
DYRK1A and Activity-Dependent Neuroprotective Protein Comparative Diagnosis Interest in Cerebrospinal Fluid and Plasma in the Context of Alzheimer-Related Cognitive Impairment in Down Syndrome Patients

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Mice
2.2. Cohorts of Patients
2.3. Cell Lines and Culture Conditions
2.4. Protein Extraction and Analysis
2.5. Preparation of Plasma and CSF Samples and Essays
2.6. Data Analysis
3. Results
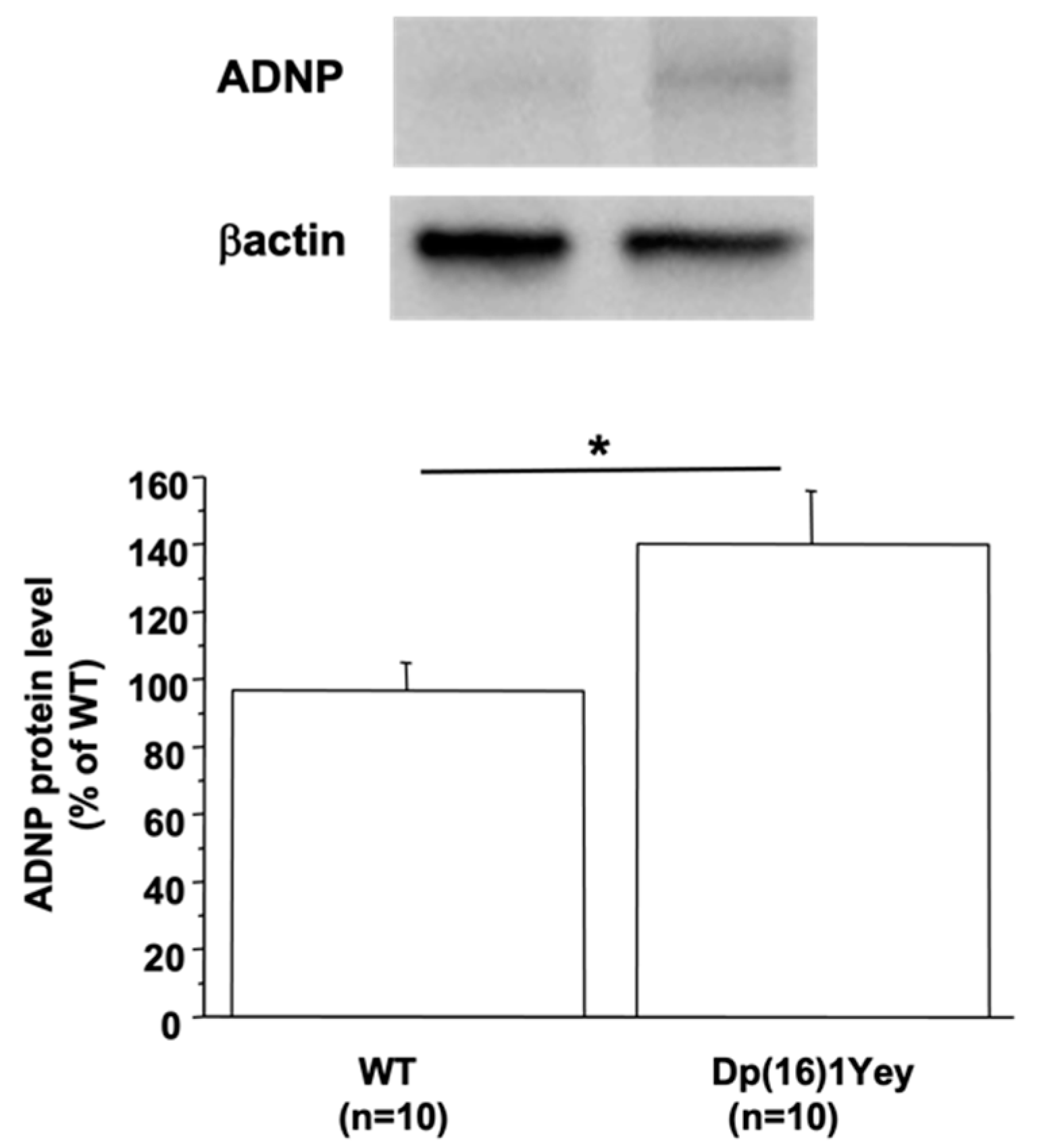
3.1. ADNP Protein Level Is Linked to DYRK1A Protein Level in the Mouse Brain
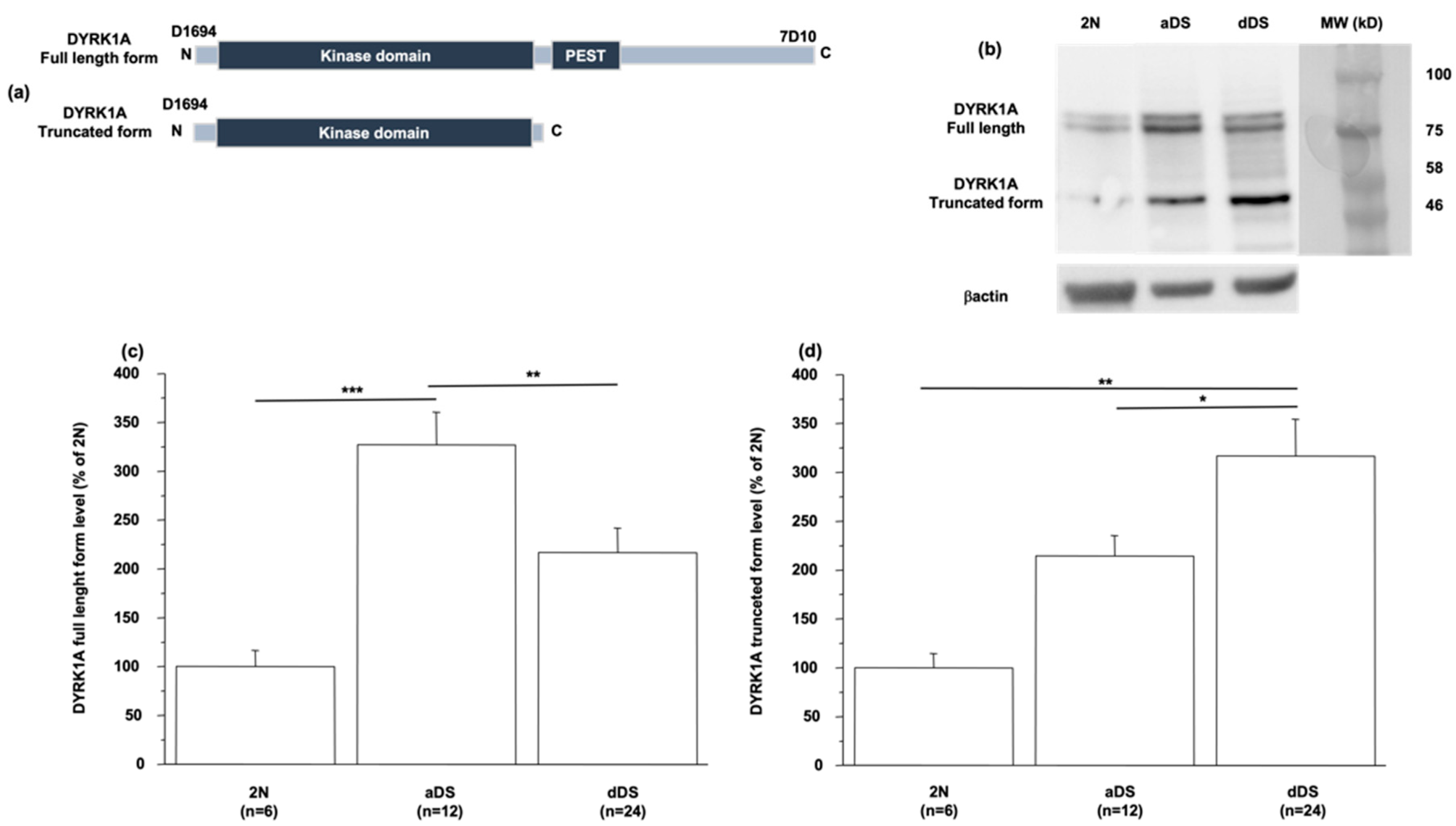
3.2. DYRK1A Protein Level Is Modified in LCLs from DS Patients with AD Dementia
3.3. DYRK1A Protein Level Is Modified in CSF and Plasma in DSAD
3.4. DYRK1A Protein Level Is Modified in CSF and Plasma from Sporadic AD Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lott, I.T.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010, 9, 623–633. [Google Scholar] [CrossRef]
- Lott, I.T. Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res. 2012, 197, 101–121. [Google Scholar] [PubMed] [Green Version]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.J.; Fisher, E.M.C.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafii, M.S.; Wishnek, H.; Brewer, J.B.; Donohue, M.C.; Ness, S.; Mobley, W.C.; Aisen, P.S.; Rissman, R.A. The down syndrome biomarker initiative (DSBI) pilot: Proof of concept for deep phenotyping of Alzheimer’s disease biomarkers in down syndrome. Front. Behav. Neurosci. 2015, 9, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafii, M.S.; Lukic, A.S.; Andrews, R.D.; Brewer, J.; Rissman, R.A.; Strother, S.C.; Wernick, M.N.; Pennington, C.; Mobley, W.C.; Ness, S.; et al. Down Syndrome Biomarker Initiative and the Alzheimer’s Disease Neuroimaging Initiative. PET Imaging of Tau Pathology and Relationship to Amyloid, Longitudinal MRI, and Cognitive Change in Down Syndrome: Results from the Down Syndrome Biomarker Initiative (DSBI). J. Alzheimer’s Dis. 2017, 60, 439–450. [Google Scholar]
- Head, E.; Lott, I.T.; Wilcock, D.M.; Lemere, C.A. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr. Alzheimer Res. 2016, 13, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Davidson, Y.S.; Robinson, A.; Prasher, V.P.; Mann, D.M.A. The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer’s disease in individuals with Down syndrome. Acta Neuropathol. Commun. 2018, 6, 56. [Google Scholar] [CrossRef]
