white protein nanoparticles protects anti-oxidant activity of curcumin. Food Chem. 2019, 280, 65–72. [Google Scholar] [CrossRef]
- Somu, P.; Paul, S. Surface conjugation of curcumin with self-assembled lysozyme nanoparticle enhanced its bioavailability and therapeutic efficacy in multiple cancer cells. J. Mol. Liq. 2021, 338, 116623. [Google Scholar] [CrossRef]
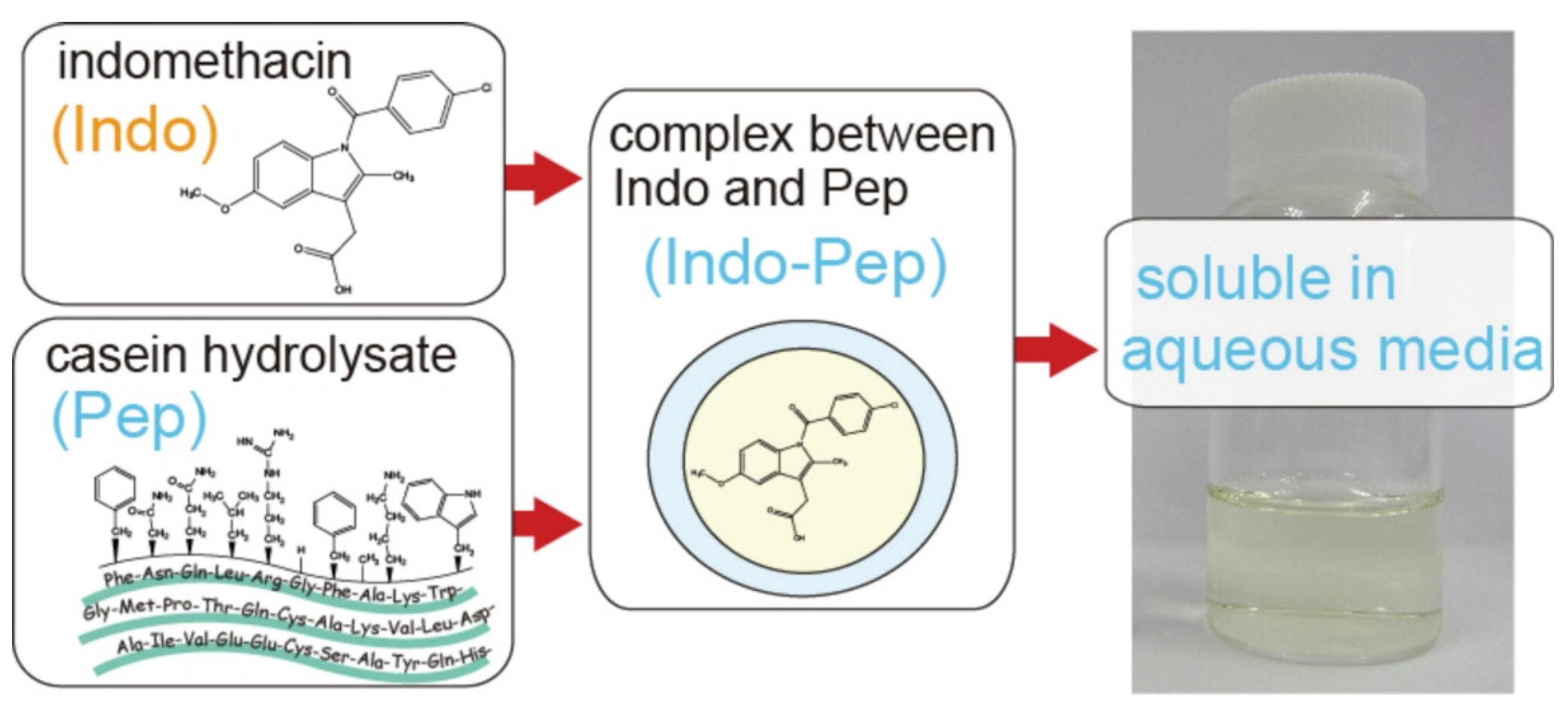
- Inada, A.; Oshima, T.; Takahashi, H.; Baba, Y. Enhancement of water solubility of indomethacin by complexation with protein hydrolysate. Int. J. Pharm. 2013, 453, 587–593. [Google Scholar] [CrossRef]
- Mumenthaler, M.; Leuenberger, H. Atmospheric spray-freeze drying: A suitable alternative in freeze-drying technology. Int. J. Pharm. 1991, 72, 97–110. [Google Scholar] [CrossRef]
- Leuenberger, H. Spray Freeze-drying—The Process of Choice for Low Water Soluble Drugs? J. Nanoparticle Res. 2002, 4, 111–119. [Google Scholar] [CrossRef]
- Williams III, R.O.; Johnston, K.P.; Young, T.J.; Rogers, T.L.; Barron, M.K.; Yu, Z.; Hu, J. Process for Production of Nanoparticles and Microparticles by Spray Freezing into Liquid. U.S. Patent US6862890B2, 8 March 2005. [Google Scholar]
- McMorland, G.H.; Douglas, M.J.; Jeffery, W.K.; Ross, P.L.; Axelson, J.E.; Kim, J.H.; Gambling, D.R.; Robertson, K. Effect of pH-adjustment of bupivacaine on onset and duration of epidural analgesia in parturients. Can. Anaesth. Soc. J. 1986, 33, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Rasool, A.A.; Hussain, A.A.; Dittert, L.W. Solubility enhancement of some water-insoluble drugs in the presence of nicotinamide and related compounds. J. Pharm. Sci. 1991, 80, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Patole, T.; Deshpande, A. Co-crystallization-a technique for solubility enhancement. Int. J. Pharm. Sci. Res. 2014, 5, 3566. [Google Scholar] [CrossRef]
- Millard, J.; Alvarez-Núñez, F.; Yalkowsky, S. Solubilization by cosolvents. Establishing useful constants for the log-linear model. Int. J. Pharm. 2002, 245, 153–166. [Google Scholar] [CrossRef]
- Paroha, S.; Chandel, A.K.S.; Dubey, R.D. Nanosystems for drug delivery of coenzyme Q10. Environ. Chem. Lett. 2018, 16, 71–77. [Google Scholar] [CrossRef]
- Gowthamarajan, K.; Singh, S.K. Dissolution testing for poorly soluble drugs: A continuing perspective. Dissolution Technol. 2010, 17, 24–32. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Morakul, B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 13–23. [Google Scholar] [CrossRef]
- Lenhardt, T.; Vergnault, G.; Grenier, P.; Scherer, D.; Langguth, P. Evaluation of nanosuspensions for absorption enhancement of poorly soluble drugs: In vitro transport studies across intestinal epithelial monolayers. AAPS J. 2008, 10, 435–438. [Google Scholar] [CrossRef]
- Lipinski, C. Poor aqueous solubility—An industry wide problem in drug discovery. Am. Pharm. Rev. 2002, 5, 82–85. [Google Scholar]
- Rosen, M. Surfactant and Interfacial Phenomena, 2nd ed.; Wiely: New York, NY, USA, 1989; Volume 151. [Google Scholar] [CrossRef]
- Liu, C.; Desai, K.G.H.; Liu, C. Solubility of valdecoxib in the presence of ethanol and sodium lauryl sulfate at (298.15, 303.15, and 308.15) K. J. Chem. Eng. Data 2004, 49, 1847–1850. [Google Scholar] [CrossRef]
- Ajewski, R.; Stella, V. Pharmaceutical application of cylodextrin. 2. In vivo drug delivery. J. Pharm. Sci. 1996, 85, 1142–1167. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Magnúsdóttir, A.; Másson, M.; Sigurjónsdóttir, J.F. Self-association and cyclodextrin solubilization of drugs. J. Pharm. Sci. 2002, 91, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Bodor, N.; Buchwald, P. Theoretical insights into the formation, structure, and energetics of some cyclodextrin complexes. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 9–14. [Google Scholar] [CrossRef]
- Loftsson, T.; Matthíasson, K.; Másson, M. The effects of organic salts on the cyclodextrin solubilization of drugs. Int. J. Pharm. 2003, 262, 101–107. [Google Scholar] [CrossRef]
- Loftsson, T.; Másson, M.; Brewster, M.E. Self-association of cyclodextrins and cyclodextrin complexes. J. Pharm. Sci. 2004, 93, 1091–1099. [Google Scholar] [CrossRef]
- Reddy, A.; Parthiban, S.; Vikneswari, A.; Senthilkumar, G. A modern review on solid lipid nanoparticles as novel controlled drug delivery system. Int. J. Res. Pharm. Nano Sci. 2014, 3, 313–325. [Google Scholar]
- Severino, P.; Pinho, S.C.; Souto, E.B.; Santana, M.H. Polymorphism, crystallinity and hydrophilic–lipophilic balance of stearic acid and stearic acid–capric/caprylic triglyceride matrices for production of stable nanoparticles. Colloids Surf. B Biointerfaces 2011, 86, 125–130. [Google Scholar] [CrossRef]
- Garud, A.; Singh, D.; Garud, N. Solid lipid nanoparticles (SLN): Method, characterization and applications. Int. Curr. Pharm. J. 2012, 1, 384–393. [Google Scholar] [CrossRef]
- Ekambaram, P.; Abdul Hasan Sathali, A.; Priyanka, K. Solid lipid nanoparticles: A review. Sci. Rev. Chem. Commun. 2012, 2, 80–102. [Google Scholar]
- Westesen, K.; Bunjes, H.; Koch, M.H.J. Physicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J. Control. Release 1997, 48, 223–236. [Google Scholar] [CrossRef]
- Westesen, K.; Siekmann, B.; Koch, M.H.J. Investigations on the physical state of lipid nanoparticles by synchrotron radiation X-ray diffraction. Int. J. Pharm. 1993, 93, 189–199. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ray, S.; Thakur, R.S. Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian J. Pharm. Sci. 2009, 71, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Park, Y.-J. Improved Antitumor Efficacy of Hyaluronic Acid-Complexed Paclitaxel Nanoemulsions in Treating Non-Small Cell Lung Cancer. Biomol. Ther. 2017, 25, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Najlah, M.; Kadam, A.; Wan, K.-W.; Ahmed, W.; Taylor, K.M.; Elhissi, A.M. Novel paclitaxel formulations solubilized by parenteral nutrition nanoemulsions for application against glioma cell lines. Int. J. Pharm. 2016, 506, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hayes, D.G.; Chen, G.; Zhong, Q. Transparent Dispersions of Milk-Fat-Based Nanostructured Lipid Carriers for Delivery of β-Carotene. J. Agric. Food Chem. 2013, 61, 9435–9443. [Google Scholar] [CrossRef]
- Mehmood, T.; Ahmad, A.; Ahmed, A.; Ahmed, Z. Optimization of olive oil based O/W nanoemulsions prepared through ultrasonic homogenization: A response surface methodology approach. Food Chem. 2017, 229, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Koutelidakis, A.E.; Argyri, K.; Sevastou, Z.; Lamprinaki, D.; Panagopoulou, E.; Paximada, E.; Sali, A.; Papalazarou, V.; Mallouchos, A.; Evageliou, V. Bioactivity of epigallocatechin gallate nanoemulsions evaluated in mice model. J. Med. Food 2017, 20, 923–931. [Google Scholar] [CrossRef]
