
Therapeutic miR-506-3p Replacement in Pancreatic Carcinoma Leads to Multiple Effects including Autophagy, Apoptosis, Senescence, and Mitochondrial Alterations In Vitro and In Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. miRNA Transfection
2.3. Measurement of Cell Proliferation, Live/Dead Cell Staining, and Hoechst33342 Staining
2.4. RNA Preparation, Reverse Transcription, and qPCR
2.5. Western Blot
2.6. Spheroid Formation
2.7. Determination of Apoptosis, Necrosis, and Autophagy
2.8. Cell Cycle Analyses and Determination of ROS
2.9. Senescence Induction and Measurement of Mitochondrial Potential and Structure
2.10. Confocal Imaging
2.11. Xenograft Mouse Model and Nanoparticle-Based Therapy
2.12. Immunohistochemistry
2.13. Statistical Analyses
3. Results
3.1. Decreased Cell Proliferation and Viability upon Transient miR-506 Transfection
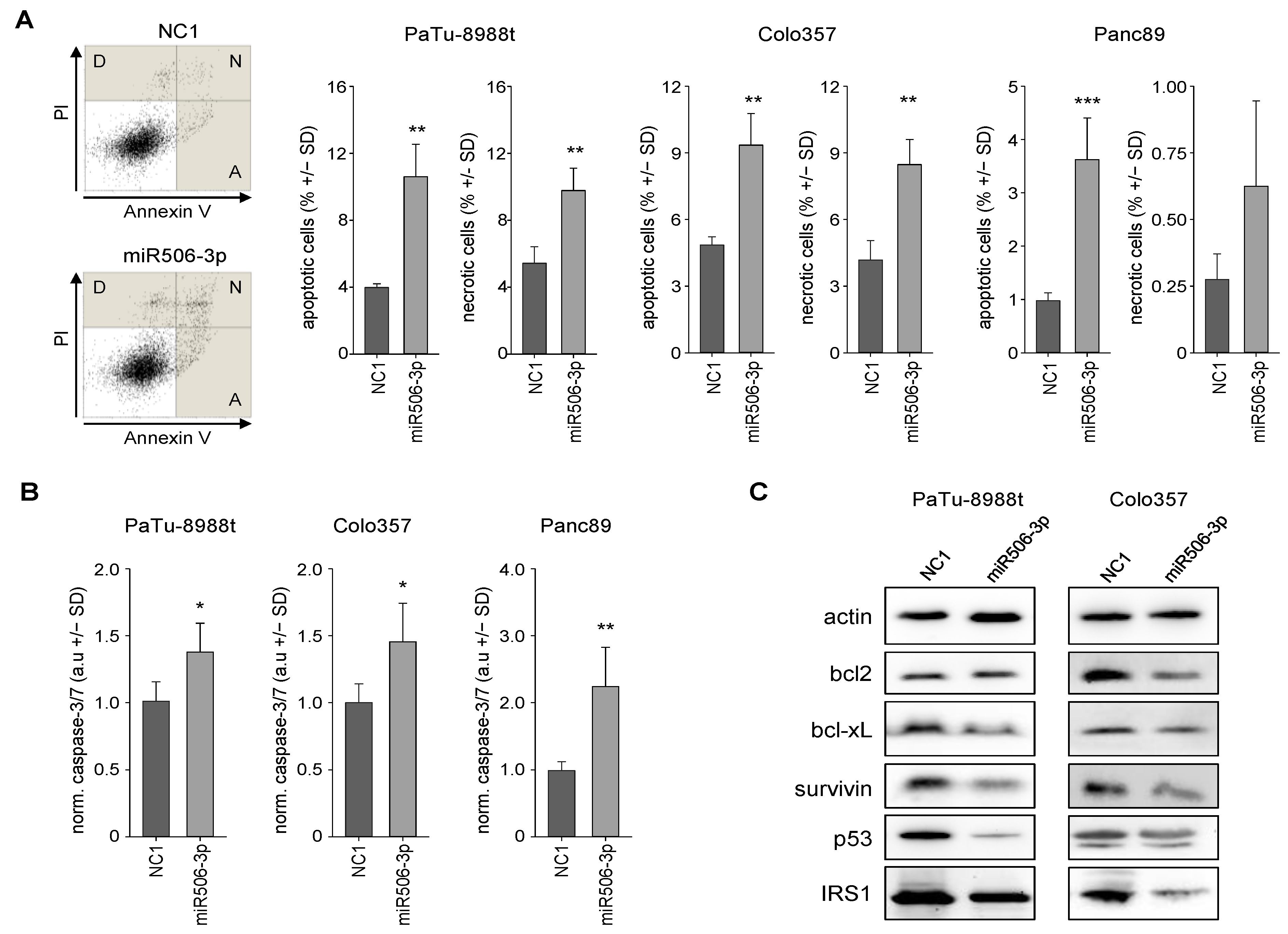
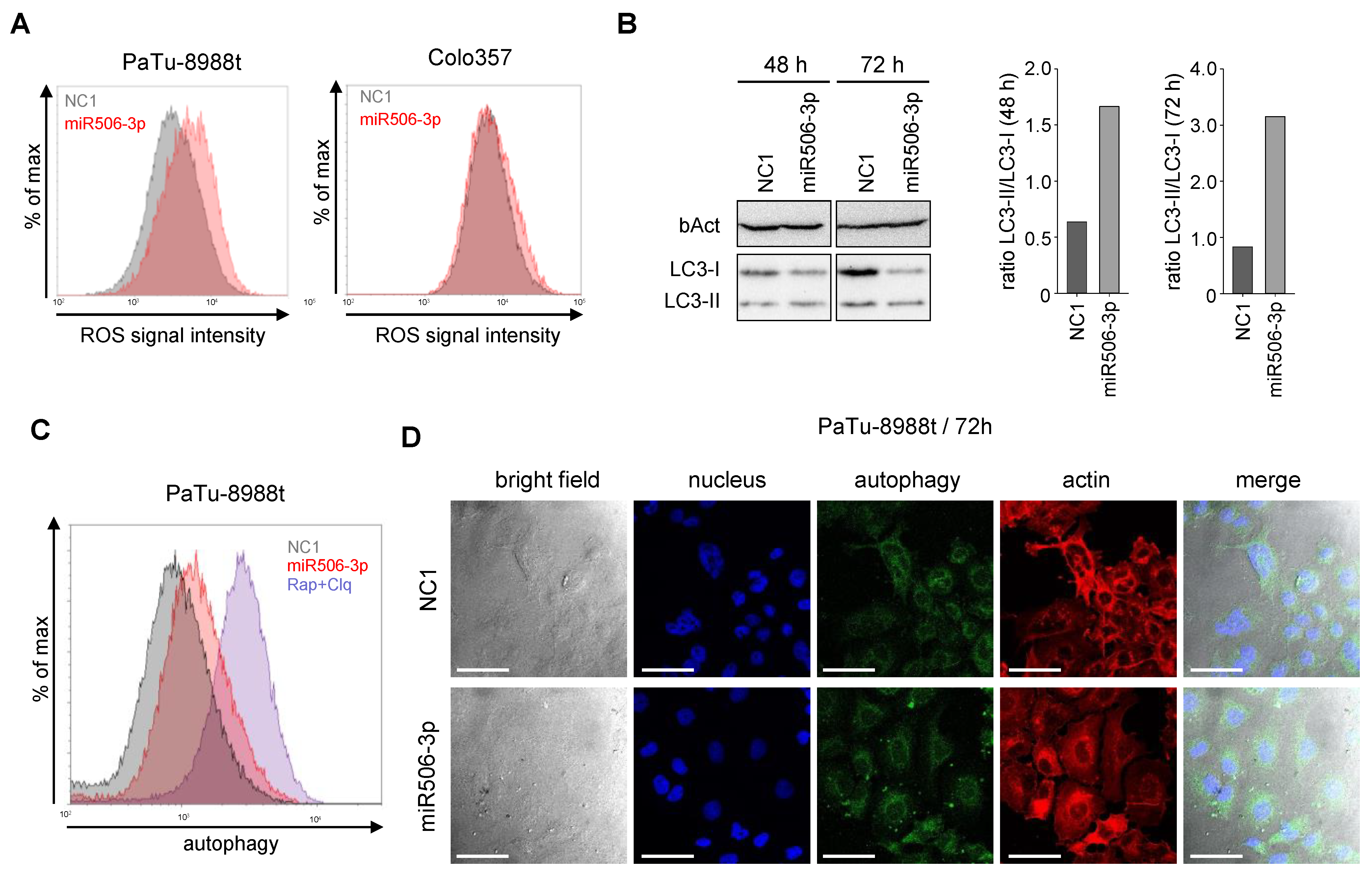
3.2. MiR-506 Induces Cell Death by Apoptosis, Autophagy and Senescence Induction, and Affects Mitochondrial Potential and Structure
3.3. Induction of Cell Cycle Alterations after miRNA-506-3p Replacement
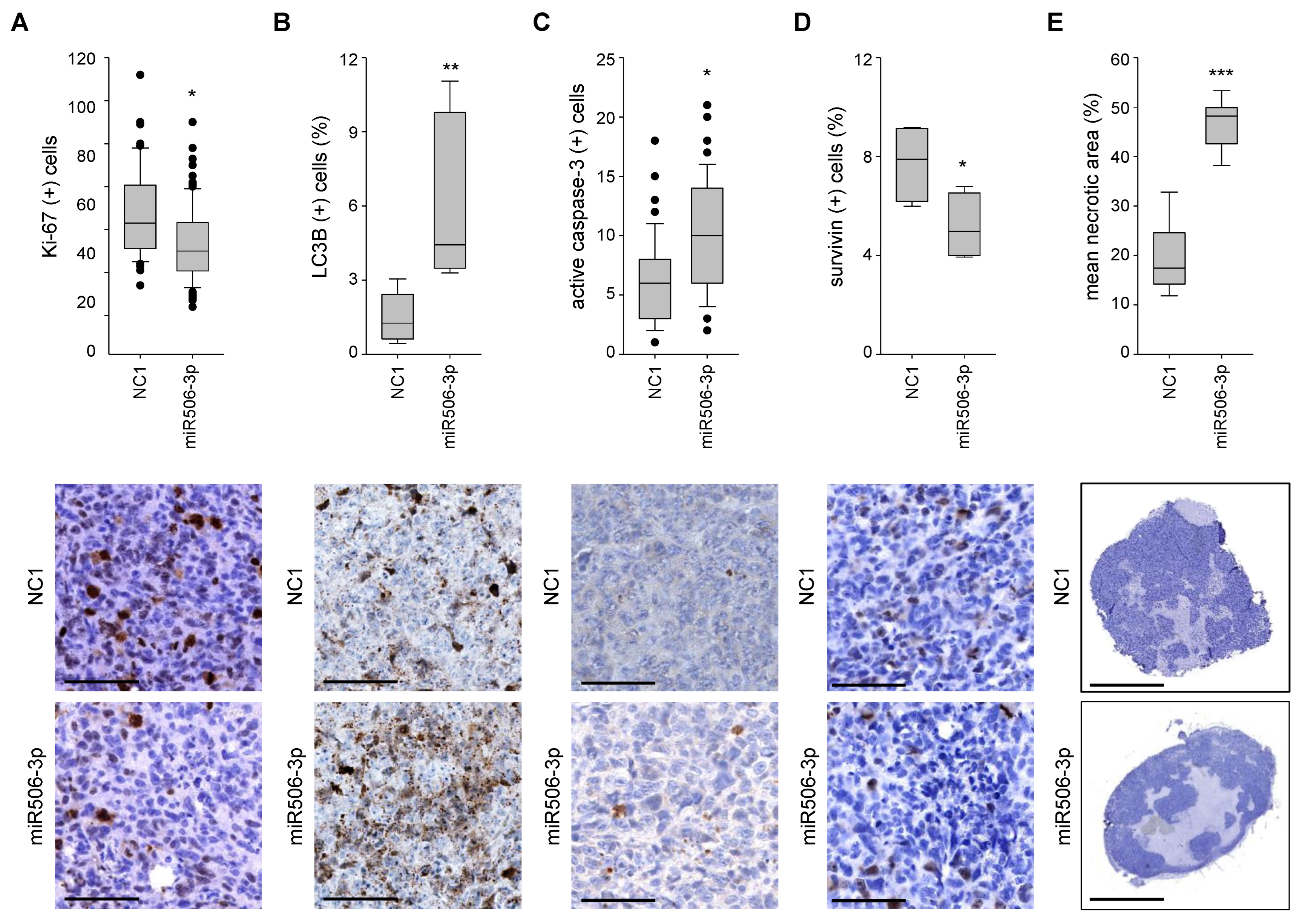
3.4. Nanoparticle-Formulated miRNA-506-3p Mimic Inhibits the Growth of Xenografts
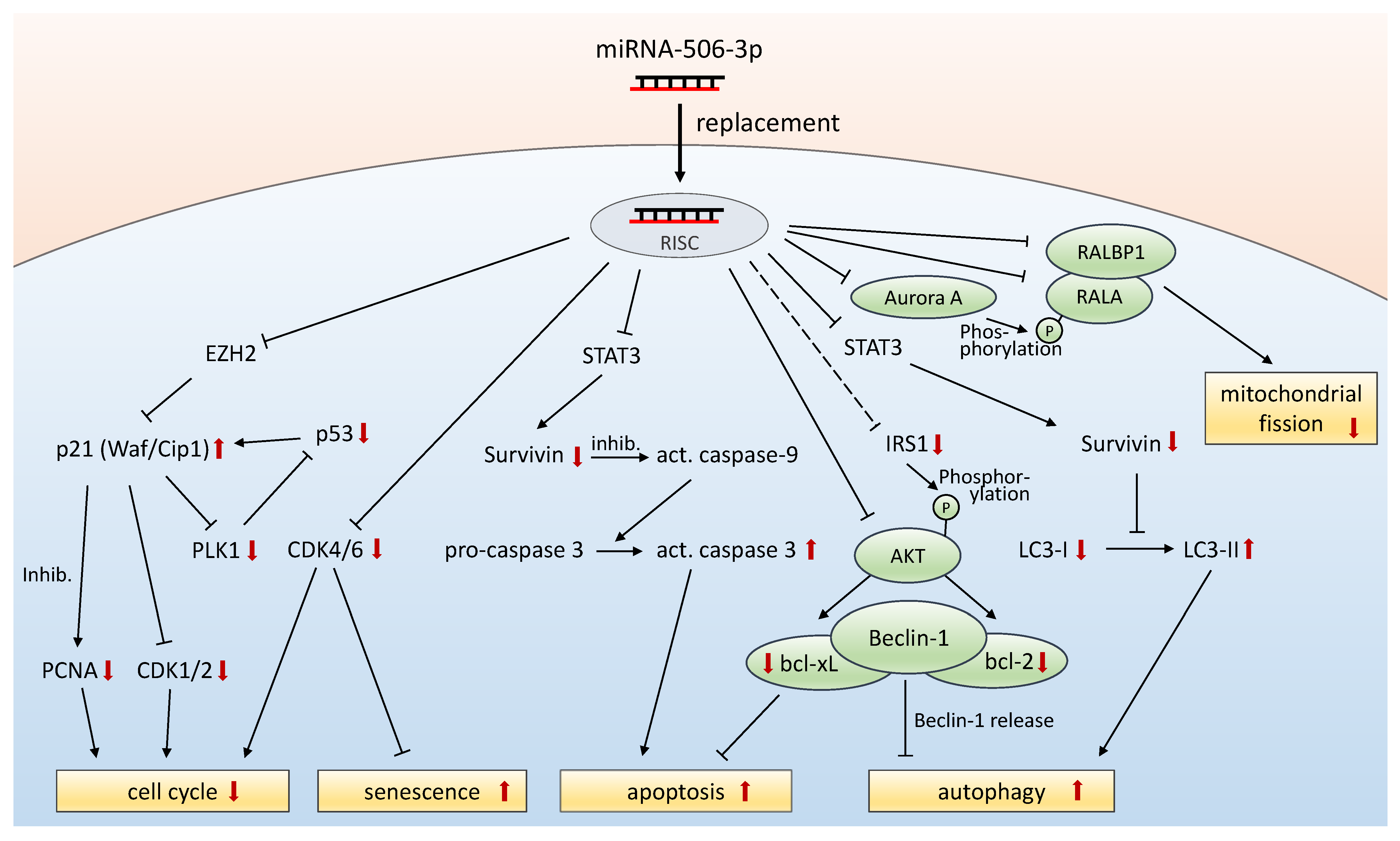