- Fortea, J.; Carmona-Iragui, M.; Benejam, B.; Fernandez, S.; Videla, L.; Barroeta, O.; Alcolea, D.; Pegueroles, J.; Munoz, L.; Berbin, O.; et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet Neurol. 2018, 17, 860–869. [Google Scholar] [CrossRef]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Gomez, W.; Morales, R.; Maracaja-Coutinho, V.; Parra, V.; Nassif, M. Down syndrome and Alzheimer’s disease: Common molecular traits beyond the amyloid precursor protein. Aging 2020, 12, 1011–1033. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.X. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortea, J.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernández, S.; Altuna, M.; Pegueroles, J.; Montal, V.; et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet 2020, 395, 1988–1997. [Google Scholar] [CrossRef]
- Fagan, A.M.; Henson, R.L.; Li, Y.; Boerwinkle, A.H.; Xiong, C.; Bateman, R.J.; Goate, A.; Ances, B.M.; Doran, E.; Christian, B.T.; et al. Alzheimer’s Biomarker Consortium–Down Syndrome; Dominantly Inherited Alzheimer Network: Comparison of CSF biomarkers in Down syndrome and autosomal dominant Alzheimer’s disease: A cross-sectional study. Lancet Neurol. 2021, 20, 615–626. [Google Scholar] [CrossRef]
- Janel, N.; Sarazin, M.; Corlier, F.; Corne, H.; de Souza, L.C.; Hamelin, L.; Aka, A.; Lagarde, J.; Blehaut, H.; Hindié, V.; et al. Plasma DYRK1A as a novel risk factor for Alzheimer’s disease. Transl. Psychiatry 2014, 4, e425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janel, N.; Alexopoulos, P.; Badel, A.; Lamari, F.; Camproux, A.C.; Lagarde, J.; Simon, S.; Feraudet-Tarisse, C.; Lamourette, P.; Arbones, M.; et al. Combined assessment of DYRK1A, BDNF and homocysteine levels as diagnostic marker for Alzheimer’s disease. Transl. Psychiatry 2017, 7, e1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Chung, K.C. New perspectives of Dyrk1A role in neurogenesis and neuropathologic features of Down syndrome. Exp. Neurobiol. 