- Rodríguez-Burneo, N.; Busquets, M.A.; Estelrich, J. Magnetic nanoemulsions: Comparison between nanoemulsions formed by ultrasonication and by spontaneous emulsification. Nanomaterials 2017, 7, 190. [Google Scholar] [CrossRef]
- Abdelwahab, S.I.; Sheikh, B.Y.; Taha, M.M.E.; How, C.W.; Abdullah, R.; Yagoub, U.; El-Sunousi, R.; Eid, E.E. Thymoquinone-loaded nanostructured lipid carriers: Preparation, gastroprotection, in vitro toxicity, and pharmacokinetic properties after extravascular administration. Int. J. Nanomed. 2013, 8, 2163. [Google Scholar] [CrossRef]
- Yin, J.; Xiang, C.; Wang, P.; Yin, Y.; Hou, Y. Biocompatible nanoemulsions based on hemp oil and less surfactants for oral delivery of baicalein with enhanced bioavailability. Int. J. Nanomed. 2017, 12, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Mikulcová, V.; Kašpárková, V.; Humpolíček, P.; Buňková, L. Formulation, characterization and properties of hemp seed oil and its emulsions. Molecules 2017, 22, 700. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Liu, S.; Wang, Q.; Li, X. Nanoemulsions as novel oral carriers of stiripentol: Insights into the protective effect and absorption enhancement. Int. J. Nanomed. 2015, 10, 4937. [Google Scholar] [CrossRef]
- Panatieri, L.F.; Brazil, N.T.; Faber, K.; Medeiros-Neves, B.; von Poser, G.L.; Rott, M.B.; Zorzi, G.K.; Teixeira, H.F. Nanoemulsions containing a coumarin-rich extract from Pterocaulon balansae (Asteraceae) for the treatment of ocular Acanthamoeba keratitis. AAPS PharmSciTech 2017, 18, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Ragelle, H.; Crauste-Manciet, S.; Seguin, J.; Brossard, D.; Scherman, D.; Arnaud, P.; Chabot, G.G. Nanoemulsion formulation of fisetin improves bioavailability and antitumour activity in mice. Int. J. Pharm. 2012, 427, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Hejri, A.; Khosravi, A.; Gharanjig, K.; Hejazi, M. Optimisation of the formulation of β-carotene loaded nanostructured lipid carriers prepared by solvent diffusion method. Food Chem. 2013, 141, 117–123. [Google Scholar] [CrossRef]
- Tan, S.; Billa, N.; Roberts, C.; Burley, J. Surfactant effects on the physical characteristics of Amphotericin B-containing nanostructured lipid carriers. Colloids Surf. A Physicochem. Eng. Asp. 2010, 372, 73–79. [Google Scholar] [CrossRef]
- Wang, L.; Luo, Q.; Lin, T.; Li, R.; Zhu, T.; Zhou, K.; Ji, Z.; Song, J.; Jia, B.; Zhang, C. PEGylated nanostructured lipid carriers (PEG–NLC) as a novel drug delivery system for biochanin A. Drug Dev. Ind. Pharm. 2015, 41, 1204–1212. [Google Scholar] [CrossRef]
- Sood, J.; Sapra, B.; Tiwary, A.K. Microemulsion transdermal formulation for simultaneous delivery of valsartan and nifedipine: Formulation by design. AAPS PharmSciTech 2017, 18, 1901–1916. [Google Scholar] [CrossRef]
- Dash, R.N.; Mohammed, H.; Humaira, T.; Ramesh, D. Design, optimization and evaluation of glipizide solid self-nanoemulsifying drug delivery for enhanced solubility and dissolution. Saudi Pharm. J. 2015, 23, 528–540. [Google Scholar] [CrossRef]
- Bondì, M.L.; Botto, C.; Amore, E.; Emma, M.R.; Augello, G.; Craparo, E.F.; Cervello, M. Lipid nanocarriers containing sorafenib inhibit colonies formation in human hepatocarcinoma cells. Int. J. Pharm. 2015, 493, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, A.; Ponnusamy, C.; Kandasamy, P.; Krishnaswami, V.; Palanichamy, R.; Kandasamy, R.; Lakshmanan, M.; Natesan, S. Development and evaluation of camptothecin loaded polymer stabilized nanoemulsion: Targeting potential in 4T1-breast tumour xenograft model. Eur. J. Pharm. Sci. 2018, 116, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pan, W.; Gan, L.; Zhu, C.; Gan, Y.; Nie, S. Preparation of a dispersible PEGylate nanostructured lipid carriers (NLC) loaded with 10-hydroxycamptothecin by spray-drying. Chem. Pharm. Bull. 2008, 56, 1645–1650. [Google Scholar] [CrossRef]
- Shah, A.V.; Desai, H.H.; Thool, P.; Dalrymple, D.; Serajuddin, A.T. Development of self-microemulsifying drug delivery system for oral delivery of poorly water-soluble nutraceuticals. Drug Dev. Ind. Pharm. 2018, 44, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Griesser, J.; Burtscher, S.; Köllner, S.; Nardin, I.; Prüfert, F.; Bernkop-Schnürch, A. Zeta potential changing self-emulsifying drug delivery systems containing phosphorylated polysaccharides. Eur. J. Pharm. Biopharm. 2017, 119, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Katiyar, S.S.; Kushwah, V.; Jain, S. Nanoemulsion loaded gel for topical co-delivery of clobitasol propionate and calcipotriol in psoriasis. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.; Shelat, P.; Lalwani, A. Statistical modeling, optimization and characterization of solid self-nanoemulsifying drug delivery system of lopinavir using design of experiment. Drug Deliv. 2016, 23, 3027–3042. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.V.; Seth, A.K.; Balaraman, R.; Aundhia, C.J.; Maheshwari, R.A.; Parmar, G.R. Nanostructured lipid carriers for oral bioavailability enhancement of raloxifene: Design and in vivo study. J. Adv. Res. 2016, 7, 423–434. [Google Scholar] [CrossRef]
- Elnaggar, Y.S.; El-Massik, M.A.; Abdallah, O.Y. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: Design and optimization. Int. J. Pharm. 2009, 380, 133–141. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Katare, O.; Singh, B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf. B Biointerfaces 2012, 100, 50–61. [Google Scholar] [CrossRef]
- Kamboj, S.; Rana, V. Quality-by-design based development of a self-microemulsifying drug delivery system to reduce the effect of food on Nelfinavir mesylate. Int. J. Pharm. 2016, 501, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Yang, G.; Ren, J.; Guo, T.; Du, Y.; Feng, N. Formulation design, preparation, and in vitro and in vivo characterizations of β-elemene-loaded nanostructured lipid carriers. Int. J. Nanomed. 2013, 8, 2533. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, O.; Kim, K.S.; Shafique, S.; Kim, D.S.; Jin, S.G.; Seo, Y.G.; Youn, Y.S.; Oh, K.T.; Lee, B.-J.; Park, Y.J. Development of novel cilostazol–loaded solid SNEDDS using a SPG membrane emulsification technique: Physicochemical characterization and in vivo evaluation. Colloids Surf. B Biointerfaces 2017, 150, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Drechsler, M.; Mariani, P.; Carducci, F.; Servadio, M.; Melancia, F.; Ratano, P.; Campolongo, P.; Trezza, V.; Cortesi, R. Lipid nanoparticles for administration of poorly water soluble neuroactive drugs. Biomed. Microdevices 2017, 19, 44. [Google Scholar] [CrossRef]
