
Low-Grade Inflammation in the Pathogenesis of Osteoarthritis: Cellular and Molecular Mechanisms and Strategies for Future Therapeutic Intervention

 , and
, and
Abstract
:1. Introduction
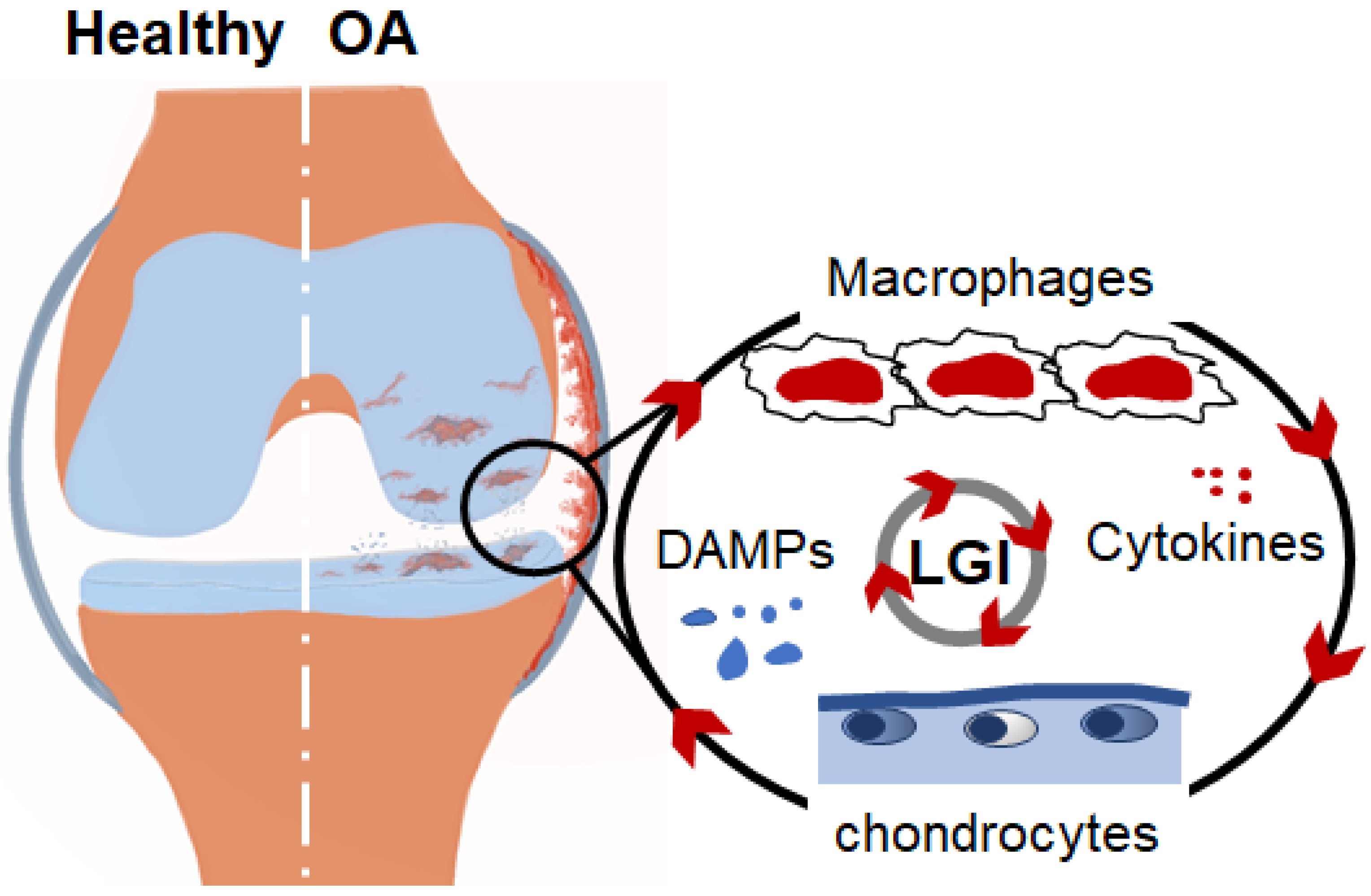
2. Interplay between Chondrocytes and Macrophages Represents Positive Feedback Loops Triggering Inflammation in the OA Joint
3. DAMPs Switch LGI and the OA Process in the Joint
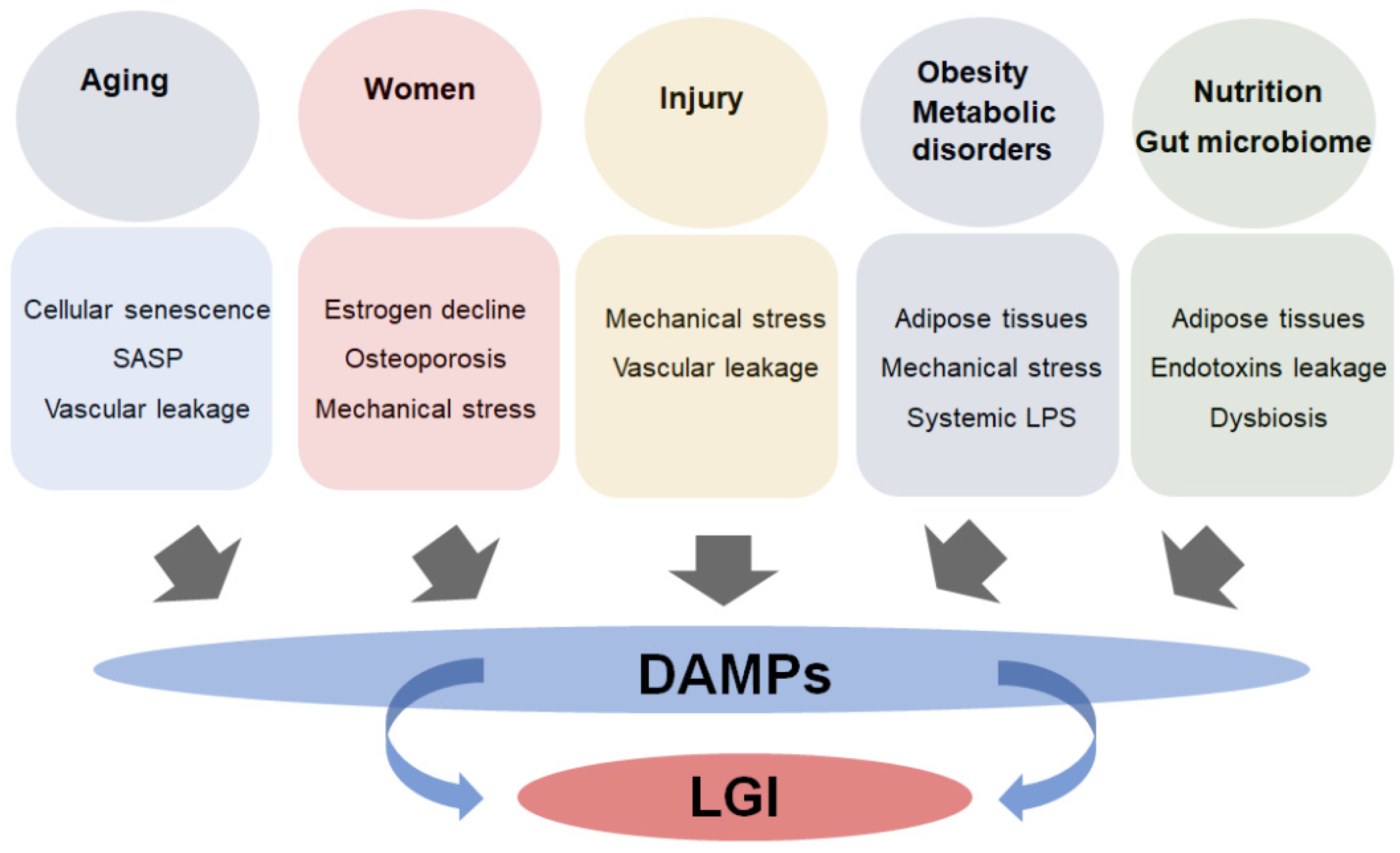