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ju, J.; Ni, B.; Wang, H. The emerging role of miR-506 in cancer. Oncotarget 2016, 7, 62778–62788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streicher, K.L.; Zhu, W.; Lehmann, K.P.; Georgantas, R.W.; Morehouse, C.A.; Brohawn, P.; Carrasco, R.A.; Xiao, Z.; Tice, D.A.; Higgs, B.W.; et al. A novel oncogenic role for the miRNA-506-514 cluster in initiating melanocyte transformation and promoting melanoma growth. Oncogene 2012, 31, 1558–1570. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Sun, Y.; Ji, P.; Li, X.; Cogdell, D.; Yang, D.; Kerrigan, B.C.P.; Shmulevich, I.; Chen, K.; Sood, A.K.; et al. MiR-506 suppresses proliferation and induces senescence by directly targeting the CDK4/6-FOXM1 axis in ovarian cancer. J. Pathol. 2014, 233, 308–318. [Google Scholar] [CrossRef]
- Deng, J.; Lei, W.; Xiang, X.; Zhang, L.; Yu, F.; Chen, J.; Feng, M.; Xiong, J. MicroRNA-506 inhibits gastric cancer proliferation and invasion by directly targeting Yap1. Tumour Biol. 2015, 36, 6823–6831. [Google Scholar] [CrossRef]
- Lv, Z.; Yu, F.; Lv, M.; Li, D.; Cai, H.; Ma, L.; Luo, Q.; Yuan, X. MiR-506 Over-Expression Inhibits Proliferation and Metastasis of Breast Cancer Cells. Med. Sci. Monit. 2015, 21, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Liu, Y.; Wang, K.; Xu, Z. miR-506 regulates breast cancer cell metastasis by targeting IQGAP1. Int. J. Oncol. 2015, 47, 1963–1970. [Google Scholar] [CrossRef]
- Yao, W.-J.; Wang, Y.-L.; Lu, J.-G.; Guo, L.; Qi, B.; Chen, Z.-J. MicroRNA-506 inhibits esophageal cancer cell proliferation via targeting CREB1. Int. J. Clin. Exp. Pathol. 2015, 8, 10868–10874. [Google Scholar]