2013, 22, 244–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchon, A.; Herault, Y. DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in Down syndrome. Front. Behav. Neurosci. 2016, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Tejedor, F.J.; Hammerle, B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011, 278, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, J.; Gong, C.X.; Hwang, Y.W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef]
- Malishkevich, A.; Marshall, G.A.; Schultz, A.P.; Sperling, R.A.; Aharon-Peretz, J.; Gozes, I. Blood-Borne Activity-Dependent Neuroprotective Protein (ADNP) is Correlated with Premorbid Intelligence, Clinical Stage, and Alzheimer’s Disease Biomarkers. J. Alzheimer’s Dis. 2016, 50, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedj, F.; Pereira, P.L.; Najas, S.; Barallobre, M.J.; Chabert, C.; Souchet, B.; Sebrie, C.; Verney, C.; Herault, Y.; Arbones, M.; et al. DYRK1A: A master regulatory protein controlling brain growth. Neurobiol. Dis. 2012, 46, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, T.; Morishima, M.; Pao, A.; LaDuca, J.; Conroy, J.; Nowak, N.; Matsui, S.; Shiraishi, I.; Yu, Y.E. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum. Mol. Genet. 2007, 16, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Tlili, A.; Hoischen, A.; Ripoll, C.; Benabou, E.; Badel, A.; Ronan, A.; Touraine, R.; Grattau, Y.; Stora, S.; van Bon, B.; et al. BDNF and DYRK1A are variable and inversely correlated in lymphoblastoid cell lines from Down syndrome patients. Mol. Neurobiol. 2012, 46, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Aït Yahya-Graison, E.; Aubert, J.; Dauphinot, L.; Rivals, I.; Prieur, M.; Golfier, G.; Rossier, J.; Personnaz, L.; Creau, N.; Bléhaut, H.; et al. Classification of human chromosome 21 gene-expression variations in Down syndrome: Impact on disease phenotypes. Am. J. Hum. Genet. 2007, 81, 475–491. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Yin, X.; Gu, J.; Zhang, X.; Shi, J.; Qian, W.; Ji, Y.; Cao, M.; Gu, X.; Ding, F.; et al. Truncation and Activation of Dual Specificity Tyrosine Phosphorylation-regulated Kinase 1A by Calpain I: A Molecular Mechanism Linked to TAU Pathology in alzheimer disease. J. Biol. Chem. 2015, 290, 15219–15237. [Google Scholar] [CrossRef] [Green Version]
