
The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
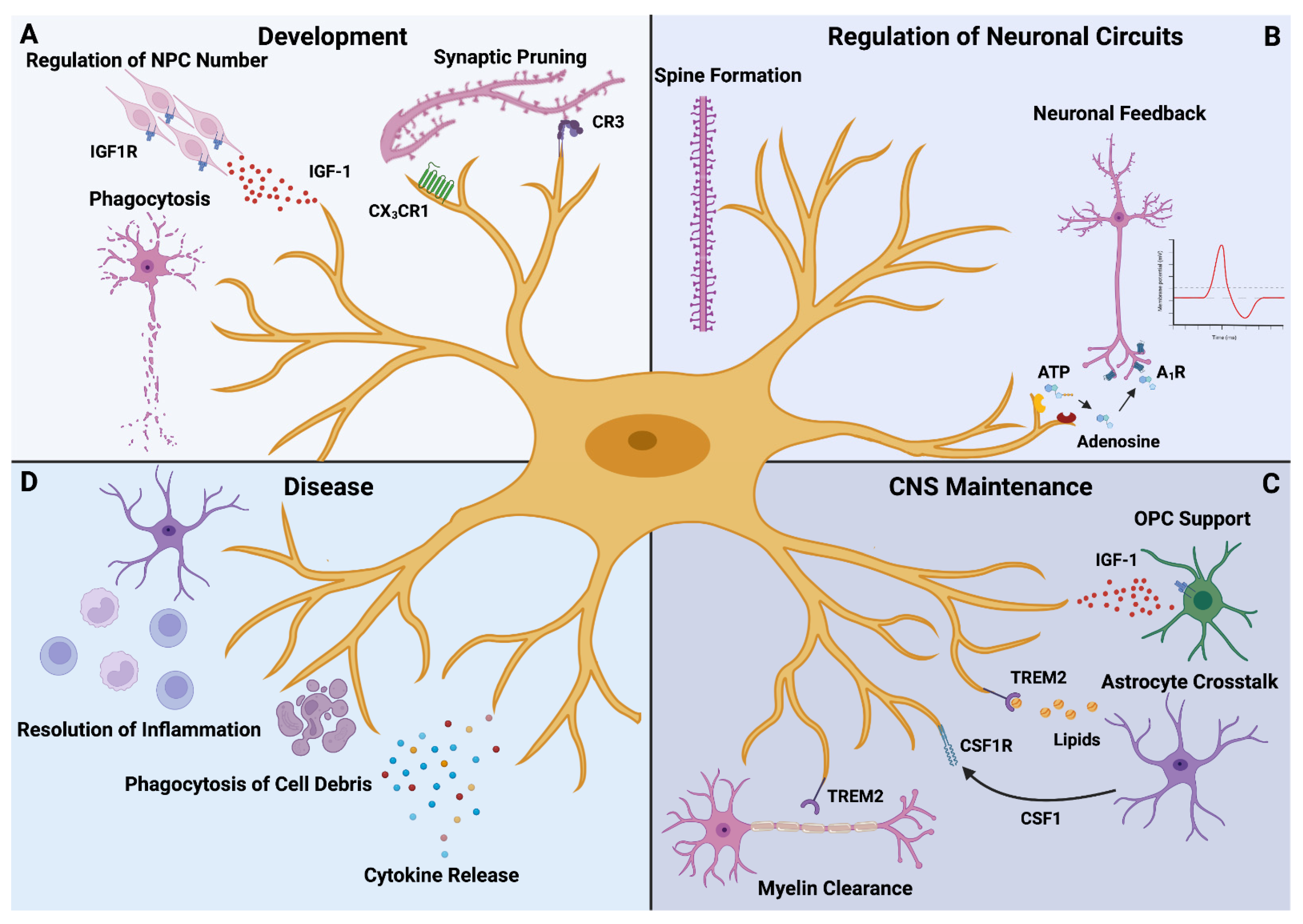
2. Microglia in the CNS–Old Dogs with New Tricks
3. The Role of Osteopontin in the Central Nervous System
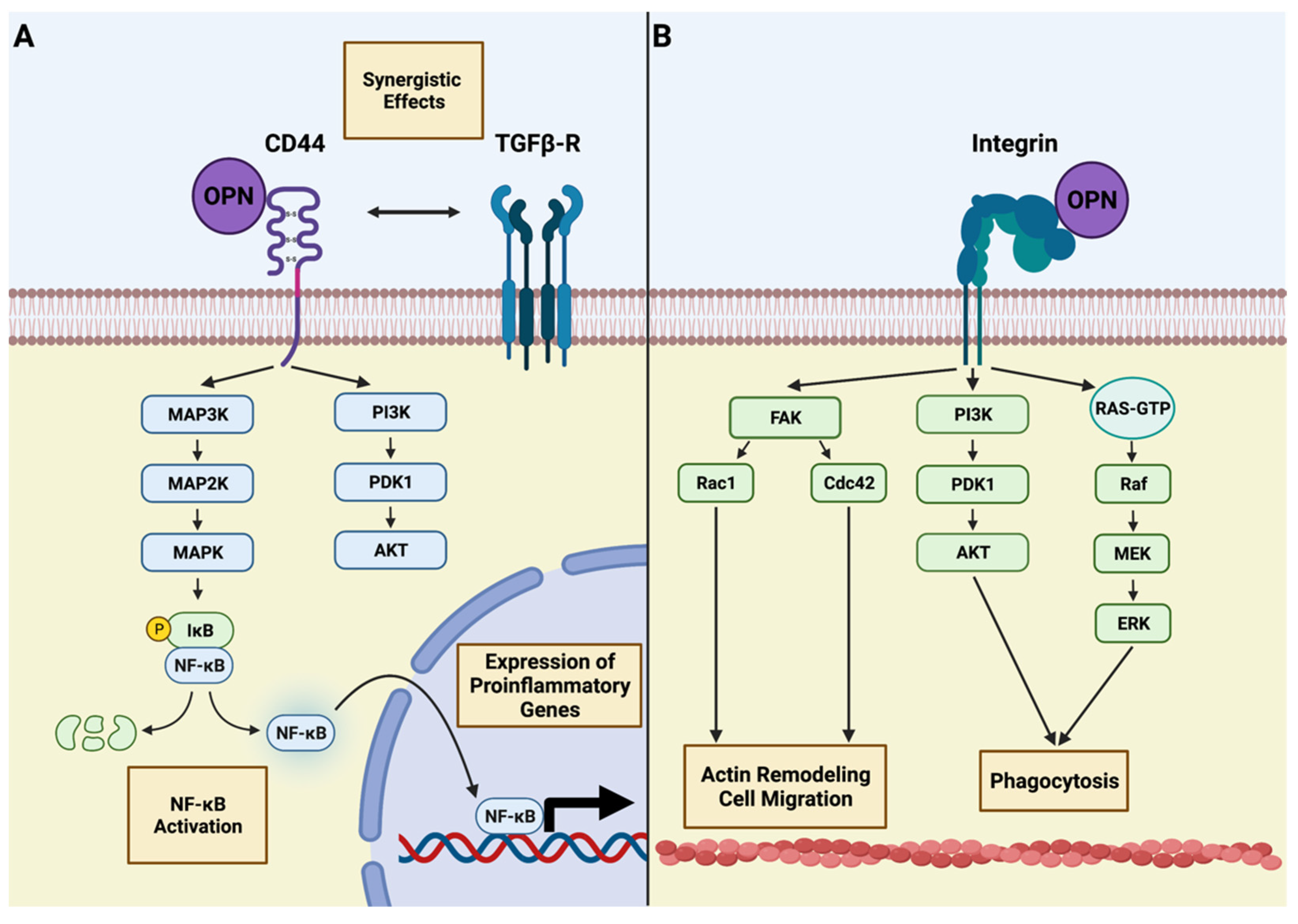
4. Osteopontin and Microglia—An Intimate Interplay
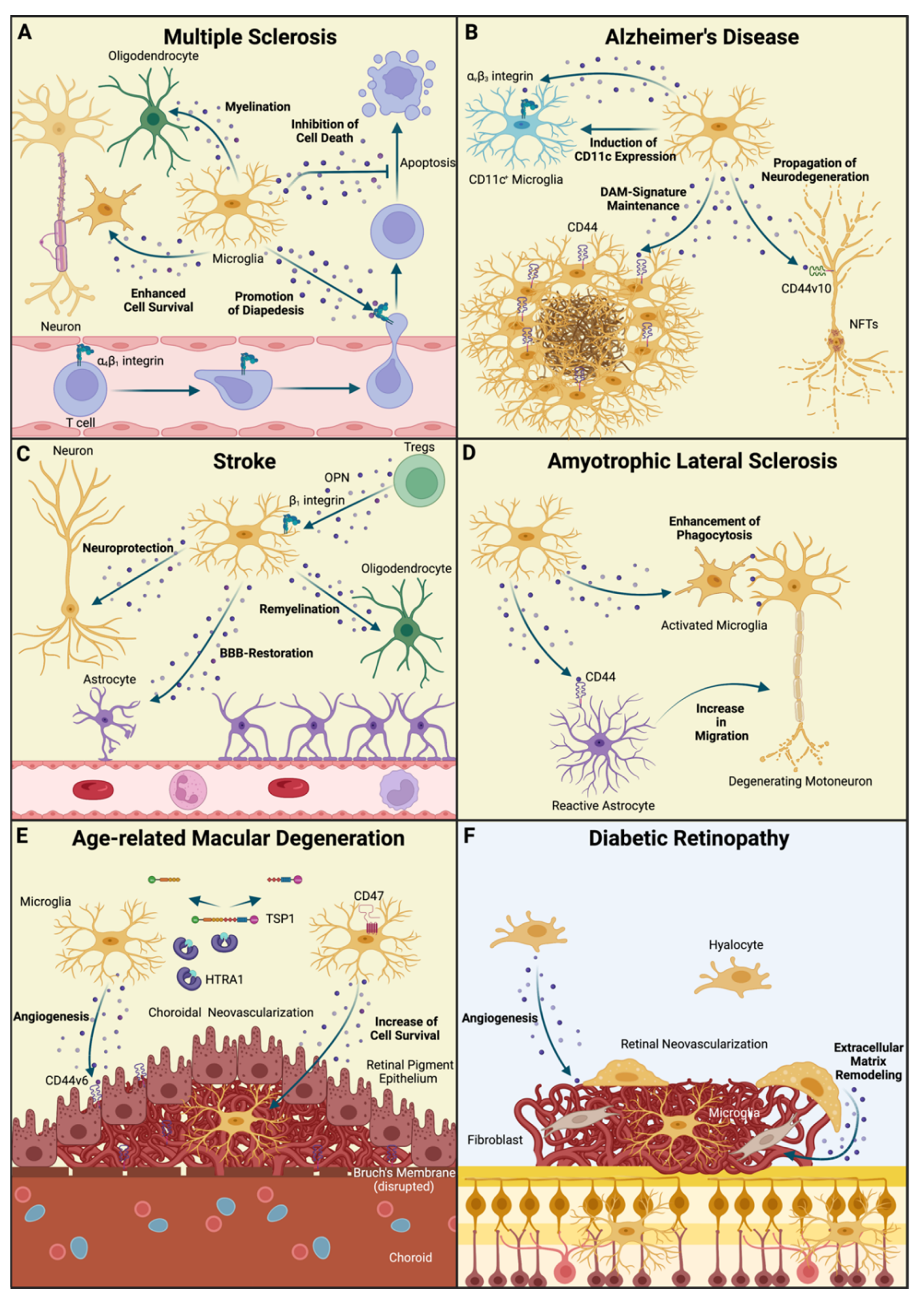
5. The Role of Osteopontin and Microglia in the Diseased CNS
6. Multiple Sclerosis
7. Alzheimer’s Disease
8. Cerebrovascular Disease
9. Amyotrophic Lateral Sclerosis
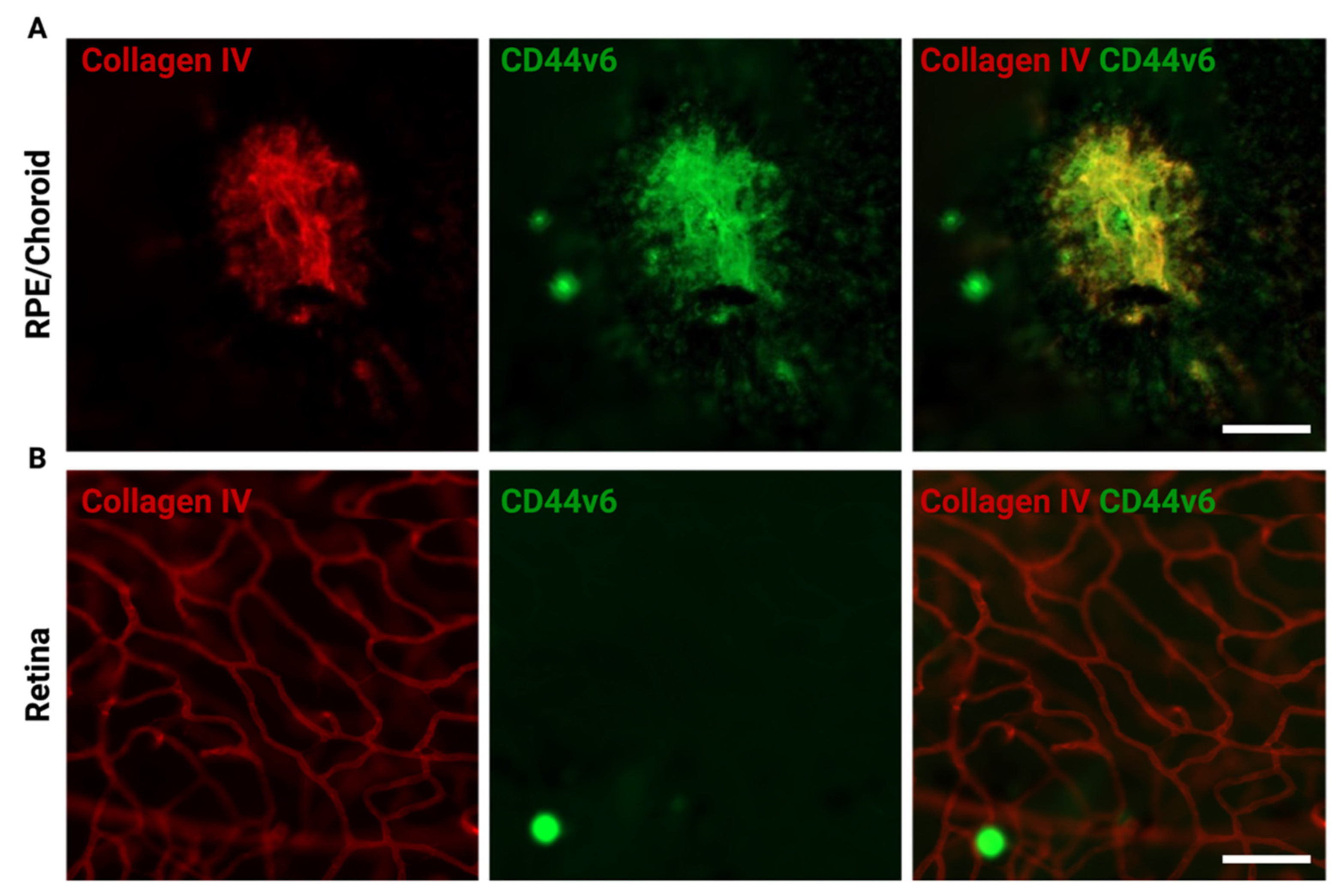
10. Age-Related Macular Degeneration
11. Diabetic Retinopathy
12. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kierdorf, K.; Masuda, T.; Jordão, M.J.C.; Prinz, M. Macrophages at CNS Interfaces: Ontogeny and Function in Health and Disease. Nat. Rev. Neurosci. 2019, 20, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, Fate and Dynamics of Macrophages at Central Nervous System Interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A Single-Cell Atlas of Mouse Brain Macrophages Reveals Unique Transcriptional Identities Shaped by Ontogeny and Tissue Environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef]
- Utz, S.G.; See, P.; Mildenberger, W.; Thion, M.S.; Silvin, A.; Lutz, M.; Ingelfinger, F.; Rayan, N.A.; Lelios, I.; Buttgereit, A.; et al. Early Fate Defines Microglia and Non-Parenchymal Brain Macrophage Development. Cell 2020, 181, 557–573.e18. [Google Scholar] [CrossRef]
- Chow, R.L.; Lang, R.A. Early Eye Development in Vertebrates. Annu. Rev. Cell Dev. Biol. 2001, 17, 255–296. [Google Scholar] [CrossRef] [Green Version]
- O’Koren, E.G.; Yu, C.; Klingeborn, M.; Wong, A.Y.W.; Prigge, C.L.; Mathew, R.; Kalnitsky, J.; Msallam, R.A.; Silvin, A.; Kay, J.N.; et al. Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 2019, 50, 723–737.e7. [Google Scholar] [CrossRef] [Green Version]
