
Comprehensive Genetic Analyses of Inherited Peripheral Neuropathies in Japan: Making Early Diagnosis Possible

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
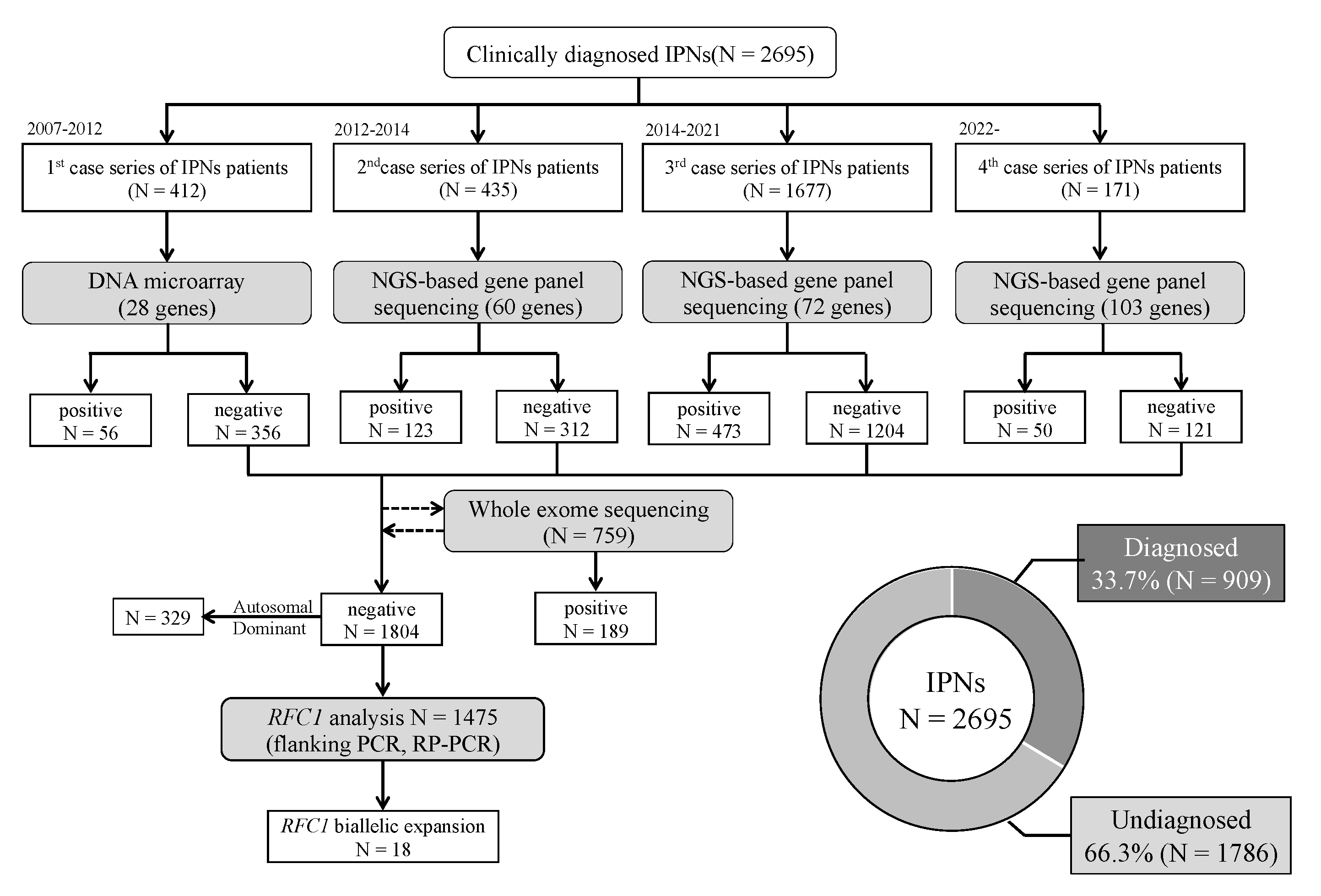
2.1. Patients
2.2. DNA Microarray Screening
2.3. NGS-Based Gene Panel Sequencing
2.4. Whole-Exome Sequencing (WES)
2.5. Data Analysis and Variant Interpretation
2.6. CNV Analysis
2.7. Flanking Polymerase Chain Reaction (PCR) and Repeat-Primed PCR (RP-PCR) of RFC1 Repeat Expansions
3. Results
3.1. Genetic Profile
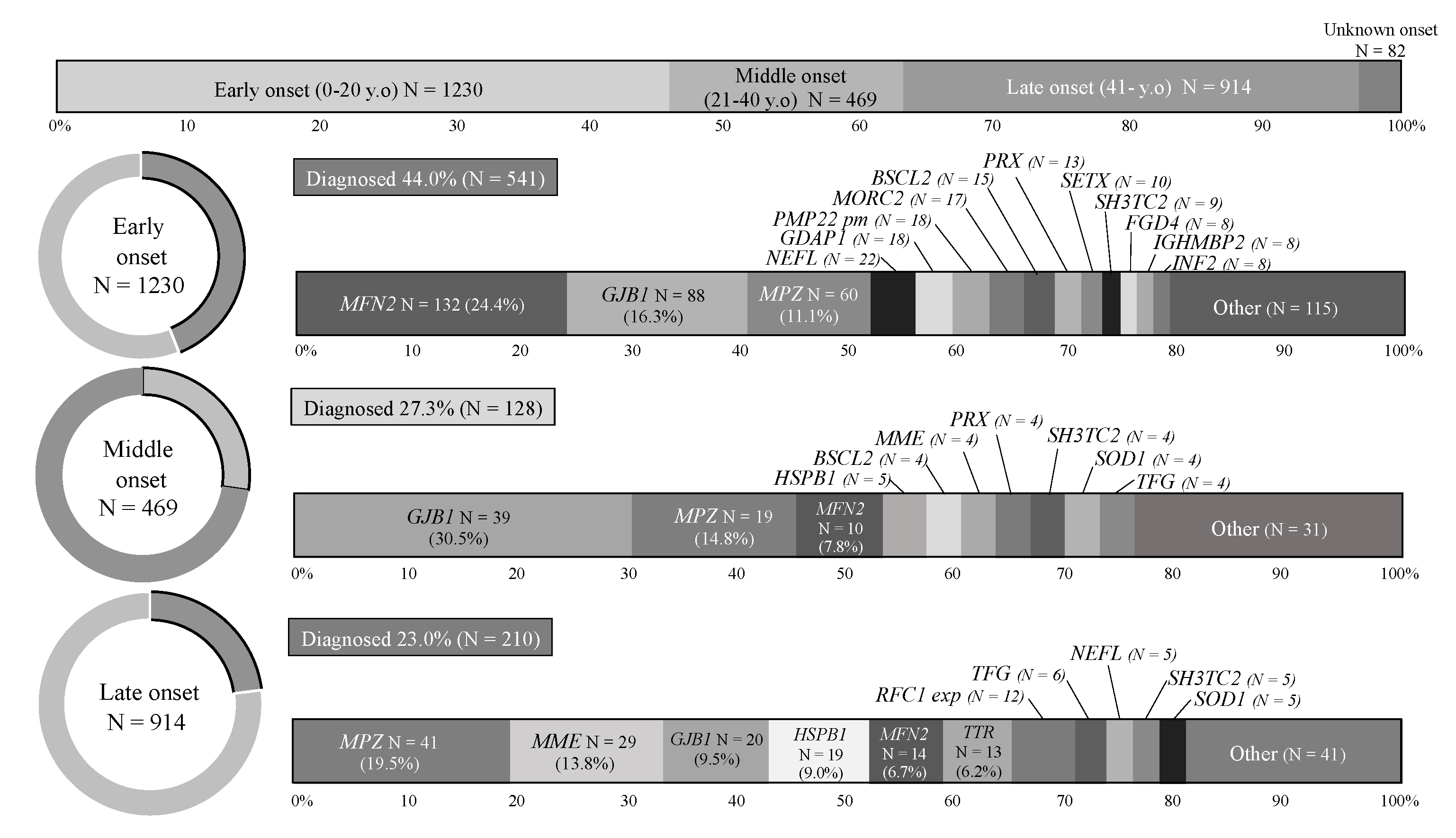
3.2. Analysis by Onset Age
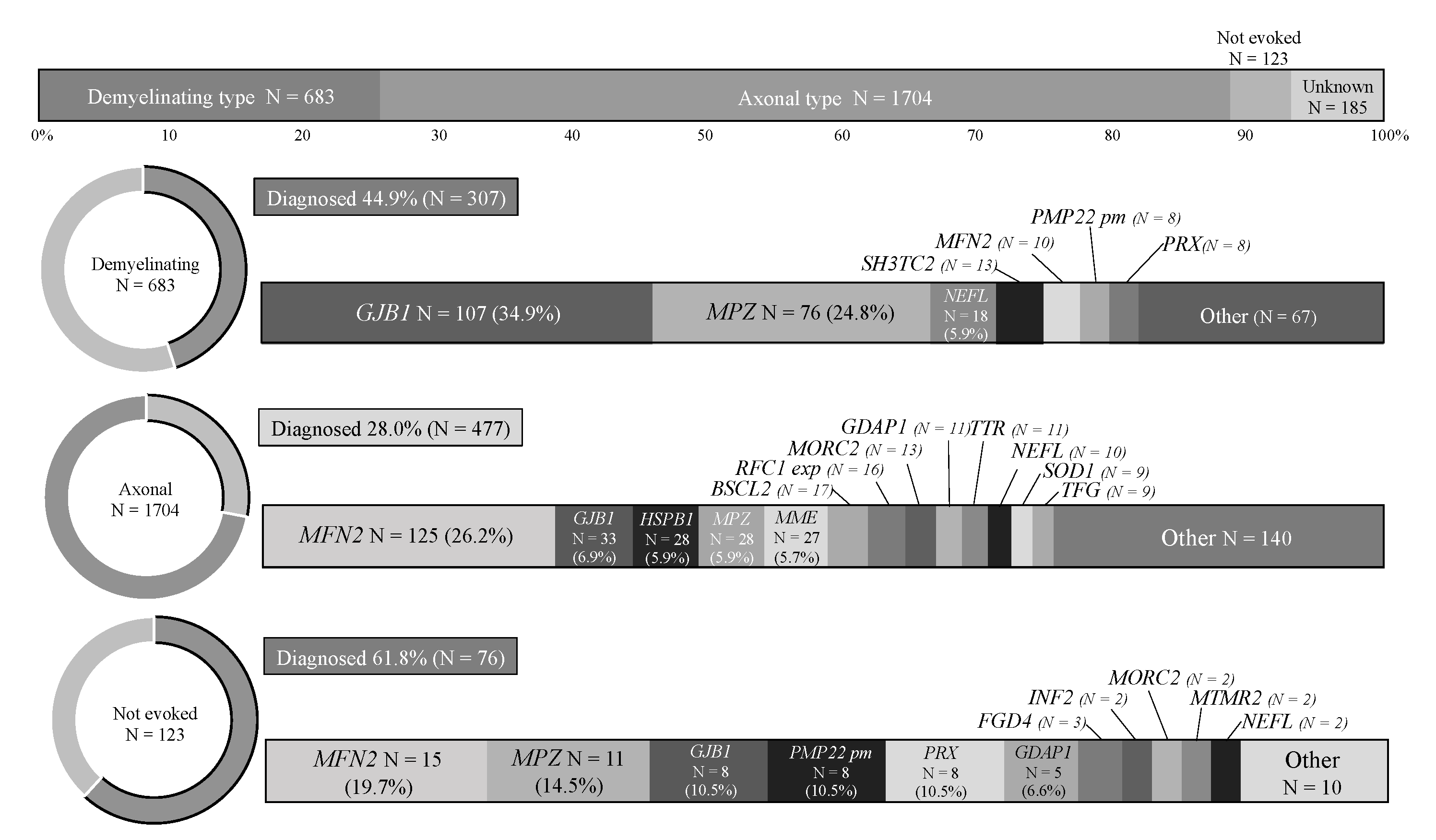
3.3. Analysis by CMT Subtypes
3.4. CNV Analysis
3.5. Bi-Allelic RFC1 Repeat Expansion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stojkovic, T. Hereditary neuropathies: An update. Rev. Neurol. 2016, 172, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Takashima, H. Clinical genetics of Charcot–Marie–Tooth disease. J. Hum. Genet. 2022, 18. epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Derouault, P.; Chauzeix, J.; Rizzo, D.; Miressi, F.; Magdelaine, C.; Bourthoumieu, S.; Durand, K.; Dzugan, H.; Feuillard, J.; Sturtz, F.; et al. CovCopCan: An efficient tool to detect Copy Number Variation from amplicon sequencing data in inherited diseases and cancer. PLoS Comput. Biol. 2020, 16, e1007503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, A.; Simone, R.; Sullivan, R.; Vandrovcova, J.; Tariq, H.; Yau, W.Y.; Humphrey, J.; Jaunmuktane, Z.; Sivakumar, P.; Polke, J.; et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 2019, 51, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Currò, R.; Salvalaggio, A.; Tozza, S.; Gemelli, C.; Dominik, N.; Deforie, V.G.; Magrinelli, F.; Castellani, F.; Vegezzi, E.; Businaro, P.; et al. RFC1 expansions are a common cause of idiopathic sensory neuropathy. Brain 2021, 144, 1542–1550. [Google Scholar] [CrossRef]
- Tagliapietra, M.; Cardellini, D.; Ferrarini, M.; Testi, S.; Ferrari, S.; Monaco, S.; Cavallaro, T.; Fabrizi, G.M. RFC1 AAGGG repeat expansion masquerading as Chronic Idiopathic Axonal Polyneuropathy. J. Neurol. 2021, 268, 4280–4290. [Google Scholar] [CrossRef]
