
Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts?


Abstract
:1. What Is Lung Fibrosis?
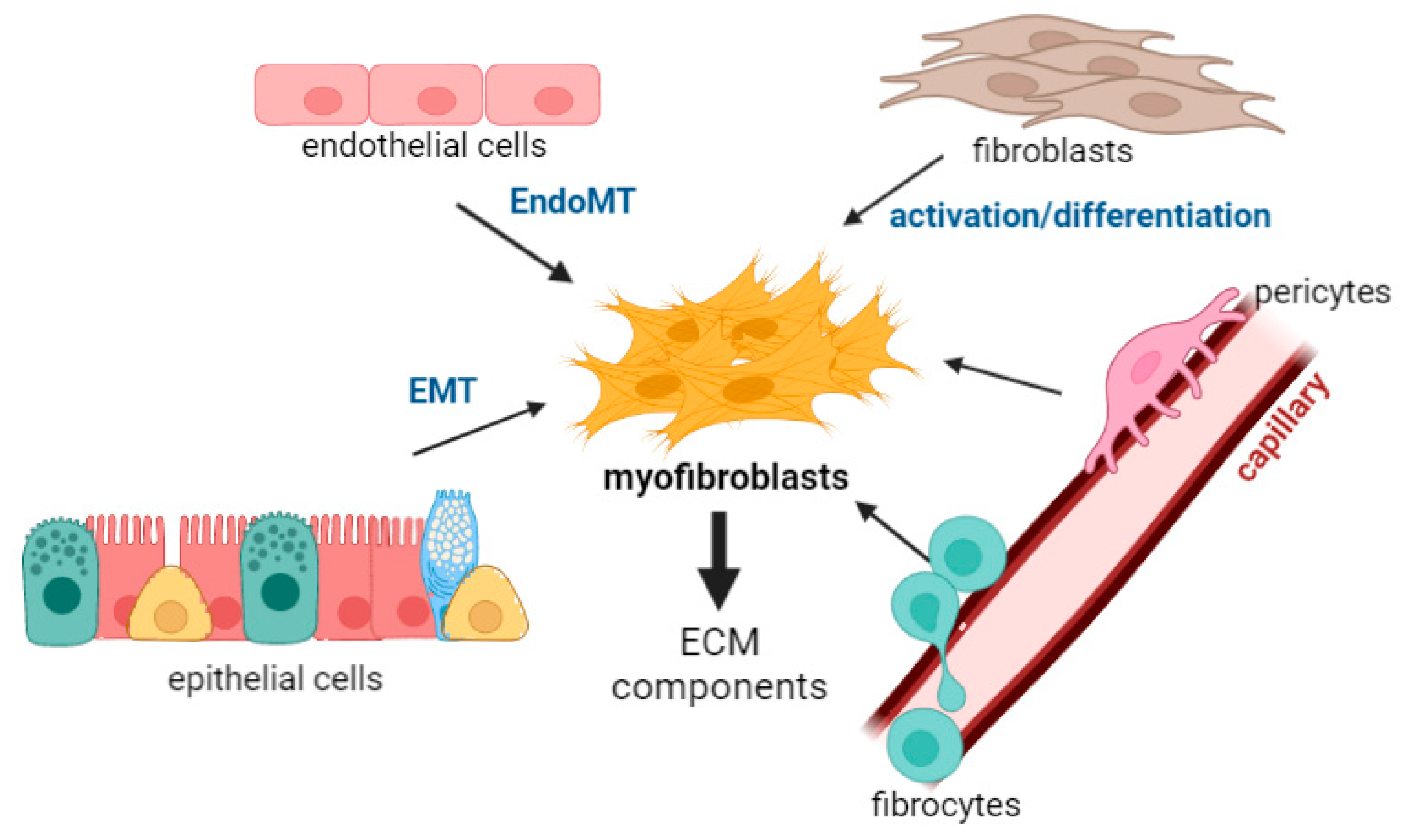
2. Myofibroblasts in the Lung
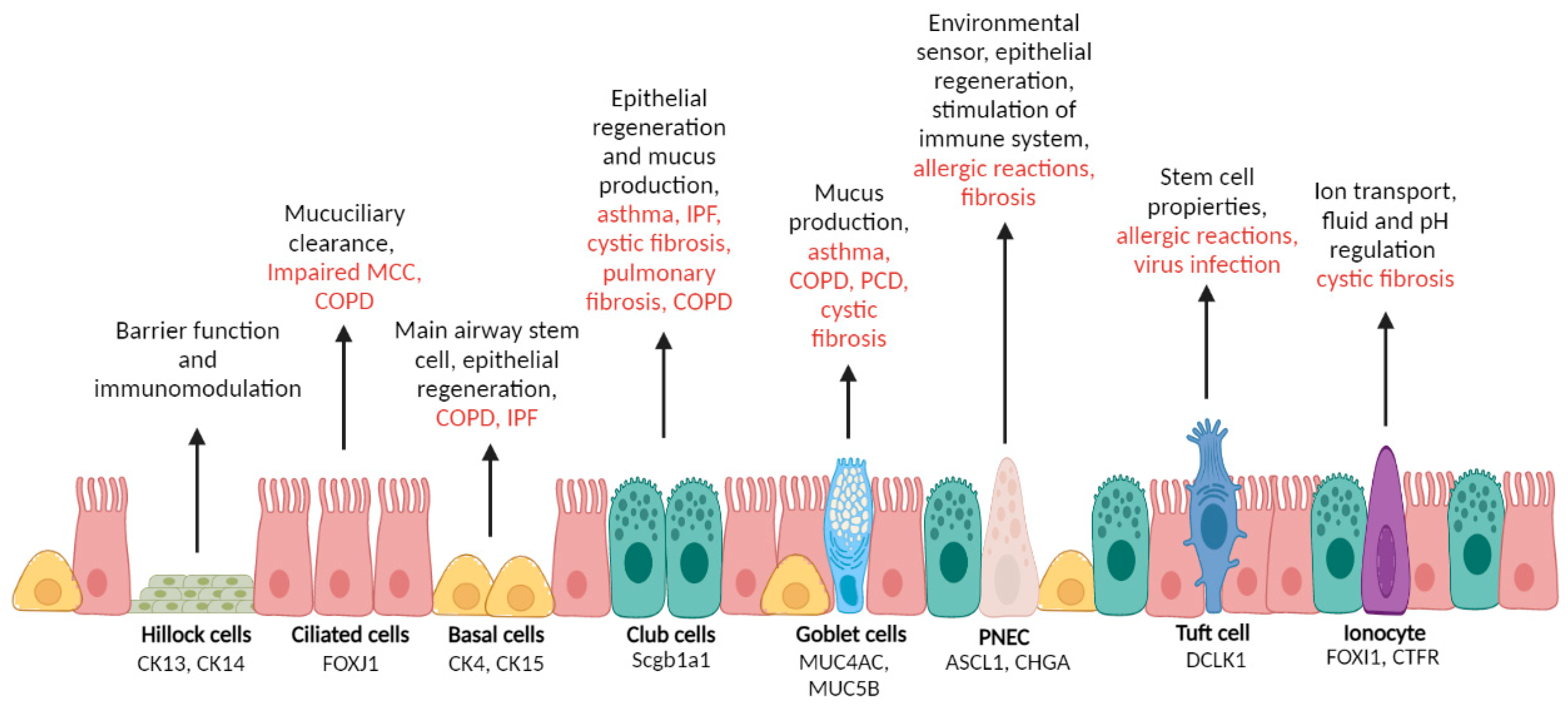
2.1. Epithelial Cells
2.2. Endothelial Cells
2.3. Resident Mesenchymal Cells: Fibroblasts
2.4. Bone Marrow/Derived Precursors: Fibrocytes
2.5. Perivascular Stromal Origin: Pericytes
3. Role of Macrophages in Myofibroblasts Differentiation and Activation
3.1. Macrophage-Derived Secretory Proteins and Fibrosis in the Lungs: The Macrophage Secretome

{kind=link}
{kind=link}
| Secretome Molecules | Macrophage Type | Study Subject | Role in Fibrosis | Reference |
|---|---|---|---|---|
| KC | Monocyte-derived AMs | LPS stimulation | Increase proliferation of ECM components | [69,77] |
| type IV collagenase | AMs | BLM-induced fibrosis mice model | Abnormal collagen degradation | [78,79] |
| MMP-1 | lung macrophages | IPF, COVID-19 | Role in IPF presently unknown. Present in early fibrotic processes in COVID-19 patients | [26,80] |
| MMP-2 | lung macrophages | IPF | Effect on fibrocytes: Tissue migration and homing | [81] |
| MMP-3 | AMs | IPF patients, lung | Induction of epithelial-mesenchymal transition through activation of β-catenin signaling | [82] |
| MMP-7 | lung macrophages | IPF, COVID-19 | Effect on AEC2 cells. Present in early fibrotic processes in COVID-19 patients | [26,80] |
| MMP-8 | lung macrophages | IPF patients, lung | Effect on fibrocytes: Tissue migration and homing. Initiation of collagen destruction and remodelling | [81,83,84] |
| MMP9 | AMs IMs | BLM-induced fibrosis mice model IPF patients, lung | Degradation and remodeling of extracellular matrix components | [78,84,85] |
| MMP-12 | macrophages | LPS stimulation | Effect on TGF-β1 signaling pathway activation | [86,87] |
| MMP-13 | Monocyte-derived macrophages e. | Lung fibrosis | Cleaves fibrillar collagens. Specific role largely unknown. | [76,87,88] |
| Periostin | lung macrophages | IPF, COVID-19 | Present in early fibrotic processes in COVID-19 patients | [80] |
| TIMP1 | IMs | IPF patients, lung | Promotes the fibrotic response | [89] |
| MT1-MMP | macrophages | IAV sensing | ECM remodelling through collagenase activity | [90] |
| CCL18 | AMs | Pulmonary fibrosis | Increases collagen production | [91] |
| IFNβ | AMs | IAV sensing | Not specified | [92] |
| IFNγ, | M1 macrophages macrophage | Lung injury | Macrophage polarization | [93] |
| TGF-β1 | M2 macrophages, AM | Lung fibrosis, BLM-induced fibrosis mice model, COPD patients | Differentiation of fibroblasts into myofibroblasts. EMT transition through the TGF-Smad2 signalling pathway | [76,94,95] |
| TNFα | AM, M1 macrophages | COPD patients, enhanced lung injury, BLM-induced fibrosis mice model | Initiates inflammation and enhaces lung injury. Induces M1 macrophages to produce proinflammatory cytokines | [76,94,96] |
| PDGF | primary macrophages | inflammatory process, Lung injury, lung fibrosis | Promotes de fibrotic process | [76,97] |
| VEGF | primary macrophages | inflammatory process, Lung injury | Promotes blood vessel development and cellular proliferation | [76,98] |
| IGF-1 | primary macrophages | inflammatory process, Lung injury | Promotes blood vessel development and cellular proliferation | [76] |
| amphiregulin | primary macrophages, AM | inflammatory process, LPS stimulation, Lung injury | Contributes to immune regulation, Protects lung tissues. Counteracts epithelial damage. Enhances EGFR signalling and proliferative responses. | [99,100,101] |
| CCL2 | macrophages | BLM-induced fibrosis mice model, IPF patients, BALF | Cellular recruitment to the lung. Enhancement of cytokine and collagen production. | [102,103] |
| CCL24 | macrophages | BLM-induced fibrosis mice model. IPF patients, BALF | Not specified. | [102,103] |
| arginase 1 | Monocyte-derived macrophages | Lung fibrosis | Effect on collagen synthesis | [76,104] |
| IL-4 | M2 macrophages | Lung fibrosis | ||
| IL-6 | AM, M1 macrophages | Viral infection, enhanced lung injury | Promotes macrophages recruitment to the lung | [76,105] |
| IL-8 | AM | COPD patients | Promotes neutrophil recrutiment | [94] |
| IL-10 | M2 macrophages | Lung fibrosis | Induces reprogramming of macrophages to the M2 phenotype. Enhances tissue repair and promotes matrix synthesis. | [76] |