- Souchet, B.; Audrain, M.; Billard, J.M.; Dairou, J.; Fol, R.; Orefice, N.S.; Tada, S.; Gu, Y.; Dufayet-Chaffaud, G.; Limanton, E.; et al. Inhibition of DYRK1A proteolysis modifies its kinase specificity and rescues Alzheimer phenotype in APP/PS1 mice. Acta Neuropathol. Commun. 2019, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Montal, V.; Barroeta, I.; Bejanin, A.; Pegueroles, J.; Carmona-Iragui, M.; Altuna, M.; Benejam, B.; Videla, L.; Fernández, S.; Padilla, C.; et al. Metabolite Signature of Alzheimer’s Disease in Adults with Down Syndrome. Ann. Neurol. 2021, 90, 407–416. [Google Scholar] [CrossRef]
- Bartus, R. The calpain hypothesis of neurodegeneration: Evidence for a common cytotoxic pathway. Neuroscientist 1997, 3, 314–327. [Google Scholar] [CrossRef]
- Shumway, S.D.; Maki, M.; Miyamoto, S. The PEST domain of IkappaBalpha is necessary and sufficient for in vitro degradation by mu-calpain. J. Biol. Chem. 1999, 274, 30874–30881. [Google Scholar] [CrossRef] [Green Version]
- Latour, A.; Gu, Y.; Kassis, N.; Daubigney, F.; Colin, C.; Gausserès, B.; Middendorp, S.; Paul, J.L.; Hindié, V.; Rain, J.C.; et al. LPS-Induced Inflammation Abolishes the Effect of DYRK1A on IkB Stability in the Brain of Mice. Mol. Neurobiol. 2019, 56, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Oda, Y.; Tomizawa, K.; Gong, C.X. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J. Biol. Chem. 2005, 280, 37755–37762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, D.M.; Hurban, J.; Helman, A.M.; Sudduth, T.L.; McCarty, K.L.; Beckett, T.L.; Ferrell, J.C.; Murphy, M.P.; Abner, E.L.; Schmitt, F.A.; et al. Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2468–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenneman, D.E.; Spong, C.Y.; Hauser, J.M.; Abebe, D.; Pinhasov, A.; Golian, T.; Gozes, I. Protective peptides that are orally active and mechanistically nonchiral. J. Pharmacol. Exp. Ther. 2004, 309, 1190–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozes, I.; Ivashko-Pachima, Y. ADNP: In search for molecular mechanisms and innovative therapeutic strategies for frontotemporal degeneration. Front. Aging Neurosci. 2015, 7, 205. [Google Scholar] [CrossRef]