4. Risk Factors Associated with LGI and Joint Destruction in OA
4.1. Aging
4.2. Injury
4.3. Sex
4.4. Obesity and Metabolic Disorders
4.5. Nutrition and Gut Microbiome Dysbiosis
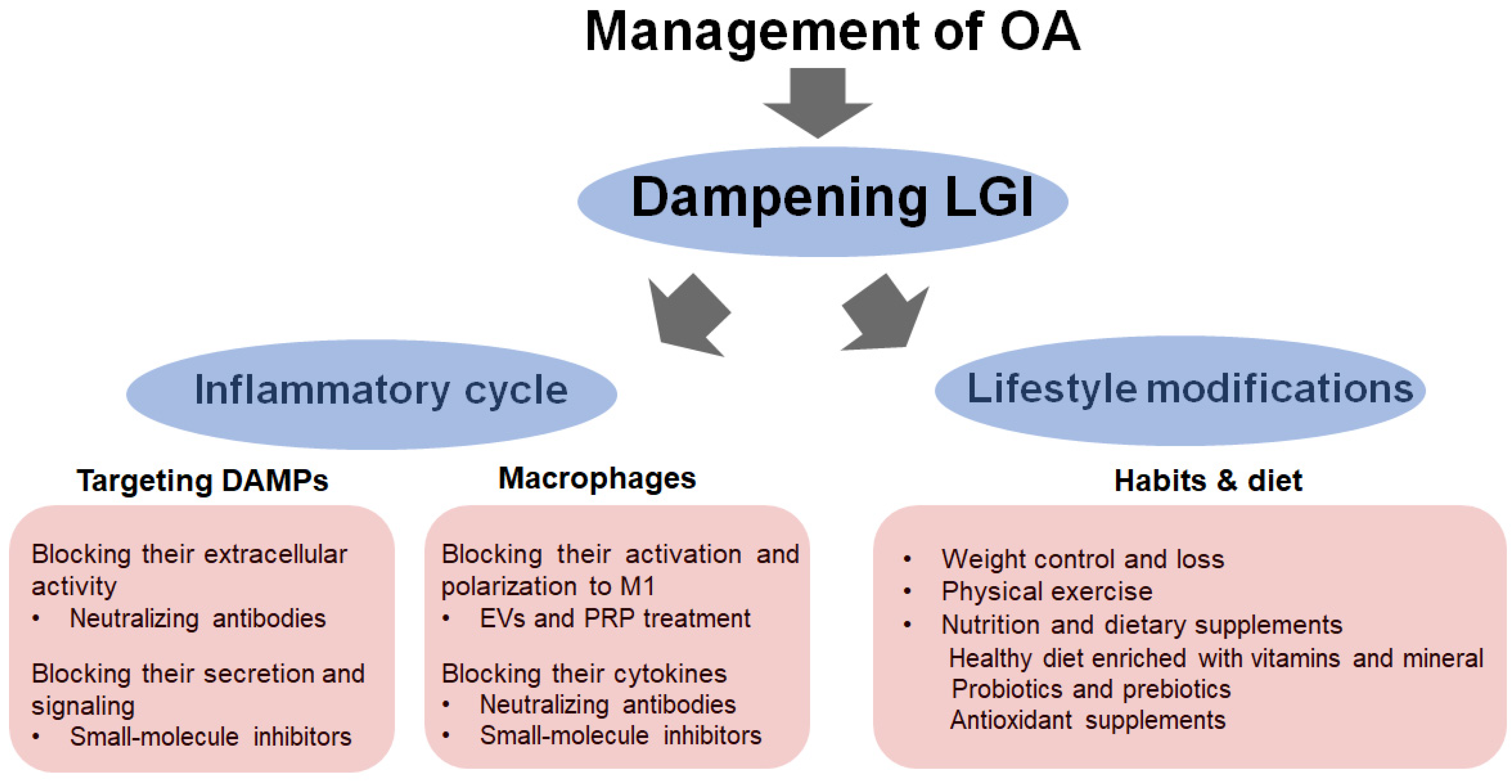
5. Dampening LGI as a Strategy for Managing OA
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Standardization of Osteoarthritis Definitions. Available online: https://www.oarsi.org/research/standardization-osteoarthritis-definitions. (accessed on 1 April 2019).