| IL-12 | M1 macrophages | Lung injury | Not specified | [93] |
| IL-1β | IMs, AMs | Uninjured lung, IPF patients, lung | Triggers the activation, proliferation and transdifferentiation of epithelia cells and resident fibroblasts into myofibroblasts. Stimulates the secretion of neutrophil-attracting CXC chemokines. | [101] |
| IL-18 | AMs | IPF patients, lung | Not specified | [106] |
3.2. Other Immune Cells
4. Signaling Pathways in Fibrosis
5. Therapeutic Targets in Lung Tissue Remodelling and Fibrosis
5.1. Pirfenidone and Nintedanib
5.2. Novel Therapies
5.2.1. Inhibition of Pro-Fibrotic Molecules
5.2.2. Drugs Targeting ECM Proteins Directly
5.2.3. Other Targets
| Therapy | Mechanism of Action | Clinical Trial Identifier | Status | Observations | Ref. |
|---|---|---|---|---|---|
| Fresolimumab | Antibody to neutralize TGFβ | NCT00125385 | Phase I completed | - | |
| Lebrikizumab | Anti-IL-13 and IL-14 antibody | NCT01872689 | Phase II completed | [147] | |
| Tralokinumab | Anti-IL-13 antibody | NCT01629667 | Phase II terminated (lack of efficacy) | The study was stopped due to lack of efficacy | [146] |
| NCT02036580 | Phase II completed | Early termination due to lack of efficacy | - | ||
| Pamrevlumab (FG-3019) | Anti-CTGF antibody | NCT01890265 | Phase II completed | Preliminary report shows overall safety and marginally favorable outcome in some patients | [149] |
| NCT03955146 | Phase III recruiting | - | |||
| BG00011 (STX-100) | Anti-integrin antibody | NCT01371305 | Phase II completed | This trial was terminated due to safety concerns | - |
| Simtuzumab | Anti-LOX antibody | NCT01769196 | Phase II terminated | The study recommended early termination due to lack of efficacy | [151] |
| PRM-151 | Recombinant human pentraxin 2 (also known as serum amyloid P) | NCT01254409 | Phase I | A preliminary report showed overall safety and a marginally favorable outcome in some patients | [157] |
| NCT02550873 | Phase II completed | Decreased pulmonary function decline and stability in 6MWD over 24 weeks | [160] | ||
| NCT04594707 | Phase III recruiting | ||||
| PBI-4050 | GPR84 antagonist/GPR40 agonist | NCT02538536 | Phase II completed | PBI-4050 alone or in combination with nintedanib or pirfenidone showed no safety concerns | [159] |
6. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vestbo, J.; Hurd, S.S.; Agustí, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease: GOLD Executive Summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Postma, D.S.; Kerstjens, H.A.M. Characteristics of Airway Hyperresponsiveness in Asthma and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 1998, 158, S187–S192. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Philp, A.M.; Corte, T.; Travis, M.A.; Schilter, H.; Hansbro, N.G.; Burns, C.J.; Eapen, M.S.; Sohal, S.S.; Burgess, J.K.; et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol. Ther. 2021, 225, 107839. [Google Scholar] [CrossRef]
- Hung, C. Origin of Myofibroblasts in Lung Fibrosis. Curr. Tissue Microenviron. Rep. 2020, 1, 155–162. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The Myofibroblast. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases: The myofibroblast. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.M.; Gabbiani, G. Recent Developments in Myofibroblast Biology. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Coen, M.; Gabbiani, G.; Bochaton-Piallat, M.L. Myofibroblast-Mediated Adventitial Remodeling: An Underestimated Player in Arterial Pathology. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2391–2396. [Google Scholar] [CrossRef] [Green Version]
- Braga, F.A.V.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef] [Green Version]
- Goldfarbmuren, K.C.; Jackson, N.D.; Sajuthi, S.P.; Dyjack, N.; Li, K.S.; Rios, C.L.; Plender, E.G.; Montgomery, M.T.; Everman, J.L.; Bratcher, P.E.; et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 2020, 11, 2485. [Google Scholar] [CrossRef]
- Hong, K.U.; Reynolds, S.D.; Watkins, S.; Fuchs, E.; Stripp, B.R. Basal Cells Are a Multipotent Progenitor Capable of Renewing the Bronchial Epithelium. Am. J. Pathol. 2004, 164, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.D.; Wypych, T.P. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol. 2021, 14, 978–990. [Google Scholar] [CrossRef]