- Dawoud, M.Z.; Nasr, M. Comparison of drug release from liquid crystalline monoolein dispersions and solid lipid nanoparticles using a flow cytometric technique. Acta Pharm. Sin. B 2016, 6, 163–169. [Google Scholar] [CrossRef]
- Brüsewitz, C.; Schendler, A.; Funke, A.; Wagner, T.; Lipp, R. Novel poloxamer-based nanoemulsions to enhance the intestinal absorption of active compounds. Int. J. Pharm. 2007, 329, 173–181. [Google Scholar] [CrossRef]
- Seo, Y.G.; Kim, D.-W.; Cho, K.H.; Yousaf, A.M.; Kim, D.S.; Kim, J.H.; Kim, J.O.; Yong, C.S.; Choi, H.-G. Preparation and pharmaceutical evaluation of new tacrolimus-loaded solid self-emulsifying drug delivery system. Arch. Pharmacal Res. 2015, 38, 223–228. [Google Scholar] [CrossRef]
- Thatipamula, R.; Palem, C.; Gannu, R.; Mudragada, S.; Yamsani, M. Formulation and in vitro characterization of domperidone loaded solid lipid nanoparticles and nanostructured lipid carriers. Daru J. Fac. Pharm. Tehran Univ. Med. Sci. 2011, 19, 23. [Google Scholar]
- Elmowafy, M.; Ibrahim, H.M.; Ahmed, M.A.; Shalaby, K.; Salama, A.; Hefesha, H. Atorvastatin-loaded nanostructured lipid carriers (NLCs): Strategy to overcome oral delivery drawbacks. Drug Deliv. 2017, 24, 932–941. [Google Scholar] [CrossRef]
- Chavan, R.B.; Modi, S.R.; Bansal, A.K. Role of solid carriers in pharmaceutical performance of solid supersaturable SEDDS of celecoxib. Int. J. Pharm. 2015, 495, 374–384. [Google Scholar] [CrossRef]
- Hsu, H.; Huang, R.; Kao, T.; Inbaraj, B.; Chen, B. Preparation of carotenoid extracts and nanoemulsions from Lycium barbarum L. and their effects on growth of HT-29 colon cancer cells. Nanotechnology 2017, 28, 135103. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Zhou, C.-L.; Chen, F.-P.; Han, D.; Wang, C.-Y.; Li, J.-X.; Chi, Z.; Liu, C.-G. Development of a carboxymethyl chitosan functionalized nanoemulsion formulation for increasing aqueous solubility, stability and skin permeability of astaxanthin using low-energy method. J. Microencapsul. 2017, 34, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Kamble, R.N.; Mehta, P.P.; Kumar, A. Efavirenz self-nano-emulsifying drug delivery system: In vitro and in vivo evaluation. AAPS PharmSciTech 2016, 17, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Safwat, S.; Ishak, R.A.; Hathout, R.M.; Mortada, N.D. Nanostructured lipid carriers loaded with simvastatin: Effect of PEG/glycerides on characterization, stability, cellular uptake efficiency and in vitro cytotoxicity. Drug Dev. Ind. Pharm. 2017, 43, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, F.; Cofelice, M.; Venditti, F.; Ceglie, A.; Miguel, M.; Lindman, B.; Lopez, F. In-vitro digestion of curcumin loaded chitosan-coated liposomes. Colloids Surf. B Biointerfaces 2018, 168, 29–34. [Google Scholar] [CrossRef]
- Freag, M.S.; Saleh, W.M.; Abdallah, O.Y. Self-assembled phospholipid-based phytosomal nanocarriers as promising platforms for improving oral bioavailability of the anticancer celastrol. Int. J. Pharm. 2018, 535, 18–26. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, T.; Zhou, X.; Liu, H.; Sun, H.; Ma, Z.; Wu, B. Enhancement of oral bioavailability of tripterine through lipid nanospheres: Preparation, characterization, and absorption evaluation. J. Pharm. Sci. 2014, 103, 1711–1719. [Google Scholar] [CrossRef]
- Wu, B.; Lu, S.-T.; Zhang, L.-J.; Zhuo, R.-X.; Xu, H.-B.; Huang, S.-W. Codelivery of doxorubicin and triptolide with reduction-sensitive lipid–polymer hybrid nanoparticles for in vitro and in vivo synergistic cancer treatment. Int. J. Nanomed. 2017, 12, 1853. [Google Scholar] [CrossRef]
- Yao, C.; Wu, M.; Zhang, C.; Lin, X.; Wei, Z.; Zheng, Y.; Zhang, D.; Zhang, Z.; Liu, X. Photoresponsive lipid-polymer hybrid nanoparticles for controlled doxorubicin release. Nanotechnology 2017, 28, 255101. [Google Scholar] [CrossRef]
- Valicherla, G.R.; Dave, K.M.; Syed, A.A.; Riyazuddin, M.; Gupta, A.P.; Singh, A.; Mitra, K.; Datta, D.; Gayen, J.R. Formulation optimization of Docetaxel loaded self-emulsifying drug delivery system to enhance bioavailability and anti-tumor activity. Sci. Rep. 2016, 6, 26895. [Google Scholar] [CrossRef]
- Gaber, D.M.; Nafee, N.; Abdallah, O.Y. Myricetin solid lipid nanoparticles: Stability assurance from system preparation to site of action. Eur. J. Pharm. Sci. 2017, 109, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.; Reddi, S.; Rinwa, V.; Balwani, G.; Saha, R. Design and in vivo evaluation of solid lipid nanoparticulate systems of Olanzapine for acute phase schizophrenia treatment: Investigations on antipsychotic potential and adverse effects. Eur. J. Pharm. Sci. 2017, 104, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.; Maestrelli, F.; Mannelli, L.D.C.; Ghelardini, C.; Almeida, A.; Mura, P. Development of solid lipid nanoparticles as carriers for improving oral bioavailability of glibenclamide. Eur. J. Pharm. Biopharm. 2016, 102, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Hu, X.; Ma, Y.; Xie, Y.; Lu, Y.; Qi, J.; Xiang, L.; Li, F.; Wu, W. Lipids-based nanostructured lipid carriers (NLCs) for improved oral bioavailability of sirolimus. Drug Deliv. 2016, 23, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446. [Google Scholar] [CrossRef]
- Elmowafy, M.; Samy, A.; Raslan, M.A.; Salama, A.; Said, R.A.; Abdelaziz, A.E.; El-Eraky, W.; El Awdan, S.; Viitala, T. Enhancement of bioavailability and pharmacodynamic effects of thymoquinone via nanostructured lipid carrier (NLC) formulation. AAPS PharmSciTech 2016, 17, 663–672. [Google Scholar] [CrossRef]
- Zhang, S.; Meng, X.; Wang, Z.; Fan, A.; Wang, G.; Zhao, Y.; Tang, Y. Engineering hot-melt extruded solid dispersion for controlled release of hydrophilic drugs. Eur. J. Pharm. Sci. 2017, 100, 109–115. [Google Scholar] [CrossRef]
- Xu, W.; Lim, S.-J.; Lee, M.-K. Cellular uptake and antitumour activity of paclitaxel incorporated into trilaurin-based solid lipid nanoparticles in ovarian cancer. J. Microencapsul. 2013, 30, 755–761. [Google Scholar] [CrossRef]
- Christophersen, P.C.; Zhang, L.; Müllertz, A.; Nielsen, H.M.; Yang, M.; Mu, H. Solid lipid particles for oral delivery of peptide and protein drugs II—The digestion of trilaurin protects desmopressin from proteolytic degradation. Pharm. Res. 2014, 31, 2420–2428. [Google Scholar] [CrossRef]
- Yao, M.; McClements, D.J.; Xiao, H. Improving oral bioavailability of nutraceuticals by engineered nanoparticle-based delivery systems. Curr. Opin. Food Sci. 2015, 2, 14–19. [Google Scholar] [CrossRef]