- Reyes-Leiva, D.; Aldecoa, I.; Gelpi, E.; Rojas-García, R. Motor neuron involvement expands the neuropathological phenotype of late-onset ataxia in RFC1 mutation (CANVAS). Brain Pathol. 2022, 9, e13051. [Google Scholar] [CrossRef]
- Hashiguchi, A.; Higuchi, Y.; Nomura, M.; Nakamura, T.; Arata, H.; Yuan, J.; Yoshimura, A.; Okamoto, Y.; Matsuura, E.; Takashima, H. Neurofilament light mutation causes hereditary motor and sensory neuropathy with pyramidal signs. J. Peripher. Nerv. Syst. 2014, 19, 311–316. [Google Scholar] [CrossRef]
- Yoshimura, A.; Yuan, J.-H.; Hashiguchi, A.; Ando, M.; Higuchi, Y.; Nakamura, T.; Okamoto, Y.; Nakagawa, M.; Takashima, H. Genetic profile and onset features of 1005 patients with Charcot–Marie–Tooth disease in Japan. J. Neurol. Neurosurg. Psychiatry 2019, 90, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Higuchi, Y.; Yuan, J.; Yoshimura, A.; Kitao, R.; Morimoto, T.; Taniguchi, T.; Takeuchi, M.; Takei, J.; Hiramatsu, Y.; et al. Novel de novo POLR3B mutations responsible for demyelinating Charcot–Marie–Tooth disease in Japan. Ann. Clin. Transl. Neurol. 2022, 9, 747–755. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E.; Project, N.E.S. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Scriba, C.K.; Beecroft, S.J.; Clayton, J.S.; Cortese, A.; Sullivan, R.; Yau, W.Y.; Dominik, N.; Rodrigues, M.; Walker, E.; Dyer, Z.; et al. A novel RFC1 repeat motif (ACAGG) in two Asia-Pacific CANVAS families. Brain 2020, 143, 2904–2910. [Google Scholar] [CrossRef]
- Abe, A.; Numakura, C.; Kijima, K.; Hayashi, M.; Hashimoto, T.; Hayasaka, K. Molecular diagnosis and clinical onset of Charcot–Marie–Tooth disease in Japan. J. Hum. Genet. 2011, 56, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.H.; Bin Hong, Y.; Hyun, Y.S.; Nam, D.E.; Kwak, G.; Hwang, S.H.; Choi, B.-O.; Chung, K.W. Identification of Genetic Causes of Inherited Peripheral Neuropathies by Targeted Gene Panel Sequencing. Mol. Cells 2016, 39, 382–388. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Dong, H.; Wei, Q.; Li, L.; Yu, H.; Li, J.; Liu, G.; Li, H.; Bai, G.; Ma, H.; et al. Genetic spectrum and clinical profiles in a southeast Chinese cohort of Charcot–Marie–Tooth disease. Clin. Genet. 2019, 96, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Lin, Z.; Liu, L.; Li, X.; Huang, S.; Zhao, H.; Wang, B.; Zeng, S.; Cao, W.; Li, L.; et al. Genotype and phenotype distribution of 435 patients with Charcot–Marie–Tooth disease from central south China. Eur. J. Neurol. 2021, 28, 3774–3783. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Lin, K.P.; Guo, Y.C.; Tsai, Y.S.; Liao, Y.C.; Lee, Y.C. Mutation spectrum of Charcot–Marie–Tooth disease among the Han Chinese in Taiwan. Ann. Clin. Transl. Neurol. 2019, 6, 1090–1101. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.; Lin, K.; Guo, Y.; Tsai, Y.; Liao, Y.; Lee, Y. Charcot–Marie–Tooth disease: Frequency of genetic subtypes and guidelines for genetic testing. J. Neurol. Neurosurg. Psychiatry 2012, 83, 706–710. [Google Scholar] [CrossRef]
- Gess, B.; Schirmacher, A.; Boentert, M.; Young, P. Charcot–Marie–Tooth disease: Frequency of genetic subtypes in a German neuromuscular center population. Neuromuscul. Disord. 2013, 23, 647–651. [Google Scholar] [CrossRef]
- Østern, R.; Fagerheim, T.; Hjellnes, H.; Nygård, B.; Mellgren, S.I.; Nilssen, Ø. Diagnostic laboratory testing for Charcot Marie Tooth disease (CMT): The spectrum of gene defects in Norwegian patients with CMT and its implications for future genetic test strategies. BMC Med. Genet. 2013, 14, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivera, R.; Sevilla, T.; Vílchez, J.J.; Martínez-Rubio, D.; Chumillas, M.J.; Vázquez, J.F.; Muelas, N.; Bataller, L.; Millán, J.M.; Palau, F.; et al. Charcot–Marie–Tooth disease: Genetic and clinical spectrum in a Spanish clinical series. Neurology 2013, 81, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganelli, F.; Tozza, S.; Pisciotta, C.; Bellone, E.; Iodice, R.; Nolano, M.; Geroldi, A.; Capponi, S.; Mandich, P.; Santoro, L. Charcot–Marie–Tooth disease: Frequency of genetic subtypes in a Southern