- Boers, J.E.; Ambergen, A.W.; Thunnissen, F.B.J.M. Number and Proliferation of Clara Cells in Normal Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 1999, 159, 1585–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Du, Y.; Gokey, J.J.; Ray, S.; Bell, S.M.; Adam, M.; Sudha, P.; Perl, A.K.; Deshmukh, H.; Potter, S.S.; et al. Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat. Commun. 2019, 10, 37. Available online: http://www.nature.com/articles/s41467-018-07770-1 (accessed on 27 April 2022). [CrossRef]
- Basil, M.C.; Katzen, J.; Engler, A.E.; Guo, M.; Herriges, M.J.; Kathiriya, J.J.; Windmueller, R.; Ysasi, A.B.; Zacharias, W.J.; Chapman, H.A.; et al. The Cellular and Physiological Basis for Lung Repair and Regeneration: Past, Present, and Future. Cell Stem Cell 2020, 26, 482–502. [Google Scholar] [CrossRef]
- Rokicki, W.; Rokicki, M.; Wojtacha, J.; Dżeljijli, A. The role and importance of club cells (Clara cells) in the pathogenesis of some respiratory diseases. Pol. J. Cardio-Thorac. Surg. 2016, 1, 26–30. [Google Scholar] [CrossRef]
- Zuo, W.L.; Shenoy, S.A.; Li, S.; O’Beirne, S.L.; Strulovici-Barel, Y.; Leopold, P.L.; Wang, G.; Staudt, M.R.; Walters, M.S.; Mason, C.; et al. Ontogeny and Biology of Human Small Airway Epithelial Club Cells. Am. J. Respir. Crit. Care Med. 2018, 198, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.T.; Veerati, P.C.; Gosens, R.; Bartlett, N.W.; Wark, P.A.; Grainge, C.L.; Stick, S.M.; Kicic, A.; Moheimani, F.; Hansbro, P.M.; et al. Persistent induction of goblet cell differentiation in the airways: Therapeutic approaches. Pharmacol. Ther. 2018, 185, 155–169. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Kalin, T.V.; Xu, Y.; Kalinichenko, V.V. Building and Regenerating the Lung Cell by Cell. Physiol. Rev. 2019, 99, 513–554. [Google Scholar] [CrossRef]
- Tilley, A.E.; Walters, M.S.; Shaykhiev, R.; Crystal, R.G. Cilia Dysfunction in Lung Disease. Annu. Rev. Physiol. 2015, 77, 379–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouadah, Y.; Rojas, E.R.; Riordan, D.P.; Capostagno, S.; Kuo, C.S.; Krasnow, M.A. Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch. Cell 2019, 179, 403–416.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Branchfield, K.; Nantie, L.; Verheyden, J.M.; Sui, P.; Wienhold, M.D.; Sun, X. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science 2016, 351, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell Signal. 2020, 66, 109482. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, T.; de Andrade, J.; Zhou, Y.; Luckhardt, T.; Thannickal, V.J. Alveolar epithelial disintegrity in pulmonary fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 311, L185–L191. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.J.; ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Nataraj, D.; Ernst, A.; Kalluri, R. Idiopathic Pulmonary Fibrosis Is Associated with Endothelial to Mesenchymal Transition. Am. J. Respir. Cell Mol. Biol. 2010, 43, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Chakraborty, D.; Distler, J.H.W. Cellular and molecular mechanisms in fibrosis. Exp. Dermatol. 2021, 30, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Zandvoort, A.; Postma, D.S.; Jonker, M.R.; Noordhoek, J.A.; Vos, J.T.W.M.; van der Geld, Y.M.; Timens, W. Altered expression of the Smad signalling pathway: Implications for COPD pathogenesis. Eur. Respir. J. 2006, 28, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.B.; McCune, B.K.; Sporn, M.B. TGF-β: Regulation of extracellular matrix. Kidney Int. 1992, 41, 557–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, M.; Javelaud, D.; Mauviel, A. TGF-β-induced SMAD signaling and gene regulation: Consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 2004, 35, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morty, R.E.; Konigshoff, M.; Eickelberg, O. Transforming Growth Factor- Signaling across Ages: From Distorted Lung Development to Chronic Obstructive Pulmonary Disease. Proc. Am. Thorac. Soc. 2009, 6, 607–613. [Google Scholar] [CrossRef]
- Sugiura, H.; Ichikawa, T.; Liu, X.; Kobayashi, T.; Wang, X.Q.; Kawasaki, S.; Togo, S.; Kamio, K.; Mao, L.; Ann, Y.; et al. N-acetyl-l-cysteine inhibits TGF-β1-induced profibrotic responses in fibroblasts. Pulm. Pharmacol. Ther. 2009, 22, 487–491. [Google Scholar] [CrossRef]