- Keck, C.M.; Kovačević, A.; Müller, R.H.; Savić, S.; Vuleta, G.; Milić, J. Formulation of solid lipid nanoparticles (SLN): The value of different alkyl polyglucoside surfactants. Int. J. Pharm. 2014, 474, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, L.N.; Franz-Montan, M.; Breitkreitz, M.C.; Alcântara, A.C.; Castro, S.R.; Guilherme, V.A.; Barbosa, R.M.; de Paula, E. Nanostructured lipid carriers as robust systems for topical lidocaine-prilocaine release in dentistry. Eur. J. Pharm. Sci. 2016, 93, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Baskaran, R.; Jang, Y.S.; Oh, S.H.; Yoo, B.K. Quercetin-loaded solid lipid nanoparticle dispersion with improved physicochemical properties and cellular uptake. AAPS PharmSciTech 2017, 18, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; Wu, P.C.; Huang, Y.B.; Chang, J.S.; Lin, C.L.; Tsai, Y.H.; Fang, J.Y. Baicalein loaded in tocol nanostructured lipid carriers (tocol NLCs) for enhanced stability and brain targeting. Int. J. Pharm. 2012, 423, 461–470. [Google Scholar] [CrossRef]
- Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical dispersion techniques for dissolution and bioavailability enhancement of poorly water-soluble drugs. Pharmaceutics 2018, 10, 74. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Nutan, B.; Raval, I.H.; Jewrajka, S.K. Self-assembly of partially alkylated dextran-graft-poly [(2-dimethylamino) ethyl methacrylate] copolymer facilitating hydrophobic/hydrophilic drug delivery and improving conetwork hydrogel properties. Biomacromolecules 2018, 19, 1142–1153. [Google Scholar] [CrossRef]
- Kumar, A.; Nutan, B.; Jewrajka, S.K. Stability and acidic pH-mediated leakage of guest molecules from self-assembly of poly (amidoamine)-graft-alkyl copolymers. Polymer 2019, 183, 121894. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Bhingradiya, N. Therapeutic Efficacy of Herbal Formulations Through Novel Drug Delivery Systems. In Enhancing the Therapeutic Efficacy of Herbal Formulations; IGI Global: Hershey, PA, USA, 2021; pp. 1–42. [Google Scholar]
- Gaucher, G.; Dufresne, M.-H.; Sant, V.P.; Kang, N.; Maysinger, D.; Leroux, J.-C. Block copolymer micelles: Preparation, characterization and application in drug delivery. J. Control. Release 2005, 109, 169–188. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Jewrajka, S.K. Designing Multi-component Biodegradable/Biocompatible Amphiphilic Polymer Co-networks for Biomedical Applications. In Amphiphilic Polymer Co-Networks; RSC Publishing: Cambridge, UK, 2020; pp. 47–76. [Google Scholar]
- Nutan, B.; Chandel, A.K.S.; Jewrajka, S.K. Liquid prepolymer-based in situ formation of degradable poly (ethylene glycol)-linked-poly (caprolactone)-linked-poly (2-dimethylaminoethyl) methacrylate amphiphilic conetwork gels showing polarity driven gelation and bioadhesion. ACS Appl. Bio Mater. 2018, 1, 1606–1619. [Google Scholar] [CrossRef]
- Kumar, A.; Nutan, B.; Jewrajka, S.K. Modulation of Properties through Covalent Bond Induced Formation of Strong Ion Pairing between Polyelectrolytes in Injectable Conetwork Hydrogels. ACS Appl. Bio Mater. 2021, 4, 3374–3387. [Google Scholar] [CrossRef]
- Kumar, A.; Biswas, A.; Nutan, B.; Yadav, A.; Jewrajka, S.K. Structural Regulation at Poly (ethylene glycol) Termini Facilitates the Formation of Injectable Hydrogels with Modulated Degradation and Release of Biomacromolecules. ACS Appl. Polym. Mater. 2022, 4, 5532–5545. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Oki, Y.; Da, X.; Chandel, A.K.S.; Ohta, S.; Ito, T. Injectable bottlebrush triblock copolymer hydrogel crosslinked with ferric ions. Polymer 2022, 240, 124519. [Google Scholar] [CrossRef]
- Goldberg, D.S.; Vijayalakshmi, N.; Swaan, P.W.; Ghandehari, H. G3.5 PAMAM dendrimers enhance transepithelial transport of SN38 while minimizing gastrointestinal toxicity. J. Control. Release 2011, 150, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Sadekar, S.; Thiagarajan, G.; Bartlett, K.; Hubbard, D.; Ray, A.; McGill, L.D.; Ghandehari, H. Poly(amido amine) dendrimers as absorption enhancers for oral delivery of camptothecin. Int. J. Pharm. 2013, 456, 175–185. [Google Scholar] [CrossRef]
- Liu, X.; Huang, H.; Wang, J.; Wang, C.; Wang, M.; Zhang, B.; Pan, C. Dendrimers-delivered short hairpin RNA targeting hTERT inhibits oral cancer cell growth in vitro and in vivo. Biochem. Pharmacol. 2011, 82, 17–23. [Google Scholar] [CrossRef]
- Yoncheva, K.; Calleja, P.; Agüeros, M.; Petrov, P.; Miladinova, I.; Tsvetanov, C.; Irache, J.M. Stabilized micelles as delivery vehicles for paclitaxel. Int. J. Pharm. 2012, 436, 258–264. [Google Scholar] [CrossRef]
- Mo, R.; Jin, X.; Li, N.; Ju, C.; Sun, M.; Zhang, C.; Ping, Q. The mechanism of enhancement on oral absorption of paclitaxel by N-octyl-O-sulfate chitosan micelles. Biomaterials 2011, 32, 4609–4620. [Google Scholar] [CrossRef]
- Bachar, M.; Mandelbaum, A.; Portnaya, I.; Perlstein, H.; Even-Chen, S.; Barenholz, Y.; Danino, D. Development and characterization of a novel drug nanocarrier for oral delivery, based on self-assembled β-casein micelles. J. Control. Release 2012, 160, 164–171. [Google Scholar] [CrossRef]
- Shapira, A.; Davidson, I.; Avni, N.; Assaraf, Y.G.; Livney, Y.D. β-Casein nanoparticle-based oral drug delivery system for potential treatment of gastric carcinoma: Stability, target-activated release and cytotoxicity. Eur. J. Pharm. Biopharm. 2012, 80, 298–305. [Google Scholar] [CrossRef]
- Duhem, N.; Rolland, J.; Riva, R.; Guillet, P.; Schumers, J.-M.; Jérome, C.; Gohy, J.-F.; Préat, V. Tocol modified glycol chitosan for the oral delivery of poorly soluble drugs. Int. J. Pharm. 2012, 423, 452–460. [Google Scholar] [CrossRef]
- Dahmani, F.Z.; Yang, H.; Zhou, J.; Yao, J.; Zhang, T.; Zhang, Q. Enhanced oral bioavailability of paclitaxel in pluronic/LHR mixed polymeric micelles: Preparation, in vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2012, 47, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Y.; Li, Q.H.; Li, Y.P.; Guo, L.; Li, Z.L.; Gong, Y.C. Pluronic P85/poly (lactic acid) vesicles as novel carrier for oral insulin delivery. Colloids Surf. B Biointerfaces 2013, 111, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Uechi, S.; Takara, K.; Asikin, Y.; Wada, K. Evaluation of an oral carrier system in rats: Bioavailability and antioxidant properties of liposome-encapsulated curcumin. J. Agric. Food Chem. 2009, 57, 9141–9146. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Chen, D.; Ren, L.; Zhao, X.; Qin, J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J. Control. Release 2006, 114, 53–59. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, W.; Lv, P.; Wang, L.; Ma, G. Preparation and evaluation of alginate–chitosan microspheres for oral delivery of insulin. Eur. J. Pharm. Biopharm. 2011, 77, 11–19. [Google Scholar] [CrossRef]