- Sun, Y.; Hu, L.; Zheng, H.; Bagnoli, M.; Guo, Y.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Ji, P.; Chen, K.; et al. MiR-506 inhibits multiple targets in the epithelial-to-mesenchymal transition network and is associated with good prognosis in epithelial ovarian cancer. J. Pathol. 2015, 235, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, Z.; Dong, S.; Zhang, J.; Tan, J.; Wang, Y.; Ge, C.; Li, R.; Xue, Y.; Li, M.; et al. miR-506 Inhibits Epithelial-to-Mesenchymal Transition and Angiogenesis in Gastric Cancer. Am. J. Pathol. 2015, 185, 2412–2420. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cai, S.; Zhu, Z.; Jiao, H.; Zheng, X.; Tan, J.; Hu, B.; Huang, Z. MicroRNA-506 participates in pancreatic cancer pathogenesis by targeting PIM3. Mol. Med. Rep. 2015, 12, 5121–5126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wu, H.; Li, W.; Yin, L.; Guo, S.; Xu, X.; Ouyang, Y.; Zhao, Z.; Liu, S.; Tian, Y.; et al. Downregulated miR-506 expression facilitates pancreatic cancer progression and chemoresistance via SPHK1/Akt/NF-κB signaling. Oncogene 2016, 35, 5501–5514. [Google Scholar] [CrossRef]
- Huang, B.; Liu, C.; Wu, Q.; Zhang, J.; Min, Q.; Sheng, T.; Wang, X.; Zou, Y. Long non-coding RNA NEAT1 facilitates pancreatic cancer progression through negative modulation of miR-506-3p. Biochem. Biophys. Res. Commun. 2017, 482, 828–834. [Google Scholar] [CrossRef]
- Han, Y.; Ma, L.; Le Zhao Feng, W.; Zheng, X. Rosmarinic inhibits cell proliferation, invasion and migration via up-regulating miR-506 and suppressing MMP2/16 expression in pancreatic cancer. Biomed. Pharmacother. 2019, 115, 108878. [Google Scholar] [CrossRef]