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Hunter, D.J.; March, L.; Chew, M. Osteoarthritis in 2020 and beyond: A Lancet Commission. Lancet 2020, 396, 1711–1712. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R. Role of low-grade inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2017, 29, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.-H.; Jain, V.; Gibson, J.; Attarian, D.E.; Haraden, C.A.; Yohn, C.B.; Laberge, R.-M.; Gregory, S.; Kraus, V.B. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci. Rep. 2020, 10, 10868. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Terkawi, M.A.; Onodera, T.; Tian, Y.; Ebata, T.; Matsumae, G.; Alhasan, H.; Takahashi, D.; Iwasaki, N. Transcriptional profiling of murine macrophages stimulated with cartilage fragments revealed a strategy for treatment of progressive osteoarthritis. Sci. Rep. 2020, 10, 7558. [Google Scholar] [CrossRef]
- Hamasaki, M.; Terkawi, M.A.; Onodera, T.; Homan, K.; Iwasaki, N. A Novel Cartilage Fragments Stimulation Model Revealed that Macrophage Inflammatory Response Causes an Upregulation of Catabolic Factors of Chondrocytes In Vitro. Cartilage 2021, 12, 354–361. [Google Scholar] [CrossRef]
- Mobasheri, A.; Barrett-Jolley, R.; Carter, S.D.; Martín-Vasallo, P.; Schulze-Tanzil, G.; Shakibaei, M. Functional Roles of Mechanosensitive Ion Channels, ß1 Integrins and Kinase Cascades in Chondrocyte Mechanotransduction. In Mechanosensitivity in Cells and Tissues; Kamkin, A., Kiseleva, I., Eds.; Academia Publishing House Ltd.: Moscow, Russia, 2005. [Google Scholar]
- Berenbaum, F.; Eymard, F.; Houard, X. Osteoarthritis, inflammation and obesity. Curr. Opin. Rheumatol. 2013, 25, 114–118. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Anderson, D.D.; Brown, T.D.; Tochigi, Y.; Martin, J.A. The Roles of Mechanical Stresses in the Pathogenesis of Osteoarthritis: Implications for Treatment of Joint Injuries. Cartilage 2013, 4, 286–294. [Google Scholar] [CrossRef]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.M.; Scanzello, C.R. Innate inflammation and synovial macrophages in osteoarthritis pathophysiology. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 120), 57–63. [Google Scholar] [PubMed]
- Thomson, A.; Hilkens, C.M.U. Synovial Macrophages in Osteoarthritis: The Key to Understanding Pathogenesis? Front. Immunol. 2021, 12, 678757. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Shimizu, T.; Matsumae, G.; Ebata, T.; Alhasan, H.; Takahashi, D.; Terkawi, M.A.; Iwasaki, N. Inflammasome Activation in the Hip Synovium of Rapidly Destructive Coxopathy Patients and Its Relationship with the Development of Synovitis and Bone Loss. Am. J. Pathol. 2022, 192, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Nefla, M.; Holzinger, D.; Berenbaum, F.; Jacques, C. The danger from within: Alarmins in arthritis. Nat. Rev. Rheumatol. 2016, 12, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.H.; Rai, V.; Dilisio, M.F.; Agrawal, D.K. Damage-associated molecular patterns in the pathogenesis of osteoarthritis: Potentially novel therapeutic targets. Mol. Cell. Biochem. 2017, 434, 171–179. [Google Scholar] [CrossRef]
- van den Bosch, M.H.J. Inflammation in osteoarthritis: Is it time to dampen the alarm(in) in this debilitating disease? Clin. Exp. Immunol. 2019, 195, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Lambert, C.; Zappia, J.; Sanchez, C.; Florin, A.; Dubuc, J.E.; Henrotin, Y. The Damage-Associated Molecular Patterns (DAMPs) as Potential Targets to Treat Osteoarthritis: Perspectives From a Review of the Literature. Front. Med. 2020, 7, 607186. [Google Scholar] [CrossRef]
- Ebata, T.; Terkawi, M.A.; Hamasaki, M.; Matsumae, G.; Onodera, T.; Aly, M.K.; Yokota, S.; Alhasan, H.; Shimizu, T.; Takahashi, D.; et al. Flightless I is a catabolic factor of chondrocytes that promotes hypertrophy and cartilage degeneration in osteoarthritis. iScience 2021, 24, 102643. [Google Scholar] [CrossRef]
- Bhosale, A.M.; Richardson, J.B. Articular cartilage: Structure, injuries and review of management. Br. Med. Bull. 2008, 87, 77–95. [Google Scholar] [CrossRef]
- Zhang, H.; Lin, C.; Zeng, C.; Wang, Z.; Wang, H.; Lu, J.; Liu, X.; Shao, Y.; Zhao, C.; Pan, J.; et al. Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through R-spondin-2. Ann. Rheum. Dis. 2018, 77, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.B.; van Lent, P.L.; Holthuysen, A.E.; van der Kraan, P.M.; Roth, J.; van Rooijen, N.; van den Berg, W.B. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthr. Cartil. 2004, 12, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, A.B.; Van Lent, P.L.; Libregts, S.; Holthuysen, A.E.; Van Der Kraan, P.M.; Van Rooijen, N.; Van Den Berg, W.B. Crucial role of macrophages in matrix metalloproteinase-mediated cartilage destruction during experimental osteoarthritis: Involvement of matrix metalloproteinase. Arthritis Rheum. 2007, 56, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, J.; Blom, A.B.; Wainwright, S.; Hughes, C.; Caterson, B.; Berg, W.B.V.D. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Care Res. 2010, 62, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef]
- Liu-Bryan, R.; Terkeltaub, R. The growing array of innate inflammatory ignition switches in osteoarthritis. Arthritis Care Res. 2012, 64, 2055–2058. [Google Scholar] [CrossRef] [Green Version]