Italy population. J. Peripher. Nerv. Syst. 2014, 19, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.; Russo, M.; Fabrizi, G.M.; Taioli, F.; Ferrarini, M.; Testi, S.; Alfonzo, A.; Aguennouz, M.; Toscano, A.; Vita, G.; et al. Charcot–Marie–Tooth disease: Experience from a large Italian tertiary neuromuscular center. Neurol. Sci. 2020, 41, 1239–1243. [Google Scholar] [CrossRef]
- Vaeth, S.; Christensen, R.; Dunø, M.; Lildballe, D.L.; Thorsen, K.; Vissing, J.; Svenstrup, K.; Hertz, J.M.; Andersen, H.; Jensen, U.B. Genetic analysis of Charcot–Marie–Tooth disease in Denmark and the implementation of a next generation sequencing platform. Eur. J. Med. Genet. 2019, 62, 1–8. [Google Scholar] [CrossRef] [Green Version]
- DiVincenzo, C.; Elzinga, C.D.; Medeiros, A.C.; Karbassi, I.; Jones, J.R.; Evans, M.C.; Braastad, C.D.; Bishop, C.M.; Jaremko, M.; Wang, Z.; et al. The allelic spectrum of Charcot–Marie–Tooth disease in over 17,000 individuals with neuropathy. Mol. Genet. Genom. Med. 2014, 2, 522–529. [Google Scholar] [CrossRef]
- Cavalcanti, E.B.U.; Santos, S.C.d.L.; Martins, C.E.S.; de Carvalho, D.R.; Rizzo, I.M.P.D.O.; Freitas, M.C.D.N.B.; Freitas, D.D.S.; de Souza, F.S.; Junior, A.M.; Nascimento, O.J.M.D. Charcot–Marie–Tooth disease: Genetic profile of patients from a large Brazilian neuromuscular reference center. J. Peripher. Nerv. Syst. 2021, 26, 290–297. [Google Scholar] [CrossRef]
- Yalcintepe, S.; Gurkan, H.; Dogan, I.G.; Demir, S.; Sag, S.O.; Kabayegit, Z.M.; Atli, E.I.; Atli, E.; Eker, D.; Temel, S.G. The Importance of Multiple Gene Analysis for Diagnosis and Differential Diagnosis in Charcot Marie Tooth Disease. Turk. Neurosurg. 2021, 31, 888–895. [Google Scholar] [CrossRef]
- Candayan, A.; Parman, Y.; Battaloğlu, E. Clinical and Genetic Survey for Charcot–Marie–Tooth Neuropathy Based on the Findings in Turkey, a Country with a High Rate of Consanguineous Marriages. Balk. Med. J. 2022, 39, 3–11. [Google Scholar] [CrossRef]