- Sun, L.; Hu, L.; Cogdell, D.; Lu, L.; Gao, C.; Tian, W.; Zhang, Z.; Kang, Y.; Fleming, J.B.; Zhang, W. MIR506 induces autophagy-related cell death in pancreatic cancer cells by targeting the STAT3 pathway. Autophagy 2017, 13, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Hossian, A.K.M.N.; Mackenzie, G.G.; Mattheolabakis, G. Combination of miR-143 and miR-506 reduces lung and pancreatic cancer cell growth through the downregulation of cyclin-dependent kinases. Oncol. Rep. 2021, 45, 1. [Google Scholar] [CrossRef]
- Cheng, R.-F.; Wang, J.; Zhang, J.-Y.; Sun, L.; Zhao, Y.-R.; Qiu, Z.-Q.; Sun, B.-C.; Sun, Y. MicroRNA-506 is up-regulated in the development of pancreatic ductal adenocarcinoma and is associated with attenuated disease progression. Chin. J. Cancer 2016, 35, 64. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.F.; Weirauch, U.; Thomas, M.; Grünweller, A.; Hartmann, R.K.; Aigner, A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011, 71, 5214–5224. [Google Scholar] [CrossRef] [Green Version]
- Abrego, J.; Gunda, V.; Vernucci, E.; Shukla, S.K.; King, R.J.; Dasgupta, A.; Goode, G.; Murthy, D.; Yu, F.; Singh, P.K. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017, 400, 37–46. [Google Scholar] [CrossRef]
- Feld, F.M.; Nagel, P.D.; Weissinger, S.E.; Welke, C.; Stenzinger, A.; Möller, P.; Lennerz, J.K. GOT1/AST1 expression status as a prognostic biomarker in pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 