- Araya, J.; Nishimura, S.L. Fibrogenic Reactions in Lung Disease. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 77–98. [Google Scholar] [CrossRef]
- Parameswaran, K.; Willems-Widyastuti, A.; Alagappan, V.K.T.; Radford, K.; Kranenburg, A.R.; Sharma, H.S. Role of Extracellular Matrix and Its Regulators in Human Airway Smooth Muscle Biology. Cell Biochem. Biophys. 2006, 44, 139–146. [Google Scholar] [CrossRef]
- Kranenburg, A.R.; Willems-Widyastuti, A.; Mooi, W.J.; Sterk, P.J.; Alagappan, V.K.T.; de Boer, W.I.; Sharma, H.S. Enhanced Bronchial Expression of Extracellular Matrix Proteins in Chronic Obstructive Pulmonary Disease. Am. J. Clin. Pathol. 2006, 126, 725–735. [Google Scholar] [CrossRef]
- Lagente, V.; Manoury, B.; Nénan, S.; Le Quément, C.; Martin-Chouly, C.; Boichot, E. Role of matrix metalloproteinases in the development of airway inflammation and remodeling. Braz. J. Med. Biol. Res. 2005, 38, 1521–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Cui, D.; Ma, N.; Lu, L.; Gao, Y.; Cui, X.; Wang, D. The effect of extracellular matrix remodeling on airflow obstruction in a rat model of chronic obstructive pulmonary disease. Zhonghua Jie He He Hu Xi Za Zhi Zhonghua Jiehe He Huxi Zazhi Chin. J. Tuberc. Respir. Dis. 2002, 25, 403–407. [Google Scholar]
- Bucala, R. Fibrocytes at 20 Years. Mol. Med. Camb. Mass 2015, 21, S3–S5. [Google Scholar] [CrossRef] [PubMed]
- Keeley, E.C.; Schutt, R.C.; Marinescu, M.A.; Burdick, M.D.; Strieter, R.M.; Mehrad, B. Circulating fibrocytes as predictors of adverse events in unstable angina. Transl. Res. 2016, 172, 73–83.e1. [Google Scholar] [CrossRef] [Green Version]
- Moore, B.B.; Kolb, M. Fibrocytes and progression of fibrotic lung disease. Ready for showtime? Am. J. Respir. Crit. Care Med. 2014, 190, 1338–1339. [Google Scholar] [CrossRef]
- Dolgachev, V.A.; Ullenbruch, M.R.; Lukacs, N.W.; Phan, S.H. Role of Stem Cell Factor and Bone Marrow-Derived Fibroblasts in Airway Remodeling. Am. J. Pathol. 2009, 174, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Borie, R.; Quesnel, C.; Phin, S.; Debray, M.P.; Marchal-Somme, J.; Tiev, K.; Bonay, M.; Fabre, A.; Soler, P.; Dehoux, M.; et al. Detection of Alveolar Fibrocytes in Idiopathic Pulmonary Fibrosis and Systemic Sclerosis. PLoS ONE 2013, 8, e53736. [Google Scholar] [CrossRef]
- Kleaveland, K.R.; Velikoff, M.; Yang, J.; Agarwal, M.; Rippe, R.A.; Moore, B.B.; Kim, K.K. Fibrocytes Are Not an Essential Source of Type I Collagen during Lung Fibrosis. J. Immunol. 2014, 193, 5229–5239. [Google Scholar] [CrossRef]
- Heukels, P.; van Hulst, J.A.C.; van Nimwegen, M.; Boorsma, C.E.; Melgert, B.N.; van den Toorn, L.M.; Boomars, K.A.T.; Wijsenbeek, M.; Hoogsteden, H.; von der Thusen, J.; et al. Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 90. [Google Scholar] [CrossRef]
- Maharaj, S.S.; Baroke, E.; Gauldie, J.; Kolb, M.R.J. Fibrocytes in chronic lung disease—Facts and controversies. Pulm. Pharmacol. Ther. 2012, 25, 263–267. [Google Scholar] [CrossRef]
- Andersson-Sjöland, A.; de Alba, C.G.; Nihlberg, K.; Becerril, C.; Ramírez, R.; Pardo, A.; Westergren-Thorsson, G.; Selman, M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Gilpin, S.E.; Ask, K.; Cox, G.; Cook, D.; Gauldie, J.; Margetts, P.J.; Farkas, L.; Dobranowski, J.; Boylan, C.; et al. Circulating Fibrocytes Are an Indicator of Poor Prognosis in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, K.; Shamskhou, E.A.; Orcholski, M.E.; Nathan, A.; Reddy, S.; Honda, H.; Mani, V.; Zeng, Y.; Ozen, M.O.; Wang, L.; et al. Loss of Endothelium-Derived Wnt5a Is Associated with Reduced Pericyte Recruitment and Small Vessel Loss in Pulmonary Arterial Hypertension. Circulation 2019, 139, 1710–1724. [Google Scholar] [CrossRef]
- Göritz, C.; Dias, D.O.; Tomilin, N.; Barbacid, M.; Shupliakov, O.; Frisén, J. A Pericyte Origin of Spinal Cord Scar Tissue. Science 2011, 333, 238–242. [Google Scholar] [CrossRef]
- Hung, C.; Linn, G.; Chow, Y.H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of Lung Pericytes and Resident Fibroblasts in the Pathogenesis of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Jiang, Q.; Bushkin, Y.; Subbian, S.; Tyagi, S. Biphasic Dynamics of Macrophage Immunometabolism during Mycobacterium tuberculosis Infection. mBio 2019, 10, e02550-18. [Google Scholar] [CrossRef] [Green Version]