- Toso, L.; Cameroni, I.; Roberson, R.; Abebe, D.; Bissell, S.; Spong, C.Y. Prevention of developmental delays in a Down syndrome mouse model. Obstet. Gynecol. 2008, 112, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Incerti, M.; Toso, L.; Vink, J.; Roberson, R.; Nold, C.; Abebe, D.; Spong, C.Y. Prevention of Learning Deficit in a Down Syndrome Model. Obstet. Gynecol. 2011, 117, 354–361. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Gray, A.J.; Hirata-Fukae, C.; Minami, S.S.; Waterhouse, E.G.; Mattson, M.P.; LaFerla, F.M.; Gozes, I.; Aisen, P.S. Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of AD at early pathological stage. J. Mol. Neurosci. 2007, 31, 165–170. [Google Scholar] [CrossRef]
- Vulih-Shultzman, I.; Pinhasov, A.; Mandel, S.; Grigoriadis, N.; Touloumi, O.; Pittel, Z.; Gozes, I. Activity- dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J. Pharmacol. Exp. Ther. 2007, 323, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Schirer, Y.; Malishkevich, A.; Ophir, Y.; Lewis, J.; Giladi, E.; Gozes, I. Novel Marker for the Onset of Frontotemporal Dementia: Early Increase in Activity-Dependent Neuroprotective Protein (ADNP) in the Face of Tau Mutation. PLoS ONE 2014, 9, e87383. [Google Scholar] [CrossRef] [Green Version]
- Gozes, I.; Iram, T.; Maryanovsky, E.; Arviv, C.; Rozenberg, L.; Schirer, Y.; Giladi, E.; Furman-Assaf, S. Novel Tubulin and Tau Neuroprotective Fragments Sharing Structural Similarities with the Drug Candidate NAP (Davuentide). J. Alzheimer’s Dis. 2014, 40, S23–S36. [Google Scholar] [CrossRef] [PubMed]
- Braitch, M.; Kawabe, K.; Nyirenda, M.; Gilles, L.J.; Robin, R.A.; Gran, B.; Murphy, S.; Showe, L.; Constantinescu, C. Expression of activity-dependent neuroprotective protein in the immune system: Possible functions and relevance to multiple sclerosis. NeuroimmunoModulation 2010, 17, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Controls | aDS | pDS | dDS | |
|---|---|---|---|---|
| Mean age at collection (years) | 45 | 40 | 52 | 55 |
| N (CSF) | 19 | 45 | 22 | 27 |
| Female/male (CSF) | 13F/6M | 22F/23M | 11F/11M | 12F/15M |
| CSF DYRK1A long form (ng/mL) | 5 ± 0.3 | 7.4 ± 0.7 * | 4 ± 0.35 $$$ | 4 ± 0.3 $$$ |
| CSF DYRK1A long and short form (ng/mL) | 7.1 ± 0.8 | 9.4 ± 1 * | 14.8 ± 2.2 $ | 19.6 ± 3.1 $$$ |