4516–4526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, P.; Komatsu, M. p62/SQSTM1—Steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werth, S.; Urban-Klein, B.; Dai, L.; Höbel, S.; Grzelinski, M.; Bakowsky, U.; Czubayko, F.; Aigner, A. A low molecular weight fraction of polyethylenimine (PEI) displays increased transfection efficiency of DNA and siRNA in fresh or lyophilized complexes. J. Control. Release 2006, 112, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Bisikirska, B.C.; Adam, S.J.; Alvarez, M.J.; Rajbhandari, P.; Cox, R.; Lefebvre, C.; Wang, K.; Rieckhof, G.E.; Felsher, D.W.; Califano, A. STK38 is a critical upstream regulator of MYC’s oncogenic activity in human B-cell lymphoma. Oncogene 2013, 32, 5283–5291. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cui, M.; Sun, B.-D.; Liu, F.-B.; Zhang, X.-D.; Ye, L.-H. MiR-506 suppresses proliferation of hepatoma cells through targeting YAP mRNA 3’UTR. Acta Pharmacol. Sin. 2014, 35, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Cosgrave, N.; Hill, A.D.K.; Young, L.S. Growth factor-dependent regulation of survivin by c-myc in human breast cancer. J. Mol. Endocrinol. 2006, 37, 377–390. [Google Scholar] [CrossRef]
- Haque, R.; Song, J.; Haque, M.; Lei, F.; Sandhu, P.; Ni, B.; Zheng, S.; Fang, D.; Yang, J.; Song, J. c-Myc-Induced Survivin Is Essential for Promoting the Notch-Dependent T Cell Differentiation from Hematopoietic Stem Cells. Genes 2017, 8, 97. [Google Scholar] [CrossRef]