- Laskin, D.L. Macrophages and Inflammatory Mediators in Chemical Toxicity: A Battle of Forces. Chem. Res. Toxicol. 2009, 22, 1376–1385. [Google Scholar] [CrossRef] [Green Version]
- Schupp, J.C.; Binder, H.; Jäger, B.; Cillis, G.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Macrophage Activation in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. PLoS ONE 2015, 10, e0116775. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Meng, X.-M.; Ng, Y.-Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.-R.; Xiao, J.; Wang, Y.-Y.; et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-Y.; Jiang, H.; Pan, J.; Huang, X.-R.; Wang, Y.-C.; Huang, H.-F.; To, K.-F.; Nikolic-Paterson, D.J.; Lan, H.-Y.; Chen, J.-H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Chang, Y.; Zhang, C.; Xiong, Y.; Wang, X.; Ma, X.; Wang, Z.; Li, H.; Shimosawa, T.; Pei, L.; et al. UUO induces lung fibrosis with macrophage-myofibroblast transition in rats. Int. Immunopharmacol. 2021, 93, 107396. [Google Scholar] [CrossRef]
- van der Veen, T.A.; de Groot, L.E.S.; Melgert, B.N. The different faces of the macrophage in asthma. Curr. Opin. Pulm. Med. 2020, 26, 62–68. [Google Scholar] [CrossRef]
- Ogger, P.P.; Byrne, A.J. Macrophage metabolic reprogramming during chronic lung disease. Mucosal Immunol. 2021, 14, 282–295. [Google Scholar] [CrossRef]
- Tamò, L.; Simillion, C.; Hibaoui, Y.; Feki, A.; Gugger, M.; Prasse, A.; Jäger, B.; Goldmann, T.; Geiser, T.; Gazdhar, A. Gene Network Analysis of Interstitial Macrophages after Treatment with Induced Pluripotent Stem Cells Secretome (iPSC-cm) in the Bleomycin Injured Rat Lung. Stem Cell Rev. Rep. 2018, 14, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbings, S.L.; Thomas, S.M.; Atif, S.M.; McCubbrey, A.L.; Desch, A.N.; Danhorn, T.; Leach, S.M.; Bratton, D.L.; Henson, P.M.; Janssen, W.J.; et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am. J. Respir. Cell Mol. Biol. 2017, 57, 66–76. [Google Scholar] [CrossRef] [PubMed]
- The Immunological Genome Consortium; Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mould, K.J.; Barthel, L.; Mohning, M.P.; Thomas, S.M.; McCubbrey, A.L.; Danhorn, T.; Leach, S.M.; Fingerlin, T.E.; O’Connor, B.P.; Reisz, J.A.; et al. Cell Origin Dictates Programming of Resident Versus Recruited Macrophages during Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2017, 57, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Machiels, B.; Dourcy, M.; Xiao, X.; Javaux, J.; Mesnil, C.; Sabatel, C.; Desmecht, D.; Lallemand, F.; Martinive, P.; Hammad, H.; et al. A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat. Immunol. 2017, 18, 1310–1320. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Ho, L.P. Contribution of innate immune cells to pathogenesis of severe influenza virus infection. Clin. Sci. 2017, 131, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, L.A.; Rosada, R.; Moreira, A.P.; Joshi, A.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Mathai, S.; Gulati, I.; Herzog, E.L.; et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE 2010, 5, e9683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, T.; Shichino, S.; Ueha, S.; Matsushima, K. Macrophages in lung fibrosis. Int. Immunol. 2021, 33, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, Y.; Zhang, Z.; He, L.; Zhu, J.; Zhang, M.; He, X.; Cheng, Z.; Ao, Q.; Cao, Y.; et al. Chop Deficiency Protects Mice Against Bleomycin-Induced Pulmonary Fibrosis by Attenuating M2 Macrophage Production. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Schauer, I.G.; Ressler, S.J.; Rowley, D.R. Keratinocyte-derived chemokine induces prostate epithelial hyperplasia and reactive stroma in a novel transgenic mouse model: KC/IL-8 Induces Prostate Epithelial Hyperplasia. Prostate 2009, 69, 373–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.; Denney, L.; An, H.; Ho, L.P.; Zheng, Y. Alveolar and lung interstitial macrophages: Definitions, functions, and roles in lung fibrosis. J. Leukoc. Biol. 2021, 110, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Gadek, J.E.; Kelman, J.A.; Fells, G.; Weinberger, S.E.; Horwitz, A.L.; Reynolds, H.Y.; Fulmer, J.D.; Crystal, R.G. Collagenase in the Lower Respiratory Tract of Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 1979, 301, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Safont, B.; Tarraso, J.; Rodriguez-Borja, E.; Fernández-Fabrellas, E.; Sancho-Chust, J.N.; Molina, V.; Lopez-Ramirez, C.; Lope-Martinez, A.; Cabanes, L.; Andreu, A.L.; et al. Lung Function, Radiological Findings and Biomarkers of Fibrogenesis in a Cohort of COVID-19 Patients Six Months after Hospital Discharge. Arch. Bronconeumol. 2022, 58, 142–149. [Google Scholar] [CrossRef]