- Gómez, R.; Villalvilla, A.; Largo, R.; Gualillo, O.; Herrero-Beaumont, G. TLR4 signalling in osteoarthritis—finding targets for candidate DMOADs. Nat. Rev. Rheumatol. 2015, 11, 159–170. [Google Scholar] [CrossRef]
- Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Rahmati, M.; Mobasheri, A.; Mozafari, M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone 2016, 85, 81–90. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Larkin, D.J.; Kartchner, J.Z.; Doxey, A.S.; Hollis, W.R.; Rees, J.L.; Wilhelm, S.K.; Draper, C.S.; Peterson, D.M.; Jackson, G.G.; Ingersoll, C.; et al. Inflammatory markers associated with osteoarthritis after destabilization surgery in young mice with and without Receptor for Advanced Glycation End-products (RAGE). Front. Physiol. 2013, 4, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Feghali, K.; Grenier, D. Priming Effect of Fibronectin Fragments on the Macrophage Inflammatory Response: Potential Contribution to Periodontitis. Inflammation 2012, 35, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Su, S.L.; Tsai, C.D.; Lee, C.H.; Salter, D.M.; Lee, H.S. Expression and regulation of Toll-like receptor 2 by IL-1beta and fibronectin fragments in human articular chondrocytes. Osteoarthr. Cartil. 2005, 13, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.S.; Park, S.J.; Cheon, E.J.; Lee, M.H.; Kim, H.A. Fibronectin fragment-induced expression of matrix metalloproteinases is mediated by MyD88-dependent TLR-2 signaling pathway in human chondrocytes. Arthritis Res. Ther. 2015, 17, 320. [Google Scholar] [CrossRef] [Green Version]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor. J. Exp. Med. 2002, 195, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, L.; Babelova, A.; Kiss, E.; Hausser, H.-J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Götte, M.; et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Investig. 2005, 115, 2223–2233. [Google Scholar] [CrossRef]
- Barreto, G.; Soininen, A.; Ylinen, P.; Sandelin, J.; Konttinen, Y.T.; Nordström, D.C.; Eklund, K.K. Soluble biglycan: A potential mediator of cartilage degradation in osteoarthritis. Arthritis Res. Ther. 2015, 17, 379. [Google Scholar] [CrossRef] [Green Version]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef]
- Pap, T.; Bertrand, J. Syndecans in cartilage breakdown and synovial inflammation. Nat. Rev. Rheumatol. 2012, 9, 43–55. [Google Scholar] [CrossRef]
- Fichter, M.; Körner, U.; Schömburg, J.; Jennings, L.; Cole, A.A.; Mollenhauer, J. Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. J. Orthop. Res. 2006, 24, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T. Type II collagen peptide stimulates Akt leading to nuclear factor-κB activation: Its inhibition by hyaluronan. Biomed. Res. 2014, 35, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, S.; Golub, S.B.; Last, K.; Zeng, W.; Jackson, D.C.; Sutton, P.; Fosang, A.J. Bioactivity in an Aggrecan 32-mer Fragment Is Mediated via Toll-like Receptor. Arthritis Rheumatol. 2015, 67, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Liu-Bryan, R.; Pritzker, K.; Firestein, G.S.; Terkeltaub, R. TLR2 Signaling in Chondrocytes Drives Calcium Pyrophosphate Dihydrate and Monosodium Urate Crystal-Induced Nitric Oxide Generation. J. Immunol. 2005, 174, 5016–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, A.K. Crystals, inflammation, and osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 170–173. [Google Scholar] [CrossRef] [Green Version]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Yan, X.X.; Lu, L.; Peng, W.H.; Wang, L.J.; Zhang, Q.; Zhang, R.Y.; Qiu, J.; Wei, F. Increased serum HMGB1 level is associated with coronary artery disease in nondiabetic and type 2 diabetic patients. Atherosclerosis 2009, 205, 544–548. [Google Scholar] [CrossRef]
- Pisetsky, D.S.; Gauley, J.; Ullal, A.J. HMGB1 and Microparticles as Mediators of the Immune Response to Cell Death. Antioxidants Redox Signal. 2011, 15, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Kurobe, H.; Higashida, M.; Fukuda, D.; Shimabukuro, M.; Tanaka, K.; Higashikuni, Y.; Kitagawa, T.; Sata, M. HMGB1 plays a critical role in vascular inflammation and lesion formation via toll-like receptor. Atherosclerosis 2013, 231, 227–233. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, Z.; Hu, X.; Shu, C. HMGB1: A critical mediator for oxidized-low density lipoproteins induced atherosclerosis. Int. J. Cardiol. 2016, 202, 956–957. [Google Scholar] [CrossRef] [Green Version]
- Pieters, B.C.H.; Cappariello, A.; Bosch, M.H.J.V.D.; Van Lent, P.L.E.M.; Teti, A.; Van De Loo, F.A.J. Macrophage-Derived Extracellular Vesicles as Carriers of Alarmins and Their Potential Involvement in Bone Homeostasis. Front. Immunol. 2019, 10, 1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terkawi, M.A.; Matsumae, G.; Shimizu, T.; Takahashi, D.; Kadoya, K.; Iwasaki, N. Interplay between Inflammation and Pathological Bone Resorption: Insights into Recent Mechanisms and Pathways in Related Diseases for Future Perspectives. Int. J. Mol. Sci. 2022, 23, 1786. [Google Scholar] [CrossRef] [PubMed]