- Ueno, H.; Kondo, E.; Yamamoto-Honda, R.; Tobe, K.; Nakamoto, T.; Sasaki, K.; Mitani, K.; Furusaka, A.; Tanaka, T.; Tsujimoto, Y.; et al. Association of insulin receptor substrate proteins with Bcl-2 and their effects on its phosphorylation and antiapoptotic function. Mol. Biol. Cell 2000, 11, 735–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Jiang, B.-H. Interplay between Reactive oxygen Species and MicroRNAs in Cancer. Curr. Pharmacol. Rep. 2016, 2, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, M.; Ren, X.; Zhang, X.; Luo, Y.; Wang, G.; Huang, K.; Feng, S.; Bao, X.; He, X.; Liang, P.; et al. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-κB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene 2015, 34, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.-C.; Kuo, C.-W.; Tsai, S.-L.; Cheng, S.M.; Chen, S.-H.; Chan, H.-H.; Lin, C.-H.; Lin, K.-Y.; Li, C.-F.; Kanwar, J.R.; et al. Inhibition of HDAC3- and HDAC6-Promoted Survivin Expression Plays an Important Role in SAHA-Induced Autophagy and Viability Reduction in Breast Cancer Cells. Front. Pharmacol. 2016, 7, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, D.F.; Lim, K.-H.; Brady, D.C.; Pershing, N.L.K.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Choi, S.; Gibson, G.A.; Watkins, S.C.; Bakkenist, C.J.; van Houten, B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 2012, 125, 5745–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemi, A.; Gorji-Bahri, G. MicroRNA: Promising Roles in Cancer Therapy. Curr. Pharm. Biotechnol. 2020, 21, 1186–1203. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microRNA replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic siRNA: State of the art. Signal Transduct. Target Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef] [Green Version]