- Italia, J.; Bhatt, D.; Bhardwaj, V.; Tikoo, K.; Kumar, M.R. PLGA nanoparticles for oral delivery of cyclosporine: Nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral®. J. Control. Release 2007, 119, 197–206. [Google Scholar] [CrossRef]
- Juère, E.; Florek, J.; Bouchoucha, M.; Jambhrunkar, S.; Wong, K.Y.; Popat, A.; Kleitz, F. In Vitro Dissolution, Cellular Membrane Permeability, and Anti-Inflammatory Response of Resveratrol-Encapsulated Mesoporous Silica Nanoparticles. Mol. Pharm. 2017, 14, 4431–4441. [Google Scholar] [CrossRef]
- Han, L.; Tang, C.; Yin, C. Oral delivery of shRNA and siRNA via multifunctional polymeric nanoparticles for synergistic cancer therapy. Biomaterials 2014, 35, 4589–4600. [Google Scholar] [CrossRef]
- Maitra, A.; Feldmann, G.; Bisht, S. Water-Dispersible Oral, Parenteral, and Topical Formulations for Poorly Water Soluble Drugs Using Smart Polymeric Nanoparticles. U.S. Patent US8715741B2, 6 May 2014. [Google Scholar]
- Moulari, B.; Béduneau, A.; Pellequer, Y.; Lamprecht, A. Lectin-decorated nanoparticles enhance binding to the inflamed tissue in experimental colitis. J. Control. Release 2014, 188, 9–17. [Google Scholar] [CrossRef]
- Kaufman, A. Rawls’s Egalitarianism; Cambridge University Press: Cambridge, UK, 2018. [Google Scholar]
- Sung, H.-W.; Sonaje, K.; Tu, H. Pharmaceutical Composition of Nanoparticles for Protein Drug Delivery. U.S. Patent US8153153B1, 10 April 2012. [Google Scholar]
- Popescu, C.; Onyuksel, H. Biodegradable Nanoparticles Incorporating Highly Hydrophilic Positively Charged Drugs. U.S. Patent US20040247683A1, 9 December 2004. [Google Scholar]
- Pai, C.M.; Min, M.H.; Hwang, J.S.; Cho, K.M. Nanoparticle Compositions of Water-Soluble Drugs for Oral Administration and Preparation Methods Thereof. U.S. Patent US7674767B2, 9 March 2010. [Google Scholar]
- Thaxton, C.S.; Gordon, L.I.; Mutharasan, R.K.; Grun, C.N.; Foit, L. Lipophilic Nanoparticles for Drug Delivery. U.S. Patent US10568898, 25 February 2020. [Google Scholar]
- Perumal, O.; Alqahtani, M.S.A. Novel Core-Shell Nanoparticles for Oral Drug Delivery. U.S. Patent US20150150822A1, 4 June 2015. [Google Scholar]
- Pena, A.I.V.; Luque, S.S.; Fernandez, M.J.A. Chitosan and Heparin Nanoparticles. U.S. Patent US20080317864A1, 25 December 2008. [Google Scholar]
- Zhang, L.; Ling, L.; Zhou, L.Y.; Wu, Z.M.; Guo, X.D.; Jiang, W.; Luo, Q.; Qian, Y. pH-Sensitive Nanoparticles for Oral Insulin Delivery. U.S. Patent US8859004B2, 14 October 2014. [Google Scholar]
- Alqahtani, M.S.A.; Alqahtani, A.S.A.; Baji, R.S.S. Method of Synthesizing Lignin-Based Nanocompositions. U.S. Patent US10420731B1, 24 September 2019. [Google Scholar]
- Lee, S.J.; Kim, Y.H.; Lee, S.H.; Kim, K.S. Oxaliplatin Nanoparticles and Method for Preparing Same. U.S. Patent US20120177728A1, 19 July 2016. [Google Scholar]
- Bonnafous, D.; Cave, G.; Dembri, A.; Lebel-Binay, S.; Ponchel, G.; Soma, E. Oral Formulations of Chemotherapeutic Agents. U.S. Patent US20110207685A1, 25 August 2011. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trade Name | Treatment | Drug (s) | Excipient (s) | Manufacturer (Year/Method) | References |
|---|---|---|---|---|---|
| ISOPTIN-SRE | Antihypertensive | Verapamil | HPC/HPMC | Abbott Laboratories, Chicago, IL, USA (1981/melt extrusion) | [2,16,17,18,19] |
| Cesamet | Anti-emetic, analgesic | Nabilone | PVP | Valeant Pharmaceuticals, Quebec, Canada (1985/melt extrusion) | [2,16,17,18,19] |
| Nivadil | Anti-hypertensive, major cerebral artery occlusion | Nilvadipine | HPMC | Fujisawa Pharmaceuticals Co., Ltd., Tokyo, Japan (1989/not available) | [2,16,17,18,19] |
| Sporanox | Antifungal | Itraconazole | HPMC | Janssen Pharmaceuticals Beerse, Belgium (1992/spray layering) | [2,16,17,18,19] |
| PROGRAF | Immunosuppressant | Tacrolimus | HPMC | Astellas Pharma Northbrook, IL, USA, Inc. (1994/spray drying) | [2,16,17,18,19] |
| REZULIN | Antidiabetic | Troglitazone | PVP | Pfizer/Parke-Davis, Detroid, MI, United States (1997/melt extrusion) | [2,16,17,19] |
| Afeditab | Anti-hypertensive | Nifedipine | PVP or poloxamer | Elan/Watson (2000/melt or absorb) | [17,19] |
| GRIS-PEG | Antifungal | Griseofulvin | PEG | Pedinol Pharmacal Inc., Novartis, Bridgewater, NJ, USA (2003/melt extrusion) | [2,16,17,19] |
| Nimotop | Anti-hypertensive | Nimodipine | PEG | Bayer Leverkusen, Germany (2006/spray drying) | [17] |
| KALETRA | HIV | Lopinavir, Ritonavir | PVP-VA | AbbVie Inc. Chicago, IL, USA (2007/melt extrusion) | [2,16,17,18,19] |
| Fenoglide | Anti-cholesterol | Fenofibrate | PEG | Veloxis Pharmaceuticals, Cary, NC, USA (2007/spray melt) | [17] |
| INETELENCE | HIV | Etravirine | HPMC | Tibotec, Janssen, Titusville, NJ, USA (2008/spray drying) | [2,16,17,18] |
| NORVIR | HIV | Ritonovir | PVP-VA | AbbVie Inc., Chicago, IL, USA (2010/melt extrusion) | [2,17,18,19] |
| ONMEL | Antifungal | Itraconazole | PVP-VA or HPMC | GlaxoSmithKline, Brentford, UK, Stiefel (2010/melt extrusion) | [17,18,19] |
| CERTICAN and ZORTRESS | Immunosuppressant | Everolimus | HPMC | Novartis Pharmaceuticals Basel, Switzerland (2010/spray drying) | [2,16,17,18,19] |
| INCIVEK | Antiviral; hepatitis C | Telaprevir | HPMCAS | Vertex Pharmaceuticals, Boston, MA, USA, Janssen (2011/spray drying) | [2,17,18,19] |
| ZELBORAF | Melanoma skin cancer | Vemurafenib | HPMCAS | Roche Basel, Switzerland (2011/co-precipitation) | [2,17,18,19] |
| KALYDECO | Cystic fibrosis | Ivacaftor | HPMCAS | Vertex Pharmaceuticals Boston, MA, USA(2012/spray drying) | [17,18,19] |
| NOXAFIL | antifungal | Posaconazole | HPMCAS | Merck, Kenilworth, NJ, USA (2013/melt extrusion) | [17,18] |
| Type | Advantages | Limitations | References | |
|---|---|---|---|---|
| Crystal Engineering | Metastatic polymorph Co-crystal formation | Minimum amounts of surfactants and polymers are required for stabilization. High drug loading and high-energy systems are beneficial in drug dissolution. | Challenges in drug/polymer miscibility and excipient compatibility for a chosen drug. Physical instability upon storage. | [32,33] |
| Chemical Modification | Pro-drug formation | Improved drug solubility, lipophilicity, transported-mediated absorption. Has the potential to achieve site-specific delivery. | Limitations in producing screening and development. Associated with a high possibility for the formation of degradation by-products and lack of chemical stability. Disruption of solid-state crystallinity and polymorphism. | [34,35] |