- García-Arnandis, I.; Guillén, M.I.; Gomar, F.; Pelletier, J.-P.; Martel-Pelletier, J.; Alcaraz, M.J. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1β in osteoarthritic synoviocytes. Arthritis Res. Ther. 2010, 12, R165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denoble, A.E.; Huffman, K.M.; Stabler, T.V.; Kelly, S.J.; Hershfield, M.S.; McDaniel, G.E.; Coleman, R.E.; Kraus, V.B. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc. Natl. Acad. Sci. USA 2011, 108, 2088–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumahashi, N.; Naitou, K.; Nishi, H.; Oae, K.; Watanabe, Y.; Kuwata, S.; Ochi, M.; Ikeda, M.; Uchio, Y. Correlation of changes in pain intensity with synovial fluid adenosine triphosphate levels after treatment of patients with osteoarthritis of the knee with high-molecular-weight hyaluronic acid. Knee 2011, 18, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-C.; Zhang, H.-Y.; Shao, L.; Chen, L.; Liu, Z.-H.; He, X.; Gong, W.-X. S100A12 levels in synovial fluid may reflect clinical severity in patients with primary knee osteoarthritis. Biomarkers 2013, 18, 216–220. [Google Scholar] [CrossRef]
- Ke, X.; Jin, G.; Yang, Y.; Cao, X.; Fang, R.; Feng, X.; Lei, B. Synovial Fluid HMGB-1 Levels are Associated with Osteoarthritis Severity. Clin. Lab. 2015, 61, 809–818. [Google Scholar] [CrossRef]
- Liu-Bryan, R.; Terkeltaub, R. Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum. 2010, 62, 2004–2012. [Google Scholar]
- Magna, M.; Pisetsky, D.S. The Role of HMGB1 in the Pathogenesis of Inflammatory and Autoimmune Diseases. Mol. Med. 2014, 20, 138–146. [Google Scholar] [CrossRef]
- Aulin, C.; Lassacher, T.; Palmblad, K.; Harris, H.E. Early stage blockade of the alarmin HMGB1 reduces cartilage destruction in experimental OA. Osteoarthr. Cartil. 2020, 28, 698–707. [Google Scholar] [CrossRef] [Green Version]
- Yammani, R.R. S100 proteins in cartilage: Role in arthritis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bosch, M.H.; Blom, A.B.; Schelbergen, R.F.; Vogl, T.; Roth, J.P.; Slöetjes, A.W.; van den Berg, W.B.; van der Kraan, P.M.; van Lent, P.L.E.M. Induction of Canonical Wnt Signaling by the Alarmins S100A8/A9 in Murine Knee Joints: Implications for Osteoarthritis. Arthritis Rheumatol. 2016, 68, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNulty, A.L.; Rothfusz, N.E.; Leddy, H.A.; Guilak, F. Synovial fluid concentrations and relative potency of interleukin-1 alpha and beta in cartilage and meniscus degradation. J. Orthop. Res. 2013, 31, 1039–1045. [Google Scholar] [CrossRef] [Green Version]
- Ryan, L.M.; Kurup, I.V.; Derfus, B.A.; Kushnaryov, V.M. ATP-induced chondrocalcinosis. Arthritis Rheum. 1992, 35, 1520–1525. [Google Scholar] [CrossRef]
- Zhao, L.; Xing, R.; Wang, P.; Zhang, N.; Yin, S.; Li, X.; Zhang, L. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP-induced pyroptosis in knee osteoarthritis. Mol. Med. Rep. 2018, 17, 5463–5469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J. IL-33 is a potential new target in OA. Nat. Rev. Rheumatol. 2021, 17, 3. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Y.; Chen, H.; Liu, X.; Li, M. Blocking Interleukin-33 Alleviates the Joint Inflammation and Inhibits the Development of Collagen-Induced Arthritis in Mice. J. Immunol. Res. 2020, 2020, 4297354. [Google Scholar] [CrossRef]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Toh, W.S.; Brittberg, M.; Farr, J.; Foldager, C.B.; Gomoll, A.H.; Hui, J.H.P.; Richardson, J.B.; Roberts, S.; Spector, M. Cellular senescence in aging and osteoarthritis. Acta Orthop. 2016, 87 (Suppl. 363), 6–14. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.A.; Buckwalter, J.A. The role of chondrocyte senescence in the pathogenesis of osteoarthritis and in limiting cartilage repair. J. Bone Jt. Surg. Am. 2003, 85 (Suppl. 2), 106–110. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Gandhi, U.; Long, D.L.; Yin, W.; Chubinskaya, S. Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol. 2014, 66, 2201–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felson, D.; Lawrence, R.C.; Dieppe, P.A.; Hirsch, R.; Helmick, C.G.; Jordan, J.M.; Kington, R.S.; Lane, N.E.; Nevitt, M.C.; Zhang, Y.; et al. Osteoarthritis: New Insights. Part 1: The Disease and Its Risk Factors. Ann. Intern. Med. 2000, 133, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, T.; Angelov, A.K. Modifiable risk factors in knee osteoarthritis: Treatment implications. Rheumatol. Int. 2019, 39, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Bigoni, M.; Sacerdote, P.; Turati, M.; Franchi, S.; Gandolla, M.; Gaddi, D.; Moretti, S.; Munegato, D.; Augusti, C.A.; Bresciani, E.; et al. Acute and late changes in intraarticular cytokine levels following anterior cruciate ligament injury. J. Orthop. Res. 2013, 31, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.J.; Bonnet, C.; Stadnik, P.; Duance, V.C.; Mason, D.; Blain, E.J. Inflammatory and degenerative phases resulting from anterior cruciate rupture in a non-invasive murine model of post-traumatic osteoarthritis. J. Orthop. Res. 2018, 36, 2118–2127. [Google Scholar] [CrossRef] [Green Version]
- Straub, R.H. The Complex Role of Estrogens in Inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Giuliani, F. The Role of Inflammation in Depression and Fatigue. Front. Immunol. 2019, 10, 1696. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Toups, M.; Nemeroff, C.B. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron 2020, 107, 234–256. [Google Scholar] [CrossRef]