- Keohane, D.; Schwartz, J.; Gundapaneni, B.; Stewart, M.; Amass, L. Tafamidis delays disease progression in patients with early stage transthyretin familial amyloid polyneuropathy: Additional supportive analyses from the pivotal trial. Amyloid 2017, 24, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.D.; Dasgupta, N.R.; Rissing, S.M.; Smith, J.; Feigenbaum, H. Safety and efficacy of a TTR specific antisense oligonucleotide in patients with transthyretin amyloid cardiomyopathy. Amyloid 2017, 24, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Nan, H.; Koh, K.; Ichinose, Y.; Gao, L.; Shimozono, K.; Hata, T.; Kim, Y.-J.; Ohtsuka, T.; Cortese, A.; et al. RFC1 repeat expansion in Japanese patients with late-onset cerebellar ataxia. J. Hum. Genet. 2020, 65, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Yoshida, K.; Koshimizu, E.; Doi, H.; Yamada, M.; Miyaji, Y.; Ueda, N.; Tsuyuzaki, J.; Kodaira, M.; Onoue, H.; et al. Repeat conformation heterogeneity in cerebellar ataxia, neuropathy, vestibular areflexia syndrome. Brain 2022, 145, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Beecroft, S.J.; Cortese, A.; Sullivan, R.; Yau, W.Y.; Dyer, Z.; Wu, T.Y.; Mulroy, E.; Pelosi, L.; Rodrigues, M.; Taylor, R.; et al. A Māori specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain 2020, 143, 2673–2680. [Google Scholar] [CrossRef]
- Beijer, D.; Dohrn, M.F.; De Winter, J.; Fazal, S.; Cortese, A.; Stojkovic, T.; Fernández-Eulate, G.; Remiche, G.; Gentile, M.; Van Coster, R.; et al. RFC1 repeat expansions: A recurrent cause of sensory and autonomic neuropathy with cough and ataxia. Eur. J. Neurol. 2022, 29, 2156–2161. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RFC1 Repeat | (AAGGG)exp | (ACAGG)exp | (AAGGG)exp | (AAGGG)exp | (ACAGG)exp | All Cases |
|---|---|---|---|---|---|---|
| (AAGGG)exp | (ACAGG)exp | (ACAGG)exp | (AAAGG)13 (AAGGG)exp | (AAAGG)12 (AAGGG)exp | ||
| Patient number | N = 6 | N = 4 | N = 6 | N = 1 | N = 1 | N = 18 |
| Onset age | 28.8 ± 22.8 | 49.75 ± 27.6 | 64.7 ± 11.6 | 25 | 50 | 46.3 ± 24.2 |
| Sex | Male 6, Female 0 | Male 4, Female 0 | Male 4, Female 2 | Male | Female | Male 15, Female 3 |
| Muscle weakness | 6/6 [100%] | 4/4 [100%] | 5/6 [83.3%] | + | + | 17/18 [94.4%] |