| Salt formation | The most commonly applied technique to increase solubility and the preferred approach for the development of liquid formulation. Enhances the dissolution rate by increasing the apparent intrinsic solubility of the drug. Ease of synthesis and low cost of raw material. | Restricted to weakly basic or acidic drugs; inappropriate for neutral-digested substances. After oral delivery, the medication is transformed back into either its free acid or basic form. Limitations in salt screening and the selection of optimal salt forms. | [36,37] | |
| Particle size reduction | Micronization and nanosized drugs, e.g., NanoCrystal, DissoCubes | Easy to scale up and time efficient. Reduced drug degradation because the drug is in the crystalline solid state. Feasibility of formulation of a drug under different pharmaceutical dosage forms. | Physicochemical-related stability issues such as aggregation or a change in the solid state of the drug. The excess use of excipients as stabilizers may change the drug’s bioavailability, and pharmacological activity. Bulking care is essential, particularly during handling and transport. | [2,38,39] |
| Amorphization | Solid dispersion | Provides extra stability and protection of the drug during formulation. Enhanced solubility and dissolution rate compared with traditional crystal habit modification; it also retards agglomeration/crystallization of drug molecules due to its molecular level dispersion and steric hindrance interactions within the polymeric matrices. | Drugs that are high-energy amorphous tend to recrystallize and change into low-energy crystalline forms. The miscibility between the selected drug and polymeric matrices is required. Limited stability is a known drawback. | [40,41,42] |
| Solvent Composition | pH adjustment | The simple and powerful strategy for solubility adjustment of ionizable drugs. The level of ionization of the drug candidates enables full solvation of the target medication dose. This method works equally well with drug salts or the corresponding free basic or acid medicines. | The long-term effect on the drug stability. The distortion on physiological pH. The precipitation tendencies and incompatibility upon dilution. | [43,44,45] |
| Co- solvent | Provides the optimum solubility for nonpolar drugs by reducing solvent polarity. The presence of a co-solvent can provide additional solubilization for drug solutions where pH manipulation is insufficient. | The use of co-solvents is limited to relatively few solvents. The risk of precipitation upon dilution. It may alter the pH and strength of the buffers that are contained in a drug formulation. | ||
| Drug carrier systems | Micelles | Its hydrophobic core acts as a reservoir for lipophilic drugs. Ease of chemical modification and can be stimuli responsive. | The disintegration of micelles due to their dilution after oral administration, in vivo instability below the critical micelle concentration. Low drug loading. | [2,46,47] |
| Nanoparticles | Increased solubility of lipophilic drugs, enhanced drug stability, sustained drug delivery, shielding of the drug cargo from enzymatic activity, prolonged retention in the gastrointestinal tract, improved mucoadhesiveness, overcoming multidrug resistance, the potential for targeting specific cells and uptake via M cells. | Challenges in biocompatibility and safety of polymeric carriers. Toxicity is a result of the high tissue accumulation of non-biodegradable NPs. Difficulties in optimizing the process parameters and scaling up the production into a pharmaceutical product. | [48,49,50] | |
| Cyclodextrins | Generally recognized as a safe (GRAS) excipient. Suitable for the generation of supersaturated drug solutions. Enhance both the physical and chemical stability of drugs and their shelf-life. | The requirement for a large amount of cyclodextrin compared to the drug to solubilize the drug. The weak binding and dissociation of complexes upon dilution in the GIT. The intact drug/cd complexes are unable to permeate the lipophilic epithelium membranes, which may result in low bioavailability, especially for BCS class III drugs. | [51,52,53] | |
| Lipid-based formulations (SLN, liposomes, SEDDS) | Non-immunogenic, biocompatible, can stimulate the secretion of bile salts, phospholipids and cholesterol, which form vesicles and micelles that then facilitate drug absorption, scalable and easily manufacturable. | Poor stability and short shelf life. | [54,55,56] |
| Major Solubility Enhancement | ||
|---|---|---|
| Physical Modification | Chemical Modification | Miscellaneous Method |
|
|
|
| Drug | Indications | Inventor Company | Drug Delivery Company | Trade Name |
|---|---|---|---|---|
| Methyl phenidate HCl | CNS stimulant | Novartis Basel, Switzerland | Elan Nanosystems | Ritalin® |
| Morphine sulfate | Psychostimulant drug | King Pharmaceuticals Bristol, UK | Elan Nanosystems | Avinza® |
| Aprepitant | Anti-emetic | Merk & Co. Kenilworth, NJ, USA | Elan Nanosystems | Emend® |
| Tizanidine HCl | Muscle relaxant | Acorda New York, NY, USA | Elan Nanosystems | Zanaflex Capsules® |
| Megestrol | Anti-anorexic | Par Pharmaceutical New York, NY, USA | Elan Nanosystems | Triglide® |
| Fenofibrate | Hypercholesterolemia | ScielePharma Inc., Atlanta, GA, USA | IDD-P Skyepharma | Trilide® |
| Dexmethylphenidate HCl | Attention deficit hyperactivity disorder (ADHD) | Novartis Basel, Switzerland | Elan Nanosystems | Focalin® |
| Fenofibrate | Hypercholesterolemia | Abbott Laboratories Chicago, IL, USA | Abbott Laboratories | Tricor® |
| Rapamycin, sirolimus | Immunosuppressant | Wyeth Madison, NJ, USA | Elan Nanosystems | Rapamune® |
| Company | Formulation Approach Based on Nanotechnology | Description |
|---|---|---|
| American Biosciences (Blauvelt, NY, USA) | Nanoparticle albumin-bound technology | Paclitaxel albumin nanoparticles |
| BioSante Pharmaceuticals (Lincolnshire, IL, USA) | For the enhancement of oral bioavailability of hormones/proteins and vaccines, nanoparticles of calcium phosphate were developed | Calcium phosphate nanoparticle |
| Baxter Pharmaceuticals (Deerfield, IL, USA) | Nanoedge technology: particle size reduction by homogenization, micro-precipitation, lipid emulsion and other dispersed systems. | Nano-lipid emulsion |
| Imbedding (Burlingame, CA, USA) | Silicon membranes were used for implantable drug delivery Membrane pore size (10–100 nm) | Stretchable silicon nanomembrane |
| Technology | Drug Molecule | BCS Class | Trade Name | Formulation | Therapeutic Use |
|---|---|---|---|---|---|
| Nanocrystal (wet media milling) | Rapamycin | II | Rapamune | Tablets | Immunosuppressant |
| Aprepitant | IV | Emend | Capsules | Antiemetic | |
| Finofibrate | II | Tricor | Tablets | Antilipidemic | |