- Ji, X.; Singleterry, S.; Kulikova, A.; Harrison, Y.; Shivakumar, G.; Brown, E.S. Association of menopause symptoms with depressive symptom severity in a diverse community-based sample. Maturitas 2021, 143, 78–80. [Google Scholar] [CrossRef]
- Coggon, D.; Reading, I.; Croft, P.; McLaren, M.; Barrett, D.; Cooper, C. Knee osteoarthritis and obesity. Int. J. Obes. 2001, 25, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anandacoomarasamy, A.; Fransen, M.; March, L. Obesity and the musculoskeletal system. Curr. Opin. Rheumatol. 2009, 21, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Blagojevic, M.; Jinks, C.; Jeffery, A.; Jordan, K. Risk factors for onset of osteoarthritis of the knee in older adults: A systematic review and meta-analysis. Osteoarthr. Cartil. 2010, 18, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.; Li, K.; Li, X.; Yu, X.; Wang, W.; Ding, L.; Liu, L. Oral Resveratrol Prevents Osteoarthritis Progression in C57BL/6J Mice Fed a High-Fat Diet. Nutrients 2016, 8, 233. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Huang, Z.; Kraus, V.B. Does lipopolysaccharide-mediated inflammation have a role in OA? Nat. Rev. Rheumatol. 2016, 12, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Stabler, T.; Pei, F.; Kraus, V. Both systemic and local lipopolysaccharide (LPS) burden are associated with knee OA severity and inflammation. Osteoarthr. Cartil. 2016, 24, 1769–1775. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, M.T.; Katz, J.D. Osteoarthritis: Another component of metabolic syndrome? Metab Syndr Relat Disord. 2010, 8, 295–305. [Google Scholar] [CrossRef]
- Zhuo, Q.; Yang, W.; Chen, J.; Wang, Y. Metabolic syndrome meets osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 729–737. [Google Scholar] [CrossRef]
- Li, H.; George, D.M.; Jaarsma, R.L.; Mao, X. Metabolic syndrome and components exacerbate osteoarthritis symptoms of pain, depression and reduced knee function. Ann. Transl. Med. 2016, 4, 133. [Google Scholar] [CrossRef] [Green Version]
- Stürmer, T.; Sun, Y.; Sauerland, S.; Zeissig, I.; Günther, K.P.; Puhl, W.; Brenner, H. Serum cholesterol and osteoarthritis. The baseline examination of the Ulm Osteoarthritis Study. J. Rheumatol. 1998, 25, 1827–1832. [Google Scholar] [PubMed]
- Papathanasiou, I.; Anastasopoulou, L.; Tsezou, A. Cholesterol metabolism related genes in osteoarthritis. Bone 2021, 152, 116076. [Google Scholar] [CrossRef] [PubMed]
- Gkretsi, V.; Simopoulou, T.; Tsezou, A. Lipid metabolism and osteoarthritis: Lessons from atherosclerosis. Prog. Lipid Res. 2011, 50, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Farnaghi, S.; Crawford, R.; Xiao, Y.; Prasadam, I. Cholesterol metabolism in pathogenesis of osteoarthritis disease. Int. J. Rheum. Dis. 2017, 20, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliddal, H.; Leeds, A.R.; Christensen, R. Osteoarthritis, obesity and weight loss: Evidence, hypotheses and horizons—A scoping review. Obes. Rev. 2014, 15, 578–586. [Google Scholar] [CrossRef]
- Rhoads, J.P.; Major, A.S.; Rathmell, J.C. Fine tuning of immunometabolism for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 313–320. [Google Scholar] [CrossRef]
- Leong, D.J.; Choudhury, M.; Hirsh, D.M.; Hardin, J.A.; Cobelli, N.J.; Sun, H.B. Nutraceuticals: Potential for Chondroprotection and Molecular Targeting of Osteoarthritis. Int. J. Mol. Sci. 2013, 14, 23063–23085. [Google Scholar] [CrossRef] [Green Version]
- Diamanti-Kandarakis, E.; Palimeri, S.; Palioura, E. Current perspectives on the health risks associated with the consumption of advanced glycation end products: Recommendations for dietary management. Diabetes Metab. Syndr. Obesity Targets Ther. 2015, 8, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Driban, J.B.; Xu, C.; Lapane, K.L.; McAlindon, T.E.; Eaton, C. Dietary Fat Intake and Radiographic Progression of Knee Osteoarthritis: Data from the Osteoarthritis Initiative. Arthritis Care Res. 2017, 69, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.-E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Front. Microbiol. 2016, 7, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ding, W.; Wang, H.; Dai, L.; Zong, W.; Wang, Y.; Bi, J.; Han, W.; Dong, G. Gut microbiota and obesity-associated osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, A.; Morales, C.I.; Câmara, N.O.S.; Almeida, R.R. Innate Sensing of the Gut Microbiota: Modulation of Inflammatory and Autoimmune Diseases. Front. Immunol. 2016, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Li, F.; Pi, G. Association Between Gut Microbiota and Osteoarthritis: A Review of Evidence for Potential Mechanisms and Therapeutics. Front. Cell. Infect. Microbiol. 2022, 12, 812596. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.B.; Davis, K.M.; Lysenko, E.S.; Zhou, A.Y.; Yu, Y.; Weiser, J.N. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 2010, 16, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Korotkyi, O.; Kyriachenko, Y.; Kobyliak, N.; Falalyeyeva, T.; Ostapchenko, L. Crosstalk between gut microbiota and osteoarthritis: A critical view. J. Funct. Foods 2020, 68, 103904. [Google Scholar] [CrossRef]