- García-de-Alba, C.; Becerril, C.; Ruiz, V.; González, Y.; Reyes, S.; García-Alvarez, J.; Selman, M.; Pardo, A. Expression of Matrix Metalloproteases by Fibrocytes: Possible Role in Migration and Homing. Am. J. Respir. Crit. Care Med. 2010, 182, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, C.M.; Dolgonos, L.; Zemans, R.L.; Young, S.K.; Robertson, J.; Briones, N.; Suzuki, T.; Campbell, M.N.; Gauldie, J.; Radisky, D.C.; et al. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am. J. Pathol. 2011, 179, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Craig, V.J.; Polverino, F.; Laucho-Contreras, M.E.; Shi, Y.; Liu, Y.; Osorio, J.C.; Tesfaigzi, Y.; Pinto-Plata, V.; Gochuico, B.R.; Rosas, I.O.; et al. Mononuclear phagocytes and airway epithelial cells: Novel sources of matrix metalloproteinase-8 (MMP-8) in patients with idiopathic pulmonary fibrosis. PLoS ONE 2014, 9, e97485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, M.T.; McMahon, K.; Mackarel, A.J.; Prikk, K.; Sorsa, T.; Maisi, P.; Sepper, R.; FitzGerald, M.; O’Connor, C. Matrix metalloproteinases and tissue inhibitor of metalloproteinase-1 in sarcoidosis and IPF. Eur. Respir. J. 2002, 20, 1220–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemjabbar, H.; Gosset, P.; Lechapt-Zalcman, E.; Franco-Montoya, M.L.; Wallaert, B.; Harf, A.; Lafuma, C. Overexpression of alveolar macrophage gelatinase B (MMP-9) in patients with idiopathic pulmonary fibrosis: Effects of steroid and immunosuppressive treatment. Am. J. Respir. Cell Mol. Biol. 1999, 20, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Sala-Newby, G.B.; Susana, A.; Johnson, J.L.; Newby, A.C. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-κB. PLoS ONE 2012, 7, e42507. [Google Scholar] [CrossRef] [Green Version]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nkyimbeng, T.; Ruppert, C.; Shiomi, T.; Dahal, B.; Lang, G.; Seeger, W.; Okada, Y.; D’Armiento, J.; Günther, A. Pivotal Role of Matrix Metalloproteinase 13 in Extracellular Matrix Turnover in Idiopathic Pulmonary Fibrosis. PLoS ONE 2013, 8, e73279. [Google Scholar] [CrossRef] [PubMed]
- Dancer, R.C.A.; Wood, A.M.; Thickett, D.R. Metalloproteinases in idiopathic pulmonary fibrosis. Eur. Respir. J. 2011, 38, 1461–1467. [Google Scholar] [CrossRef] [Green Version]
- Talmi-Frank, D.; Altboum, Z.; Solomonov, I.; Udi, Y.; Jaitin, D.A.; Klepfish, M.; David, E.; Zhuravlev, A.; Keren-Shaul, H.; Winter, D.; et al. Extracellular Matrix Proteolysis by MT1-MMP Contributes to Influenza-Related Tissue Damage and Mortality. Cell Host Microbe 2016, 20, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Prasse, A.; Pechkovsky, D.V.; Toews, G.B.; Jungraithmayr, W.; Kollert, F.; Goldmann, T.; Vollmer, E.; Müller-Quernheim, J.; Zissel, G. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am. J. Respir. Crit. Care Med. 2006, 173, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Jaworska, J.; Coulombe, F.; Downey, J.; Tzelepis, F.; Shalaby, K.; Tattoli, I.; Berube, J.; Rousseau, S.; Martin, J.G.; Girardin, S.E.; et al. NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc. Natl. Acad. Sci. USA 2014, 111, E2110–E2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtan, P.; Mierzejewski, M.; Osińska, I.; Domagała-Kulawik, J. Macrophage polarization in interstitial lung diseases. Cent. Eur. J. Immunol. 2016, 2, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, L.; Dong, C. Roles of Myeloid and Lymphoid Cells in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Front. Immunol. 2018, 9, 1431. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2018.01431 (accessed on 12 April 2022). [CrossRef] [PubMed]
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol. Int. 2017, 41, 960–968. [Google Scholar] [CrossRef]
- Pulmonary Macrophages: Key Players in the Innate Defence of the Airways—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/26286722/ (accessed on 13 April 2022).