- Aigner, A. Perspectives, issues and solutions in RNAi therapy: The expected and the less expected. Nanomedicine 2019, 14, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, C.P.; Dwyer, R.M. Nanoparticle-Based Delivery of Tumor Suppressor microRNA for Cancer Therapy. Cells 2020, 9, 521. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borchardt, H.; Kogel, A.; Kalwa, H.; Weirauch, U.; Aigner, A. Therapeutic miR-506-3p Replacement in Pancreatic Carcinoma Leads to Multiple Effects including Autophagy, Apoptosis, Senescence, and Mitochondrial Alterations In Vitro and In Vivo. Biomedicines 2022, 10, 1692. https://doi.org/10.3390/biomedicines10071692
Borchardt H, Kogel A, Kalwa H, Weirauch U, Aigner A. Therapeutic miR-506-3p Replacement in Pancreatic Carcinoma Leads to Multiple Effects including Autophagy, Apoptosis, Senescence, and Mitochondrial Alterations In Vitro and In Vivo. Biomedicines. 2022; 10(7):1692. https://doi.org/10.3390/biomedicines10071692
Chicago/Turabian StyleBorchardt, Hannes, Alexander Kogel, Hermann Kalwa, Ulrike Weirauch, and Achim Aigner. 2022. "Therapeutic miR-506-3p Replacement in Pancreatic Carcinoma Leads to Multiple Effects including Autophagy, Apoptosis, Senescence, and Mitochondrial Alterations In Vitro and In Vivo" Biomedicines 10, no. 7: 1692. https://doi.org/10.3390/biomedicines10071692