- Wieghofer, P.; Hagemeyer, N.; Sankowski, R.; Schlecht, A.; Staszewski, O.; Amann, L.; Gruber, M.; Koch, J.; Hausmann, A.; Zhang, P.; et al. Mapping the Origin and Fate of Myeloid Cells in Distinct Compartments of the Eye by Single-Cell Profiling. EMBO J. 2021, 40, e105123. [Google Scholar] [CrossRef]
- Rosmus, D.-D.; Wieghofer, P. Guardians of the Eye: New Tales about Retinal Microglia and Other Ocular Macrophages. Neural Regen. Res. 2022, 17, 1275–1277. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP Mediates Rapid Microglial Response to Local Brain Injury in Vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and Non-Immune Functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Jakovac, H.; Grubić Kezele, T.; Šućurović, S.; Mulac-Jeričević, B.; Radošević-Stašić, B. Osteopontin–Metallothionein I/II Interactions in Experimental Autoimmunune Encephalomyelitis. Neuroscience 2017, 350, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Chabas, D.; Baranzini, S.E.; Mitchell, D.; Bernard, C.C.; Rittling, S.R.; Denhardt, D.T.; Sobel, R.A.; Lock, C.; Karpuj, M.; Pedotti, R.; et al. The Influence of the Proinflammatory Cytokine, Osteopontin, on Autoimmune Demyelinating Disease. Science 2001, 294, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Rentsendorj, A.; Sheyn, J.; Fuchs, D.-T.; Daley, D.; Salumbides, B.C.; Schubloom, H.E.; Hart, N.J.; Li, S.; Hayden, E.Y.; Teplow, D.B.; et al. A Novel Role for Osteopontin in Macrophage-Mediated Amyloid-β Clearance in Alzheimer’s Models. Brain Behav. Immun. 