- Shimokado, K.; Raines, E.W.; Madtes, D.K.; Barrett, T.B.; Benditt, E.P.; Ross, R. A significant part of macrophage-derived growth factor consists of at least two forms of PDGF. Cell 1985, 43, 277–286. [Google Scholar] [CrossRef]
- Berse, B.; Brown, L.F.; Van de Water, L.; Dvorak, H.F.; Senger, D.R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell. 1992, 3, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226. Available online: https://pubmed.ncbi.nlm.nih.gov/25692699/ (accessed on 13 April 2022). [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Meng, C.; Liu, G.; Yang, D.; Fu, L.; Zhang, M.; Zhang, Z.; Xia, H.; Yao, S.; Zhang, S. Classically Activated Macrophages Protect against Lipopolysaccharide-Induced Acute Lung Injury by Expressing Amphiregulin in Mice. Anesthesiology 2016, 124, 1086–1099. [Google Scholar] [CrossRef]
- Allard, B.; Panariti, A.; Martin, J.G. Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Front. Immunol. 2018, 9, 1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, C.P.; Opalek, J.M.; McMaken, S.; Newland, C.A.; O’Brien, J.M.; Hunter, M.G.; Bringardner, B.D.; Monick, M.M.; Brigstock, D.R.; Stromberg, P.C.; et al. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Arisi, I.; Puxeddu, E.; Mramba, L.K.; Amicosante, M.; Swaisgood, C.M.; Pallante, M.; Brantly, M.L.; Sköld, C.M.; Saltini, C. Bronchoalveolar lavage (BAL) cells in idiopathic pulmonary fibrosis express a complex pro-inflammatory, pro-repair, angiogenic activation pattern, likely associated with macrophage iron accumulation. PLoS ONE 2018, 13, e0194803. [Google Scholar] [CrossRef]
- Minutti, C.M.; Knipper, J.A.; Allen, J.E.; Zaiss, D.M.W. Tissue-specific contribution of macrophages to wound healing. Semin. Cell Dev. Biol. 2017, 61, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyle, C.J.; Uwadiae, F.I.; Swieboda, D.P.; Harker, J.A. Early IL-6 signalling promotes IL-27 dependent maturation of regulatory T cells in the lungs and resolution of viral immunopathology. PLoS Pathog. 2017, 13, e1006640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayan, M.; Mossman, B.T. The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part. Fibre Toxicol. 2015, 13, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinder, B.W.; Brown, K.K.; Schwarz, M.I.; Ix, J.H.; Kervitsky, A.; King, T.E. Baseline BAL Neutrophilia Predicts Early Mortality in Idiopathic Pulmonary Fibrosis. Chest 2008, 133, 226–232. [Google Scholar] [CrossRef]
- Xaubet, A.; Agustí, C.; Luburich, P.; Barberá, J.A.; Carrión, M.; Ayuso, M.C.; Roca, J.; Rodriguez-Roisin, R. Interleukin-8 expression in bronchoalveolar lavage cells in the evaluation of alveolitis in idiopathic pulmonary fibrosis. Respir. Med. 1998, 92, 338–344. [Google Scholar] [CrossRef] [Green Version]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts: NETs induce fibrosis via differentiation of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. Available online: http://journal.frontiersin.org/Article/10.3389/fmed.2015.00059/abstract (accessed on 2 May 2022). [CrossRef]
- Coker, R.K. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax 2001, 56, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekelmann, T.J.; Limper, A.H.; Colby, T.V.; McDonald, J.A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, G.; Huang, S.K.; Okunishi, K.; Scott, J.P.; Kumar Penke, L.R.; Scruggs, A.M.; Penke, L.R.K.; Scruggs, A.M.; Peters-Golden, M. Reversal of Myofibroblast Differentiation by Prostaglandin E2. Am. J. Respir. Cell Mol. Biol. 2013, 48, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Bauman, K.A.; Wettlaufer, S.H.; Okunishi, K.; Vannella, K.M.; Stoolman, J.S.; Huang, S.K.; Courey, A.J.; White, E.S.; Hogaboam, C.M.; Simon, R.H.; et al. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Investig. 2010, 120, 1950–1960. [Google Scholar] [CrossRef] [Green Version]
- Redington, A.E.; Madden, J.; Frew, A.J.; Djukanovic, R.; Roche, W.R.; Holgate, S.T.; Howarth, P.H. Transforming Growth Factor- β 1 in Asthma: Measurement in Bronchoalveolar Lavage Fluid. Am. J. Respir. Crit. Care Med. 1997, 156, 642–647. [Google Scholar] [CrossRef]
- Vignola, A.M.; Chanez, P.; Chiappara, G.; Merendino, A.; Pace, E.; Rizzo, A.; La Rocca, A.M.; Bellia, V.; Bonsignore, G.; Bousquet, J. Transforming Growth Factor- β Expression in Mucosal Biopsies in Asthma and Chronic Bronchitis. Am. J. Respir. Crit. Care Med. 1997, 156, 591–599. [Google Scholar] [CrossRef]
- Takizawa, H.; Tanaka, M.; Takami, K.; Ohtoshi, T.; Ito, K.; Satoh, M.; Okada, Y.; Yamasawa, F.; Nakahara, K.; Umeda, A. Increased Expression of Transforming Growth Factor- β 1 in Small Airway Epithelium from Tobacco Smokers and Patients with Chronic Obstructive Pulmonary Disease (COPD). Am. J. Respir. Crit. Care Med. 2001, 163, 1476–1483. [Google Scholar] [CrossRef]
- Hanna, A.; Humeres, C.; Frangogiannis, N.G. The role of Smad signaling cascades in cardiac fibrosis. Cell Signal. 2021, 77, 109826. [Google Scholar] [CrossRef]
- Gowripalan, A.; Abbott, C.R.; McKenzie, C.; Chan, W.S.; Karupiah, G.; Levy, L.; Newsome, T.P. Cell-to-cell spread of vaccinia virus is promoted by TGF-β-independent Smad4 signalling. Cell. Microbiol. 2020, 22, e13206. Available online: https://onlinelibrary.wiley.com/doi/10.1111/cmi.13206 (accessed on 25 May 2022). [CrossRef]
- Schunk, S.J.; Floege, J.; Fliser, D.; Speer, T. WNT–β-catenin signalling—A versatile player in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/β-Catenin Pathway Activation in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Kühl, S.J.; Kühl, M. On the role of Wnt/β-catenin signaling in stem cells. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. CCS 2018, 16, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/β-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci. Rep. 2018, 8, 13644. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair: Wnt1/βcatenin injury response regulates cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Dai, C.; Li, Y.; Zeng, G.; Monga, S.P.; Liu, Y. Wnt/β-Catenin Signaling Promotes Renal Interstitial Fibrosis. J. Am. Soc. Nephrol. 2009, 20, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Fang, F.; Lam, A.P.; Sargent, J.L.; Hamburg, E.; Hinchcliff, M.E.; Gottardi, C.J.; Atit, R.; Whitfield, M.L.; Varga, J. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012, 64, 2734–2745. [Google Scholar] [CrossRef] [Green Version]