| CSF ADNP (ng/mL) | 34.9 ± 1.3 | 40.1 ± 2 * | 33.1 ± 2.1 $ | 26.3 ± 2.5 $$$, § |
| N (Plasma) | 19 | 77 | 22 | 57 |
| Female/male (Plasma) | 10F/9M | 36F/41M | 10F/12M | 28F/29M |
| Plasma DYRK1A long form (ng/mL) | 2.8 ± 0.4 | 3.4 ± 0.2 * | 2.1 ± 0.3 $$ | 2.4 ± 0.2 $$ |
| Plasma DYRK1A long and short form (ng/mL) | 3.7± 0.5 | 5.2 ± 0.35 * | 6.5 ± 1 | 7.65 ± 0.6 $$$ |
| Plasma ADNP (ng/mL) | 1.5 ± 0.1 | 2 ± 0.1 * | 0.6 ± 0.1 $$$ | 1.5 ± 0.1 $$, §§ |
| Group | Controls | AD |
|---|---|---|
| N | 9 | 18 |
| Mean age at collection (years) | 67 | 64 |
| Female/male | 5F/4M | 12F/6M |
| CSF DYRK1A long form (ng/mL) | 5.7± 0.4 | 3.7 ± 0.6 * |
| CSF DYRK1A long and short form (ng/mL) | 9.8 ± 2.4 | 21 ± 3.6 * |
| CSF ADNP (ng/mL) | 35.5 ± 4.6 | 24.2 ± 2.2 * |
| Plasma DYRK1A long form (ng/mL) | 2 ± 0.3 | 1.4 ± 0.1 * |
| Plasma DYRK1A long and short form (ng/mL) | 3.5 ± 0.5 | 4.6 ± 0.3 |
| Plasma ADNP (ng/mL) | 1.4 ± 0.1 | 2.5 ± 0.2 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreau, M.; Carmona-Iragui, M.; Altuna, M.; Dalzon, L.; Barroeta, I.; Vilaire, M.; Durand, S.; Fortea, J.; Rebillat, A.-S.; Janel, N. DYRK1A and Activity-Dependent Neuroprotective Protein Comparative Diagnosis Interest in Cerebrospinal Fluid and Plasma in the Context of Alzheimer-Related Cognitive Impairment in Down Syndrome Patients. Biomedicines 2022, 10, 1380. https://doi.org/10.3390/biomedicines10061380
Moreau M, Carmona-Iragui M, Altuna M, Dalzon L, Barroeta I, Vilaire M, Durand S, Fortea J, Rebillat A-S, Janel N. DYRK1A and Activity-Dependent Neuroprotective Protein Comparative Diagnosis Interest in Cerebrospinal Fluid and Plasma in the Context of Alzheimer-Related Cognitive Impairment in Down Syndrome Patients. Biomedicines. 2022; 10(6):1380. https://doi.org/10.3390/biomedicines10061380
Chicago/Turabian StyleMoreau, Manon, Maria Carmona-Iragui, Miren Altuna, Lorraine Dalzon, Isabel Barroeta, Marie Vilaire, Sophie Durand, Juan Fortea, Anne-Sophie Rebillat, and Nathalie Janel. 2022. "DYRK1A and Activity-Dependent Neuroprotective Protein Comparative Diagnosis Interest in Cerebrospinal Fluid and Plasma in the Context of Alzheimer-Related Cognitive Impairment in Down Syndrome Patients" Biomedicines 10, no. 6: 1380. https://doi.org/10.3390/biomedicines10061380