- Amdekar, S.; Singh, V.; Singh, R.; Sharma, P.; Keshav, P.; Kumar, A. Lactobacillus casei reduces the inflammatory joint damage associated with collagen-induced arthritis (CIA) by reducing the pro-inflammatory cytokines: Lactobacillus casei: COX-2 inhibitor. J. Clin. Immunol. 2011, 31, 147–154. [Google Scholar] [CrossRef]
- Korotkyi, O.; Ostapchenko, L.; Vovk, A.; Galenova, T.; Vovk, T.; Dvorschenko, K.; Luzza, F.; Abenavoli, L.; Kobyliak, N.; Falalyeyeva, T.; et al. Effect of probiotic on serum cytokines and matrix metalloproteinases profiles during monoiodoacetate-induced osteoarthritis in rats. Minerva Biotecnol. 2019, 2019, 68–73. [Google Scholar] [CrossRef]
- Ohlsson, C.; Engdahl, C.; Fåk, F.; Andersson, A.; Windahl, S.H.; Farman, H.H.; Movérare-Skrtic, S.; Islander, U.; Sjögren, K. Probiotics Protect Mice from Ovariectomy-Induced Cortical Bone Loss. PLoS ONE 2014, 9, e92368. [Google Scholar] [CrossRef]
- Lei, M.; Guo, C.; Wang, D.; Zhang, C.; Hua, L. The effect of probiotic Lactobacillus casei Shirota on knee osteoarthritis: A randomised double-blind, placebo-controlled clinical trial. Benef. Microbes. 2017, 8, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.L.; Bomhof, M.R.; Reimer, R.A.; Hart, D.A.; Collins, K.H.; Herzog, W. Protective effect of prebiotic and exercise intervention on knee health in a rat model of diet-induced obesity. Sci. Rep. 2019, 9, 3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortuna, R.; Hart, D.A.; Sharkey, K.A.; Schachar, R.A.; Johnston, K.; Reimer, R.A. Effect of a prebiotic supplement on knee joint function, gut microbiota, and inflammation in adults with co-morbid obesity and knee osteoarthritis: Study protocol for a randomized controlled trial. Trials 2021, 22, 255. [Google Scholar] [CrossRef] [PubMed]
- Schierbeck, H.; Lundbäck, P.; Palmblad, K.; Klevenvall, L.; Erlandsson-Harris, H.; Andersson, U.; Ottosson, L. Monoclonal Anti-HMGB1 (High Mobility Group Box Chromosomal Protein 1) Antibody Protection in Two Experimental Arthritis Models. Mol. Med. 2011, 17, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Lambert, C.; Borderie, D.; Dubuc, J.-E.; Rannou, F.; Henrotin, Y. Type II collagen peptide Coll2-1 is an actor of synovitis. Osteoarthr. Cartil. 2019, 27, 1680–1691. [Google Scholar] [CrossRef]
- Kokkola, R.; Li, J.; Sundberg, E.; Aveberger, A.-C.; Palmblad, K.; Yang, H.; Tracey, K.J.; Andersson, U.; Harris, H.E. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Care Res. 2003, 48, 2052–2058. [Google Scholar] [CrossRef]
- Hamada, T.; Torikai, M.; Kuwazuru, A.; Tanaka, M.; Horai, N.; Fukuda, T.; Yamada, S.; Nagayama, S.; Hashiguchi, K.; Sunahara, N.; et al. Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthritis Care Res. 2008, 58, 2675–2685. [Google Scholar] [CrossRef]
- Björk, P.; Björk, A.; Vogl, T.; Stenström, M.; Liberg, D.; Olsson, A.; Roth, J.; Ivars, F.; Leanderson, T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009, 7, e97. [Google Scholar] [CrossRef] [Green Version]
- Schelbergen, R.F.; Geven, E.J.; Bosch, M.H.J.V.D.; Eriksson, H.; Leanderson, T.; Vogl, T.; Roth, J.; Loo, F.A.J.V.D.; Koenders, M.I.; van der Kraan, P.M.; et al. Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase-induced osteoarthritis. Ann. Rheum. Dis. 2015, 74, 2254–2258. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, G.; Si, Z.; Yu, L.; Hou, L. Glycyrrhizin, an HMGB1 inhibitor, Suppresses Interleukin-1β-Induced Inflammatory Responses in Chondrocytes from Patients with Osteoarthritis. Cartilage 2021, 13 (Suppl. 2), 947S–955S. [Google Scholar] [CrossRef]
- Jung, S.M.; Lee, J.; Baek, S.Y.; Lee, J.; Jang, S.G.; Hong, S.-M.; Park, J.-S.; Cho, M.-L.; Park, S.-H.; Kwok, S.-K. Ethyl pyruvate ameliorates inflammatory arthritis in mice. Int. Immunopharmacol. 2017, 52, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Ogawa, H.; Kuramitsu, N.; Akaike, K.; Goto, A.; Aoki, H.; Lassar, A.; Suehara, Y.; Hara, A.; Matsumoto, K.; et al. Colchicine protects against cartilage degeneration by inhibiting MMP13 expression via PLC-γ1 phosphorylation. Osteoarthr. Cartil. 2021, 29, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Das, S.K.; Goel, G.; Asthana, A.; Agarwal, G.G. Does long term colchicine prevent degradation of collagen fiber network in osteoarthritis? Int. J. Rheum. Dis. 2018, 21, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.-Y.; Thumboo, J.; Wong, B.S.; Haaland, B.; Chowbay, B.; Chakraborty, B.; Tan, M.H.; Kraus, V.B. Colchicine effectiveness in symptom and inflammation modification in knee osteoarthritis (COLKOA): Study protocol for a randomized controlled trial. Trials 2015, 16, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, B.; Pei, W.; Qu, Y.; Zhang, R.; Chu, X.; Wang, Y.; Huang, X.; You, H. MCC950, the NLRP3 Inhibitor, Protects against Cartilage Degradation in a Mouse Model of Osteoarthritis. Oxidative Med. Cell. Longev. 2021, 2021, 4139048. [Google Scholar] [CrossRef]
- Chen, Q.-L.; Yin, H.-R.; He, Q.-Y.; Wang, Y. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 2021, 138, 111442. [Google Scholar] [CrossRef]
- Sun, Y.; Zuo, Z.; Kuang, Y. An Emerging Target in the Battle against Osteoarthritis: Macrophage Polarization. Int. J. Mol. Sci. 2020, 21, 8513. [Google Scholar] [CrossRef]