- Henderson, W.R.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.-T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/β-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo Signaling Pathway Coordinately Regulates Cell Proliferation and Apoptosis by Inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [Green Version]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 Increases Organ Size and Expands Undifferentiated Progenitor Cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, K.-C.; Meng, Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front. Cell Dev. Biol. 2021, 9, 673599. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Mikami, Y.; Urushiyama, H.; Horie, M.; Matsuzaki, H.; Takeshima, H.; Makita, K.; Miyashita, N.; Mitani, A.; et al. TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci. Rep. 2017, 7, 42595. [Google Scholar] [CrossRef]
- Gokey, J.J.; Patel, S.D.; Kropski, J.A. The Role of Hippo/YAP Signaling in Alveolar Repair and Pulmonary Fibrosis. Front. Med. 2021, 8, 752316. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Togami, K.; Kanehira, Y.; Tada, H. Possible involvement of pirfenidone metabolites in the antifibrotic action of a therapy for idiopathic pulmonary fibrosis. Biol. Pharm. Bull. 2013, 36, 1525–1527. [Google Scholar] [CrossRef] [Green Version]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bizargity, P.; Liu, K.; Wang, L.; Hancock, W.W.; Visner, G.A. Inhibitory effects of pirfenidone on dendritic cells and lung allograft rejection. Transplantation 2012, 94, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Györfi, A.H.; Matei, A.E.; Distler, J.H.W. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Belperio, J.A.; Dy, M.; Burdick, M.D.; Xue, Y.Y.; Li, K.; Elias, J.A.; Keane, M.P. Interaction of IL-13 and C10 in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2002, 27, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.M.; Glaspole, I.N.; Lancaster, L.H.; Haddad, T.J.; She, D.; Roseti, S.L.; Fiening, J.P.; Grant, E.P.; Kell, C.M.; Flaherty, K.R. A Phase 2 Randomized Controlled Study of Tralokinumab in Subjects with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Costabel, U.; Glassberg, M.K.; Kondoh, Y.; Ogura, T.; Scholand, M.B.; Kardatzke, D.; Howard, M.; Olsson, J.; Neighbors, M.; et al. Phase 2 trial to assess lebrikizumab in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2021, 57, 1902442. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.; Dong, J.; D’Agostino, G.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Ko, N.S.J.; Tan, J.; Chothani, S.P.; Huang, B.; et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaaw1237. [Google Scholar] [CrossRef]
- Richeldi, L.; Pérez, E.R.F.; Costabel, U.; Albera, C.; Lederer, D.J.; Flaherty, K.R.; Ettinger, N.; Perez, R.; Scholand, M.B.; Goldin, J.; et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2020, 8, 25–33. [Google Scholar] [CrossRef]
- Horan, G.S.; Wood, S.; Ona, V.; Li, D.J.; Lukashev, M.E.; Weinreb, P.H.; Simon, K.J.; Hahm, K.; Allaire, N.E.; Rinaldi, N.J.; et al. Partial Inhibition of Integrin αvβ6 Prevents Pulmonary Fibrosis without Exacerbating Inflammation. Am J. Respir. Crit. Care Med. 2008, 177, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Kim, E.C.; Kim, Y. The human lysyl oxidase-like 2 protein functions as an amine oxidase toward collagen and elastin. Mol. Biol. Rep. 2011, 38, 145–149. [Google Scholar] [CrossRef]
- Chien, J.W.; Richards, T.J.; Gibson, K.F.; Zhang, Y.; Lindell, K.O.; Shao, L.; Lyman, S.K.; Adamkewicz, J.I.; Smith, V.; Kaminski, N.; et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur. Respir. J. 2014, 43, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- Findlay, A.D.; Foot, J.S.; Buson, A.; Deodhar, M.; Jarnicki, A.G.; Hansbro, P.M.; Liu, G.; Schilter, H.; Turner, C.I.; Zhou, W. Identification and Optimization of Mechanism-Based Fluoroallylamine Inhibitors of Lysyl Oxidase-like 2/3. J. Med. Chem. 2019, 62, 9874–9889. [Google Scholar] [CrossRef] [PubMed]
- Pilling, D.; Buckley, C.D.; Salmon, M.; Gomer, R.H. Inhibition of Fibrocyte Differentiation by Serum Amyloid P. J. Immunol. 2003, 171, 5537–5546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilling, D.; Roife, D.; Wang, M.; Ronkainen, S.D.; Crawford, J.R.; Travis, E.L.; Gomer, R.H. Reduction of Bleomycin-Induced Pulmonary Fibrosis by Serum Amyloid P. J. Immunol. 2007, 179, 4035–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillingh, M.R.; van den Blink, B.; Moerland, M.; van Dongen, M.G.J.; Levi, M.; Kleinjan, A.; Wijsenbeek, M.S.; Lupher Jr, M.R.; Harper, D.M.; Getsy, J.A.; et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm. Pharmacol. Ther. 2013, 26, 672–676. [Google Scholar] [CrossRef] [PubMed]
- van den Blink, B.; Dillingh, M.R.; Ginns, L.C.; Morrison, L.D.; Moerland, M.; Wijsenbeek, M.; Trehu, E.G.; Bartholmai, B.; Burggraaf, J. Recombinant human pentraxin-2 therapy in patients with idiopathic pulmonary fibrosis: Safety, pharmacokinetics and exploratory efficacy. Eur. Respir. J. 2016, 47, 889–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnon, L.; Leduc, M.; Thibodeau, J.-F.; Zhang, M.-Z.; Grouix, B.; Sarra-Bournet, F.; Gagnon, W.; Hince, K.; Tremblay, M.; Geerts, L.; et al. A Newly Discovered Antifibrotic Pathway Regulated by Two Fatty Acid Receptors. Am. J. Pathol. 2018, 188, 1132–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, N.; Manganas, H.; Ryerson, C.J.; Shapera, S.; Cantin, A.M.; Hernandez, P.; Turcotte, E.E.; Parker, J.M.; Moran, J.E.; Albert, G.R.; et al. Phase 2 clinical trial of PBI-4050 in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 53, 1800663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Effect of Recombinant Human Pentraxin 2 vs. Placebo on Change in Forced Vital Capacity in Patients with Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Zapater, E.; Signes-Costa, J.; Montero, P.; Roger, I. Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts? Biomedicines 2022, 10, 1423. https://doi.org/10.3390/biomedicines10061423
Ortiz-Zapater E, Signes-Costa J, Montero P, Roger I. Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts? Biomedicines. 2022; 10(6):1423. https://doi.org/10.3390/biomedicines10061423
Chicago/Turabian StyleOrtiz-Zapater, Elena, Jaime Signes-Costa, Paula Montero, and Inés Roger. 2022. "Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts?" Biomedicines 10, no. 6: 1423. https://doi.org/10.3390/biomedicines10061423