| Muscle atrophy | 3/4 [75%] | 2/3 [66.7%] | 5/6 [83.3%] | + | + | 12/15 [80%] |
| Hyporeflexia | 5/5 [100%] | 3/4 [75%] | 6/6 [100%] | − | + | 15/17 [88/2%] |
| Sensory disturbance | 5/5 [100%] | 4/4 [100%] | 6/6 [100%] | − | + | 16/17 [94.1%] |
| Cerebellar ataxia | 1/4 [25%] | 1/4 [25%] | 2/3 [66.7%] | − | + | 5/16 [31.3%] |
| Cerebellar atrophy | 1/3 [33.3%] | 0/2 [0%] | 2/3, 66.7% | − | − | 3/10 [30%] |
| Vestibular dysfunction | 0/3 [0%] | 1/4 [25%] | 0/3 [0%] | NA | + | 2/11 [9.1%] |
| Chronic cough | 0/1 [0%] | 1/3 [33.3%] | 2/3 [66.7%] | NA | NA | 3/7 [42.9%] |
| Pyramidal sign | 0/5 [0%] | 1/4 [25%] | 0/6 [0%] | − | − | 1/17 [5.9%] |
| Parkinsonism | 1/4 [25%] | 0/4 [0%] | 0/6 [0%] | − | − | 1/16 [6.3%] |
| Cognitive impairment | 0/2 [0%] | 1/3 [33.3%] | 0/5 [0%] | − | − | 1/12 [8.3%] |
| Involuntary movement | 0/2 [0%] | 0/4 [0%] | 1/6 [16.7%] | − | − | 1/14 [7.1%] |
| Autonomic dysfunction | 1/3 [33.3%] | 2/4 [50%] | 1/6 [16.7%] | + | − | 5/15 [33.3%] |
| Muscle cramp | 1/1 [100%] | 1/3 [33.3%] | 2/4 [50%] | NA | − | 4/9 [44.4%] |
| Hyper CKemia | 3/3 [100%] | 1/3 [33.3%] | 2/5 [40%] | + | − | 7/13 [53.8%] |
| Median MNCV (m/s) | 54.2 ± 6.1 | 47.5 ± 1.3 | 53.6 ± 5.8 | 50.6 | 56.5 | 52.5 ± 6.1 |
| Median CMAP (mV) | 10.8 ± 3.9 | 4.5 ± 4.2 | 6.2 ± 2.7 | 6.2 | 8.48 | 8.0 ± 4.1 |
| Median SCV (m/s) | 44.5 ± 12.6 | 31 | 39.8 | 48 | 58 | 45.8 ± 11.3 |
| Median SNAP (μV) | 23.8 ±25.3 | 0.7 ± 1.1 | 2.6 ± 5.7 | 0.4 | 2.6 | 9.1 ± 15.6 |
| Tibial MNCV (m/s) | 43.9 ± 2.2 | 37.5 ± 2.4 | 39.0 ± 6.4 | 40.2 | 43.8 | 40.9 ± 4.5 |
| Tibial CMAP (mV) | 7.8 ± 6.3 | 2.5 ± 2.9 | 4.8 ± 4.1 | 3.2 | 6.15 | 5.6 ± 5.0 |
| Sural SCV (m/s) | 50.4 ± 5.9 | NA | NA | NE | NE | 50.4 ± 5.9 |
| Sural SNAP (μV) | 8.3± 4.5 | 0 | 0 | NE | NE | 3.0 ± 4.8 |
| PMP22CNVs | MFN2 | GJB1 | MPZ | MME | NEFL | HSPB1 | PMP22 pm | BSCL2 | GDAP1 | MORC2 | PRX | SH3TC2 | RFC1 exp | TTR | DiagnosticRate (%) | Demyelinating Type: Axonal Type | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Our study (N = 2695) | *1 | 160 [5.9%] | 151 [5.6%] | 121 [4.5%] | 35 [1.3%] | 30 [1.1%] | 28 [1.0%] | 22 [0.8%] | 21 [0.8%] | 21 [0.8%] | 19 [0.7%] | 19 [0.7%] | 19 [0.7%] | 18 [0.7%] | 13 [0.5%] | 33.7 | 1:2.49 |