- Wenham, C.Y.; McDermott, M.; Conaghan, P. Biological Therapies in Osteoarthritis. Curr. Pharm. Des. 2015, 21, 2206–2215. [Google Scholar] [CrossRef]
- Gallelli, L.; Galasso, O.; Falcone, D.; Southworth, S.; Greco, M.; Ventura, V.; Romualdi, P.; Corigliano, A.; Terracciano, R.; Savino, R.; et al. The effects of nonsteroidal anti-inflammatory drugs on clinical outcomes, synovial fluid cytokine concentration and signal transduction pathways in knee osteoarthritis. A randomized open label trial. Osteoarthr. Cartil. 2013, 21, 1400–1408. [Google Scholar] [CrossRef] [Green Version]
- Wenham, C.Y.J.; Grainger, A.; Hensor, E.; Caperon, A.R.; Ash, Z.R.; Conaghan, P. Methotrexate for pain relief in knee osteoarthritis: An open-label study. Rheumatology 2013, 52, 888–892. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Yan, G.; Huang, H.; Zheng, M.; Ma, K.; Cui, X.; Lu, D.; Zheng, L.; Zhu, B.; Cheng, J.; et al. Anti-inflammatory and immunomodulatory effects of the extracellular vesicles derived from human umbilical cord mesenchymal stem cells on osteoarthritis via M2 macrophages. J. Nanobiotechnol. 2022, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, R.; Toyoda, E.; Maehara, M.; Wasai, S.; Omura, H.; Watanabe, M.; Sato, M. Effect of Platelet-Rich Plasma on M1/M2 Macrophage Polarization. Int. J. Mol. Sci. 2021, 22, 2336. [Google Scholar] [CrossRef] [PubMed]
- Nishio, H.; Saita, Y.; Kobayashi, Y.; Takaku, T.; Fukusato, S.; Uchino, S.; Wakayama, T.; Ikeda, H.; Kaneko, K. Platelet-rich plasma promotes recruitment of macrophages in the process of tendon healing. Regen. Ther. 2020, 14, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; Han, C.; Zhao, L.; Ren, Y. Is local platelet-rich plasma injection clinically superior to hyaluronic acid for treatment of knee osteoarthritis? A systematic review of randomized controlled trials. Arthritis Res. Ther. 2018, 20, 128. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, S.; Doherty, M. Lifestyle changes in the management of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2001, 15, 559–568. [Google Scholar] [CrossRef]
- Garver, M.J.; Focht, B.; Taylor, S. Integrating lifestyle approaches into osteoarthritis care. J. Multidiscip. Healthc. 2015, 8, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.; McConnell, S.; Harmer, A.R.; Van Der Esch, M.; Simic, M.; Bennell, K.L. Exercise for osteoarthritis of the knee: A Cochrane systematic review. Br. J. Sports Med. 2015, 49, 1554–1557. [Google Scholar] [CrossRef]
- Bonewald, L. Osteocytes, Muscle and Exercise: Role in Healthy Aging. FASEB J. 2017, 31 (Suppl. 1), 7.2. [Google Scholar]
- Mcalindon, T.; Felson, D.T. Nutrition: Risk factors for osteoarthritis. Ann. Rheum. Dis. 1997, 56, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Ameye, L.G.; Chee, W.S. Osteoarthritis and nutrition. From nutraceuticals to functional foods: A systematic review of the scientific evidence. Arthritis Res. Ther. 2006, 8, R127. [Google Scholar] [CrossRef] [Green Version]
- McAlindon, T.E.; Jacques, P.; Zhang, Y.; Hannan, M.T.; Aliabadi, P.; Weissman, B.; Rush, D.; Levy, D.; Felson, D.T. Do antioxidant micronutrients protect against the development and progression of knee osteoarthritis? Arthritis Rheum. 1996, 39, 648–656. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAMPs | Signaling | References |
|---|---|---|
| ECM compositions | ||
| Decorin and biglycan | C1q; Complement system | [19,27] |
| Fibronectin | TLR2, 4/JUK2-MAPK | [35] |
| Hyaluronan | TLR4/MyD88 | [38] |
| Biglycan | TLR2, 4/ERK/NF-κB | [39,40] |
| Tenascin c | TLR4/MAPK | [41] |
| Syndecan-4 | MAPK | [42] |
| Type II collagen 29-mer fragment | TLRs/NF-κB | [43,44] |
| Aggrecan 32-mer fragment | TLR2/NF-κB | [45] |
| Crystals | ||
| CPPD, BCP | TLR2/NF-κB NLR/NLRP3 | [46,47] |
| Alarmins | ||
| HMGB1 | TLR2, 4/ERK/NF-κB | [60,62] |
| s100sA8, S100A9 | TLR4/NF-κB | [63,64] |
| UA | NLR/NLRP3 | |
| ATP | NLR/NLRP3 | [66,67] |
| IL-1α | IL-1R1/MAPK | [65] |
| IL-33 | MyD88/NF-κB/MAPK | [68,69] |
| FliI | TLR4/ERK1 | [21] |
| Plasma proteins | ||
| Fibrinogen | TLR4/NF-κB | [19,27] |
| Gc-globulin | C5a; Complement system | [19,27] |
| α1-microglobulin, α2-macroglobulin | MAPK-ERK/NF-κB | [19,27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terkawi, M.A.; Ebata, T.; Yokota, S.; Takahashi, D.; Endo, T.; Matsumae, G.; Shimizu, T.; Kadoya, K.; Iwasaki, N. Low-Grade Inflammation in the Pathogenesis of Osteoarthritis: Cellular and Molecular Mechanisms and Strategies for Future Therapeutic Intervention. Biomedicines 2022, 10, 1109. https://doi.org/10.3390/biomedicines10051109
Terkawi MA, Ebata T, Yokota S, Takahashi D, Endo T, Matsumae G, Shimizu T, Kadoya K, Iwasaki N. Low-Grade Inflammation in the Pathogenesis of Osteoarthritis: Cellular and Molecular Mechanisms and Strategies for Future Therapeutic Intervention. Biomedicines. 2022; 10(5):1109. https://doi.org/10.3390/biomedicines10051109
Chicago/Turabian StyleTerkawi, M Alaa, Taku Ebata, Shunichi Yokota, Daisuke Takahashi, Tsutomu Endo, Gen Matsumae, Tomohiro Shimizu, Ken Kadoya, and Norimasa Iwasaki. 2022. "Low-Grade Inflammation in the Pathogenesis of Osteoarthritis: Cellular and Molecular Mechanisms and Strategies for Future Therapeutic Intervention" Biomedicines 10, no. 5: 1109. https://doi.org/10.3390/biomedicines10051109