2018, 67, 163–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beguier, F.; Housset, M.; Roubeix, C.; Augustin, S.; Zagar, Y.; Nous, C.; Mathis, T.; Eandi, C.; Benchaboune, M.; Drame-Maigné, A.; et al. The 10q26 Risk Haplotype of Age-Related Macular Degeneration Aggravates Subretinal Inflammation by Impairing Monocyte Elimination. Immunity 2020, 53, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, A.; Zhang, P.; Wolf, J.; Thien, A.; Rosmus, D.-D.; Boneva, S.; Schlunck, G.; Lange, C.; Wieghofer, P. Secreted Phosphoprotein 1 Expression in Retinal Mononuclear Phagocytes Links Murine to Human Choroidal Neovascularization. Front. Cell Dev. Biol. 2021, 8, 618598. [Google Scholar] [CrossRef]
- Lekwuwa, M.; Choudhary, M.; Lad, E.M.; Malek, G. Osteopontin Accumulates in Basal Deposits of Human Eyes with Age-Related Macular Degeneration and May Serve as a Biomarker of Aging. Mod. Pathol. 2021, 35, 165–176. [Google Scholar] [CrossRef]
- Steinman, L. A Molecular Trio in Relapse and Remission in Multiple Sclerosis. Nat. Rev. Immunol. 2009, 9, 440–447. [Google Scholar] [CrossRef]
- Rabenstein, M.; Vay, S.U.; Flitsch, L.J.; Fink, G.R.; Schroeter, M.; Rueger, M.A. Osteopontin Directly Modulates Cytokine Expression of Primary Microglia and Increases Their Survival. J. Neuroimmunol. 2016, 299, 130–138. [Google Scholar] [CrossRef]
- Cappellano, G.; Vecchio, D.; Magistrelli, L.; Clemente, N.; Raineri, D.; Barbero Mazzucca, C.; Virgilio, E.; Dianzani, U.; Chiocchetti, A.; Comi, C. The Yin-Yang of Osteopontin in Nervous System Diseases: Damage versus Repair. Neural Regen. Res. 2021, 16, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- De Rio-Hortega, P. El Tercer Elemento de Los Centros Nerviosos. I. La Microglia En Estado Normal. II. Intervencion de La Microglia En Los Processos Patologic Os. III. Naturaleza Probable de La Microglia. Biol. Soc. Esp. Biol. 1919, 9, 68–120. [Google Scholar]