| Megestrol acetate | II | Megace ES | Oral suspension | Hormonal Therapy | |
| High-pressure homogenization | Fenofibrate | II | Triglide | Tablets | Antilipidemic |
| Melt extrusion | Verapamil HCL | I | Isoptin SRE | Tablets | Antihypertensive |
| Nifedipine | II | Adalat SL | Capsules | Antihypertensive | |
| Troglitazone | II | Rezulin | Tablets | Antilipidemic | |
| Melt Adsorption | Nifedipine | II | Afeditab | Tablets | Antihypertensive |
| Melt granulation | Fenofibrate | II | Fenoglide | Tablets | Antilipidemic |
| Tacrolimus | II | LCP- Tacro | Tablets | Immunosuppressant | |
| Spray drying | Intellence | IV | Etravirine | Tablets | Antiviral |
| Itraconazole | II | Sporanox | Capsules | Antifungal | |
| Nilvadipine | II | Nivadil | Capsules | Antihypertensive | |
| Tacrolimus | II | Prograf | Capsules | Immunosuppressant | |
| Lyophilization | Olanzapine | II | Zyprexa | Tablets | Antipsychotic |
| Ondansetron | II | Zofran ODT | Tablets | Antiemetic | |
| Piroxicam | II | Proxalyoc | Tablets | Anti-inflammatory |
| Drug | Therapeutic Area | Product Name | Company |
|---|---|---|---|
| Cyclosporine A | Immunomodulation | Restasis | Allergan (Dublin, Ireland) |
| Prostaglandin-E1 | Vasodilator | Liple | Green Cross (Tokyo, Japan) |
| Diazepam | Sedation | Diazemuls | Braun Melsungen (Melsungen, Germany) |
| Propofol | Anesthesia | Propofol Diprivan | Baxter (Illinois, United States) |
| Dexamethasone Palmitate | Corticosteroid | Limethason | Green Cross (Tokyo, Japan) |
| Perflurodecalinþ Perflurotripropylamine | Analgesia | Fluosol-DA | Green Cross (Tokyo, Japan) |
| Etomidate | Anesthesia | Etomidat | Dumex (Lillerød, Denmark) |
| Vitamins A, D, E and K | Nutrition | Vitalipid | Kabi (Bad Homburg, Germany) |
| Flurbiprofen | Analgesia | Lipfen | Green Cross (Tokyo, Japan) |
| Drug | Lipid Utilized | Biopharmaceutical Application |
|---|---|---|
| 5-Fluoro uracil | Dynasan 114 and Dynasan 118 | Prolonged release in simulated colonic media |
| Ibuprofen | Stearic acid, triluarin and tripalmitin | Stable formulation with low toxicity |
| Apomorphine | Glycerylmonostearate, polyethylene glycol monostearate | Enhanced bioavailability in rats |
| Idarubicin | Emulsifying wax | Delivery of oral proteins |
| Calcitonin | Trimyristin | Improvement of the efficacy of proteins |
| Lopinavir | Campritol 888 ATO | Bioavailability enhanced |
| Clozapine | Trimyristin, tristearin and tripalmitin | Improvement of bioavailability |
| Nimesulide | Glycerylbehanate, glyceryltristearate, palmitostearate | Sustained release of the drug |
| Cyclosporin A | Glycerylmonostearate and glycerylpalmitostearate | Controlled release |
| Progesterone | Monostearin, oleic acid and stearic acid | Potential for oral drug delivery |
| Gonadotropin release hormone | Monostearin | Prolonged release |
| Repaglinide | Glycerylmonostearate and tristearin | Reduced toxicity |
| Excipient | Chemical | Type of Carrier | Comments | References |
|---|---|---|---|---|
| Soybean oil | Triglycerides (long-chain) | Nanoemulsions | Liquid, good biocompatibility, minimal physiological impact, weak solubilizing capacity | [125,126,127] |
| Olive oil | Triglycerides (long-chain) | Nanoemulsions | Liquid, healthy, high monounsaturated fatty acids, and simple to emulsify | [126,128,129,130,131] |
| Hemp oil | Medium/long-chain triglycerides blended with low-molecular weight lipids | Nanoemulsions | The liquid contains tocopherols, tocotrienols, phyrosterols, phospholipids, and other important fatty acids, good hydrophilicity, and self-emulsifiability. | [132,133] |
| Caprylic/capric triglycerides | Triglycerides (medium-chain) | Nanoemulsions | Liquid, solubilizing capacity, compatible with other lipids, easy to emulsify. | [134,135,136,137,138,139] |
| Captex® Series | Triglycerides (medium-chain) | Nanoemulsions | Liquid, fine solubilizing and emulsifying capacities, miscible with other lipids | [140,141,142] |
| Capmul MCM | Mono/diglycerides (medium-chain) | Nanoemulsions | Liquid, an excellent solvent powder for many organic compounds, can use as an emulsifier. | [143,144,145,146] |
| Capmul MCM C8 | Glycerol monocaprylate | Nanoemulsions | Liquids, property similar to that of Capmul MCM. | [147,148,149] |
| MaisineTM 35-1 | Glycerol monolinoleate | SEDDS | Liquid, solubilizer, bioavailability enhancer, oil phase in SEDDS | [150,151,152,153] |
| PeceolTM | Glyceryl monolete | SEDDS; NLCs; Cubosomes | Liquid, lipid dispersion agent, oil-soluble surfactant, moisturizer | [154,155,156] |
| Lauroglycol® 90 | Propylene glycol monolaurate | Nanoemulsions; SEDDS; NLCs | Liquid, water-insoluble surfactant of SEDDS, solubilizer, bioavailability enhancer, skin penetration solubilizer enhancer. | ([157,158,159] |
| CapryolTM series | Propylene glycol monocaprylate | Nanoemulsions; SEDDS; NLCs | Liquid, properties similar to that of Lauraglycol® 90 | [160,161,162] |
| Labrafil M 1944 CS | Oleoyl polyoxyl-6 glycerides | Nanoemulsions; SEDDS; NLCs | Liquid, water dispersible surfactant, able to self-emulsify good miscibility with other lipids, bioavailability enhancer, solubilizer, co-emulsifier. | [163,164,165] |
| Lecithin | Phosphatidylcholine blended with a small amount of other lipid components | Liposomes, phytosomes, lipid nanoparticles | Semi-solid, an amphiphilic lipid, used as a vesicle-forming material, solubilizing, emulsifying and stabilizing agents. | [166,167,168,169,170] |
| Gelacire® series | Lipid blends consisting of mono-, di-, or triglycerides and fatty acid macrogolglycerides | SEDDS, SLNs, NLCs | Semi-solid, non-ionic water soluble surfactant for solid/semi-solid dispersions and SEDDS, bioavailability enhancer, micelle-forming material, solubilizing and wetting agent | [160,171,172] |
| Monostearin | Glyceryl monostearate | SLNs, NLCs | Solid, lipid matrix for SLNs and NLCs; thickening, solidifying and control release adjusting agent. | [149,173] |
| Precirol® ATO 5 | Glyceryl distearate | SLNs; NLCs | Solid, lipid matrix for SLNs and NLCs, hydrophobicity and melting point greater than glyceryl monostearate. | [174,175] |
| Compritol® 888 ATO | Glyceryl behenate | SLNs, NLCs solid lipid dispersions | Solid, high-melting point lipid, used for the preparation of SLNs and NLCs, lipid matrix for sustained release, used as atomized powders. | [176,177,178] |
| Trilaurin | Glyceryl trilaurate | SLNs, NLCs | Solid, lipid matrix for SLNs and NLCs, sustained release material, thickening agent. | [179,180,181] |
| Cetyl palmitate | Palmityl palmitate | SLNs, NLCs | A solid, wax-like substance, used for preparation of SLNs and NLCs | [182,183] |
| Tripalmitin | Glyceryl tripalmitate | SLNs; NLCs | Solid, lipid matrix of SLNs and NLCs, skin-conducting agent. | [184,185] |