| Japan, 2011 (N = 354) [14] | 53 [15.0%] | 14 [4.0%] | 25 [7.1%] | 25 [7.1%] | / | 8 [2.3%] | 0 [0%] | 10 [2.8%] | / | 1 [0.3%] | / | 5 [1.4%] | / | / | / | 40.3 | 1.79:1 |
| Japan, 2018 (N = 1005) [9] | *1 | 66 [6.6%] | 66 [6.6%] | 51 [5.1%] | 8 [0.8%] | 9 [0.9%] | 14 [1.4%] | 13 [1.3%] | 6 [0.6%] | 8 [0.8%] | / | 4 [0.4%] | 5 [0.5%] | / | / | 30.0 | 1:2.42 |
| Korea, 2016 (N-78) [15] | 15 [19.2%] | 1 [1.3%] | 9 [11.5%] | 2 [2.6%] | / | 0 [0%] | 0 [0%] | 1 [1.3%] | 0 [0%] | 0 [0%] | / | 0 [0%] | 1 [1.3%] | / | / | 21.8 | 1.1:1 |
| China, 2019 (N = 150) [16] | 52 [34.7%] | 9 [7%] | 19 [14%] | [3%] *2 | / | / | / | 1% *2 | / | 1% *2 | / | / | / | / | / | 66.7 | 1.59:1 |
| China, 2021 (N = 435) [17] | 99 [22.8%] | [10.1%] *2 | [13.5%] *2 | [5.0%] *2 | [0.5%] *2 | [0.7%] *2 | [0.9%] *2 | [3.2%] *2 | 0 [0%] | [2.1%] *2 | / | / | [2.1%] *2 | / | / | 70 | 1.22:1 |
| Taiwan, 2019 (N = 427) [18] | 208 [48.7%] | 14 [3.3%] | 40 [9.4%] | 14 [3.3%] | / | 8 [1.9%] | 2 [0.5%] | 4 [0.9%] | 2 [0.5%] | 2 [0.5%] | 1 [0.2%] | 0 [0%] | 3 [0.7%] | / | 0 [0%] | 73.1 | 2.8:1 |
| UK, 2012 (N = 1607) [19] | 415 [25.8%] | 60 [3.7%] | 147 [9.1%] | 31 [1.9%] | / | 4 [0.2%] | 3 [0.2%] | 11 [0.7%] | 2 [0.1%] | 12 [0.7%] | / | / | 9 [0.6%] | / | / | 44.3 | 1.52:1 |
| Germany, 2013 (N = 776) [20] | 180 [23.2%] | 12 [1.5%] | 47 [6.1%] | 21 [2.7%] | / | 0 [0%] | / | 8 [1.0%] | / | 0 [0%] | / | 0 [0%] | 0 [0%] | / | / | 58 | 2.35:1 |
| Norway, 2013 (N = 435) [21] | 20 [4.6%] *3 | 11 [2.5%] | 12 [2.8%] | 10 [2.3%] | / | 2 [0.5%] | / | 0 [0%] | / | / | / | / | / | / | / | 16.6 | 1:2.08 |
| Spain, 2013 (N-438) [22] | 184 [42.0%] | 4 [0.9%] | 56 [12.8%] | 19 [4.3%] | / | 4 [0.9%] | 7 [1.6%] | 2 [0.5%] | / | 42 [9.6%] | / | 4 [0.9%] | 28 [6.4%] | / | / | 83.3 | 1.69:1 |
| Italy, 2014 (N = 197) [23] | 100 [50.8%] | 2 [1.0%] | 14 [7.1%] | 7 [3.6%] | / | 1 [0.5%] | 1 [0.5%] | 7 [3.6%] | / | 8 [4.1%] | / | / | 3 [1.5%] | / | / | 75.1 | 1.98:1 |