- Tremblay, M.-È.; Lecours, C.; Samson, L.; Sánchez-Zafra, V.; Sierra, A. From the Cajal Alumni Achúcarro and Río-Hortega to the Rediscovery of Never-Resting Microglia. Front. Neuroanat. 2015, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia Emerge from Erythromyeloid Precursors via Pu.1- and Irf8-Dependent Pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada González, F.; Perrin, P.; et al. Microglia Development Follows a Stepwise Program to Regulate Brain Homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and Temporal Heterogeneity of Mouse and Human Microglia at Single-Cell Resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Marín-Teva, J.L.; Dusart, I.; Colin, C.; Gervais, A.; van Rooijen, N.; Mallat, M. Microglia Promote the Death of Developing Purkinje Cells. Neuron 2004, 41, 535–547. [Google Scholar] [CrossRef]
- Peri, F.; Nüsslein-Volhard, C. Live Imaging of Neuronal Degradation by Microglia Reveals a Role for V0-ATPase A1 in Phagosomal Fusion in Vivo. Cell 2008, 133, 916–927. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.-È.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 Is Necessary for Synaptic Plasticity in Mouse Visual Cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Faust, T.E.; Gunner, G.; Schafer, D.P. Mechanisms Governing Activity-Dependent Synaptic Pruning in the Developing Mammalian CNS. Nat. Rev. Neurosci. 2021, 22, 657–673. [Google Scholar] [CrossRef]