| Nano-System | Composition | Drug Molecule Size (nm) | Size (nm) | Cell Line/Animal Model | Disease or Targeted Organ | References |
|---|---|---|---|---|---|---|
| Dendrimers | G3.5 PAMAM | SN38 | - | Caco-2 cells and HT-29/female CD-1 mice | Colorectal cancer metastases | [196] |
| Ethylene diamine and methyl acrylate | SN38 camptothecin | 13 | CD-1 mice | Oral chemotherapy of hepatic colorectal cancer metastases | [197] | |
| PAMAM | Short hairpin RNA | 107–315 | Tca8113 cells/BALB/c nude mice | Oral cancer therapy | [198] | |
| Micelles | Polyethylene oxide-polypropylene oxide-polyethylene oxide (PEO-PPO-PEO) | Paclitaxel | 180 | Female C57BL/6J mice | Oral cancer therapy | [199] |
| N-octyl-O-sulfate chitosan (NOSC) | Paclitaxel | Caco-2/SD rats | Improved oral bioavailability | [200] | ||
| Bovine-casein | Celecoxib, Paclitaxel | 20 | Human N-87 gastric cancer cells | Rheumatoid arthritis, osteoarthritis, and gastric carcinoma | [201,202] | |
| Tocopherol succinate glycol chitosan conjugates | Ketoconazole | 101 | Caco-2 cell monolayer | Improved oral bioavailability | [203] | |
| Mixed micelles | Pluronic copolymers and LHR conjugate | Paclitaxel | 140 | MCF-7 cells | Oral anticancer delivery system | [204] |
| Vesicles | PLA-P85-PLA | Insulin | 178 | OVCAR-3 cells/diabetic mice | Oral insulin delivery | [205] |
| Liposomes | Lecithins | Curcumin | 263 | Sprague-Dawley (SD) rats | Improved oral bioavailability | [206] |
| SLN | Iyceryl monostearate (GMS) | Vinpocetine | 70–200 | Male Wistar rats | Improved oral bioavailability | [207] |
| Polymeric microspheres | Chitosan and alginate | Insulin | 5–7 µm | Male SD rats | Diabetes mellitus | [208] |
| Polymeric nanoparticles | PLGA | Cyclosporine | 143 nm | Male SD rats | Improved oral bioavailability | [209] |
| Silica | Resveratol | 90 nm | Caco-2 cell monolayer | Enhanced the solubility, permeability and anti- inflammatory activity of resveratrol encapsulated in NPs | [210] | |
| Multifunctional polymeric nanoparticles | Galactose-modified trimethyl chitosan-cysteine conjugates with various galactose grafting densities | shRNA and siRNA | 130–160 nm | Caco-2 cells/tumour-bearing mice | Targeted treatment of hepatoma | [211] |
| Mannose-modified trimethyl chitosan-cysteine (MTC) conjugates | Tumor necrosis factor-α (TNF-α) siRNA | 152.9 nm | Caco-2 cells, RAW 264.7 (monocyte/macrophage-like cells)/acute hepatic injury-induced mice | Treatment of systemic inflammatory conditions | [212] | |
| Lectin-conjugated PLGA-NPs | Betamethasone | 475 nm | TNBS- induced colitis mice | Treatments of ulcerative colitis and inflammatory bowel disease | [213] |
| Patent Number | Assignee | Invention | References |
|---|---|---|---|
| WO2008073558A2 | Johns Hopkins University, USA | The invention provided new orally bioavailable smart NPs for the delivery of poorly soluble drugs, showing improved pharmacokinetics and bioavailability. | [212] |
| WO2015067751A1 | NanoSphere Health Sciences Inc., USA | Investigation disclosed the composition and development method for nutraceuticals encapsulated with phospholipid-based NPs by the emulsification method. | [214] |
| US20120003306A1 | NancMega Medical Co., USA | The report disclosed a protein/peptide delivery system composed of chitosan and poly-y-glutamic acid (y-PGA). The NPs were suggested to enhance the epithelial permeability and thus are efficient for oral drug delivery. | [215] |
| WO2004098564A2 | University of Illinois, USA | Reported the development of biodegradable NPs containing streptomycin with high loading efficiency of 50% or higher for tuberculosis treatment. The NPs can also contain other aminoglycoside drugs, which are a known substrate for the multidrug efflux mediated by P-glycoprotein (Pgp). | [216] |
| US7674767B2 | Samyang Biopharmaceuticals Co., Korea | The invention described the composition and preparation of orally administrable NPs containing complexes of water-soluble drugs and counter-ion substances. The NPs enhanced drug entrapping and resistance against lipases, thereby increasing drug absorption. | [217] |
| WO2015023797A9 | Northwestern University, USA | The patent disclosed the development and evaluation of drug-loaded nanostructures comprising an inorganic core and a lipid layer shell. The NPs showed potential in the treatment of cancer, vascular diseases and infectious diseases. | [218] |
| WO2014197640A1 | South Dakota State University, USA | Disclosed the composition and preparation method of core-shell NPs. These NPs comprise food-grade proteins along with therapeutic agents suitable for pedatrics. | [219] |
| WO2007042572A1 | Advanced In Vitro Cell Technologies S.A., Spain | The insertion described NPs comprising chitosan and heparin prepared by ionic gelation method. The NPs were stable in gastrointestinal fluids and presented excellent in vivo effectiveness and bioavailability. | [220] |
| CN102120781B | China Pharmaceutical University, China | The invention related to the preparation of oral insulin NPs. The NPs mainly contained N-amino acid chitosan as a carrier and insulin for the treatment of diabetes. The NPs were stable after oral administration with a better effect of reducing blood sugar in vivo. | [221] |
| US10420731B1 | King Saud University, Saudi Arabia | The invention described the synthesis and preparation method of lignin NPs cross-linked and stabilized by citric acid for oral administration. The NPs improved the oral bioavailability of curcumin by increasing curcumin solubility and stability, sustaining its release, enhancing intestinal permeability, and inhibiting Pgp-mediated efflux. | [222] |
| WO2011034394A2 | JW Pharmaceuticals Co., Korea | The invention reported the preparation of oxaliplatin-loaded NPs using supercritical fluid gas technology for oral chemotherapy. | [223] |
| WO2010015688A1 | BioAlliance Pharma Co., USA | The patent disclosed the composition and preparation method of a chemotherapeutic formulation containing polymer and cyclic oligosaccharide capable of complexing and delivering anticancer drugs for effective cancer treatments. | [224] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. https://doi.org/10.3390/biomedicines10092055
Bhalani DV, Nutan B, Kumar A, Singh Chandel AK. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines. 2022; 10(9):2055. https://doi.org/10.3390/biomedicines10092055
Chicago/Turabian StyleBhalani, Dixit V., Bhingaradiya Nutan, Avinash Kumar, and Arvind K. Singh Chandel. 2022. "Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics" Biomedicines 10, no. 9: 2055. https://doi.org/10.3390/biomedicines10092055