| Italy, 2019 (N = 566) [24] | 233 [41.2%] | 8 [1.4%] | 33 [5.8%] | 36 [6.4%] | / | 1 [0.2%] | 14 [2.5%] | 7 [1.2%] | 8 [1.4%] | 4 [0.7%] | / | / | 0 [0%] | / | / | 62.2 | / |
| Denmark, 2019 (N = 1442) [25] | 236 [16.4%] | 24 [1.7%] | 32 [2.2%] | 27 [1.9%] | / | 1 [0.07%] | 0 [0%] | 3 [0.2%] | 0 [0%] | 0 [0%] | / | 0 [0%] | 1 [0.07%] | / | / | 23.1 | / |
| USA, 2014 (N = 17,880) [26] | 1823 [10.2%] | 138 [0.8%] | 215 [1.2%] | 170 [1.0%] | / | 22 [0.1%] | 10 [0.06%] | 30 [0.2%] | / | 22 [0.1%] | / | 1 [0.005%] | 26 [0.1%] | / | / | 18.5 | / |
| Brazil, 2021 (N = 503) [27] | 116 [23.1%] | 12 [2.4%] | 23 [4.6%] | 6 [1.2%] | 0 [0%] | 3 [0.6%] | 0 [0%] | 6 [1.2%] | 0 [0%] | 7 [1.4%] | / | 2 [0.4%] | 3 [0.6%] | / | / | 78.3 | 3.18:1 |
| Turkey, 2021 (N = 55) [28] | 0 [0%] | 1 [1.8%] | 2 [3.6%] | 0 [0%] | / | 0 [0%] | 0 [0%] | 0 [0%] | 0 [0%] | 2 [3.6%] | / | 1 [1.8%] | 2 [3.6%] | / | / | 23.6 | / |
| Turkey, 2022 (N-649) [29] | 151 [23.3%] | 15 [2.3%] | 47 [7.2%] | 6 [0.9%] | / | 2 [0.3%] | 2 [0.3%] | 3 [0.5%] | / | 18 [2.8%] | / | 6 [0.9%] | 17 [2.6%] | / | / | 46.5 | 2.47:1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ando, M.; Higuchi, Y.; Yuan, J.; Yoshimura, A.; Taniguchi, T.; Kojima, F.; Noguchi, Y.; Hobara, T.; Takeuchi, M.; Takei, J.; et al. Comprehensive Genetic Analyses of Inherited Peripheral Neuropathies in Japan: Making Early Diagnosis Possible. Biomedicines 2022, 10, 1546. https://doi.org/10.3390/biomedicines10071546
Ando M, Higuchi Y, Yuan J, Yoshimura A, Taniguchi T, Kojima F, Noguchi Y, Hobara T, Takeuchi M, Takei J, et al. Comprehensive Genetic Analyses of Inherited Peripheral Neuropathies in Japan: Making Early Diagnosis Possible. Biomedicines. 2022; 10(7):1546. https://doi.org/10.3390/biomedicines10071546
Chicago/Turabian StyleAndo, Masahiro, Yujiro Higuchi, Junhui Yuan, Akiko Yoshimura, Takaki Taniguchi, Fumikazu Kojima, Yutaka Noguchi, Takahiro Hobara, Mika Takeuchi, Jun Takei, and et al. 2022. "Comprehensive Genetic Analyses of Inherited Peripheral Neuropathies in Japan: Making Early Diagnosis Possible" Biomedicines 10, no. 7: 1546. https://doi.org/10.3390/biomedicines10071546