- Favuzzi, E.; Huang, S.; Saldi, G.A.; Binan, L.; Ibrahim, L.A.; Fernández-Otero, M.; Cao, Y.; Zeine, A.; Sefah, A.; Zheng, K.; et al. GABA-Receptive Microglia Selectively Sculpt Developing Inhibitory Circuits. Cell 2021, 184, 4048–4063.e32. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative Feedback Control of Neuronal Activity by Microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.-M.; Akriditou, M.-A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia Contribute to Normal Myelinogenesis and to Oligodendrocyte Progenitor Maintenance during Adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Safaiyan, S.; Besson-Girard, S.; Kaya, T.; Cantuti-Castelvetri, L.; Liu, L.; Ji, H.; Schifferer, M.; Gouna, G.; Usifo, F.; Kannaiyan, N.; et al. White Matter Aging Drives Microglial Diversity. Neuron 2021, 109, 1100–1117.e10. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.-H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-Cell Profiling Identifies Myeloid Cell Subsets with Distinct Fates during Neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Šimončičová, E.; de Andrade, E.G.; Vecchiarelli, H.A.; Awogbindin, I.O.; Delage, C.I.; Tremblay, M.-È. Present and Future of Microglial Pharmacology. Trends Pharmacol. Sci. 2022. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef] [PubMed]
- Oldberg, A.; Franzén, A.; Heinegård, D. Cloning and Sequence Analysis of Rat Bone Sialoprotein (Osteopontin) CDNA Reveals an Arg-Gly-Asp Cell-Binding Sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 8819–8823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.P.; Patarca, R.; Schwartz, J.; Singh, P.; Cantor, H. Definition of a Specific Interaction between the Early T Lymphocyte Activation 1 (Eta-1) Protein and Murine Macrophages in Vitro and Its Effect upon Macrophages in Vivo. J. Exp. Med. 1990, 171, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Liaw, L.; Lindner, V.; Schwartz, S.M.; Chambers, A.F.; Giachelli, C.M. Osteopontin and Beta 3 Integrin Are Coordinately Expressed in Regenerating Endothelium in Vivo and Stimulate Arg-Gly-Asp-Dependent Endothelial Migration in Vitro. Circ. Res. 1995, 77, 665–672. [Google Scholar] [CrossRef]
- Scatena, M.; Liaw, L.; Giachelli, C.M. Osteopontin: A Multifunctional Molecule Regulating Chronic Inflammation and Vascular Disease. Arter. Thromb. Vasc. Biol. 2007, 27, 2302–2309. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Kon, S.; Nakayama, Y.; Kurotaki, D.; Saito, Y.; Kanayama, M.; Kimura, C.; Diao, H.; Morimoto, J.; Matsui, Y.; et al. The Differential Amino Acid Requirement within Osteopontin in A4 and A9 Integrin-Mediated Cell Binding and Migration. Matrix Biol. 2009, 28, 11–19. [Google Scholar] [CrossRef]
- Weber, G.F.; Ashkar, S.; Glimcher, M.J.; Cantor, H. Receptor-Ligand Interaction Between CD44 and Osteopontin (Eta-1). Science 1996, 271, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, R.; Crawford, H.C.; Haro, H.; Matrisian, L.M.; Havrda, M.C.; Liaw, L. Osteopontin, a Novel Substrate for Matrix Metalloproteinase-3 (Stromelysin-1) and Matrix Metalloproteinase-7 (Matrilysin) *. J. Biol. Chem. 2001, 276, 28261–28267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves DaSilva, A.; Liaw, L.; Yong, V.W. Cleavage of Osteopontin by Matrix Metalloproteinase-12 Modulates Experimental Autoimmune Encephalomyelitis Disease in C57BL/6 Mice. Am. J. Pathol. 2010, 177, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.L.; Kim, H.-J.; Kim, J.-H.; Garcia, V.A.; Cantor, H. Alternative Translation of Osteopontin Generates Intracellular and Secreted Isoforms That Mediate Distinct Biological Activities in Dendritic Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 7235–7239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantor, H.; Shinohara, M.L. Regulation of T-Helper-Cell Lineage Development by Osteopontin: The inside Story. Nat. Rev. Immunol. 2009, 9, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Lit, L.C.W.; Tam, L.S.; Li, E.K.; Lam, C.W.K. Elevation of Plasma Osteopontin Concentration Is Correlated with Disease Activity in Patients with Systemic Lupus Erythematosus. Rheumatology 2005, 44, 602–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Nakai, T.; Tamura, N.; Okamoto, S.; Matsuoka, K.; Sakuraba, A.; Fukushima, T.; Uede, T.; Hibi, T. Osteopontin/Eta-1 Upregulated in Crohn’s Disease Regulates the Th1 Immune Response. Gut 2005, 54, 1254–1262. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Chen, Q.; Alam, A.; Cui, J.; Suen, K.C.; Soo, A.P.; Eguchi, S.; Gu, J.; Ma, D. The Role of Osteopontin in the Progression of Solid Organ Tumour. Cell Death Dis. 2018, 9, 356. [Google Scholar] [CrossRef]
- Hur, E.M.; Youssef, S.; Haws, M.E.; Zhang, S.Y.; Sobel, R.A.; Steinman, L. Osteopontin-Induced Relapse and Progression of Autoimmune Brain Disease through Enhanced Survival of Activated T Cells. Nat. Immunol. 2007, 8, 74–83. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395. [Google Scholar] [CrossRef] [Green Version]
- Martegani, M.P.; Del Prete, F.; Gasbarri, A.; Natali, P.G.; Bartolazzi, A. Structural Variability of CD44v Molecules and Reliability of Immunodetection of CD44 Isoforms Using MAbs Specific for CD44 Variant Exon Products. Am. J. Pathol. 1999, 154, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Screaton, G.R.; Bell, M.V.; Jackson, D.G.; Cornelis, F.B.; Gerth, U.; Bell, J.I. Genomic Structure of DNA Encoding the Lymphocyte Homing Receptor CD44 Reveals at Least 12 Alternatively Spliced Exons. Proc. Natl. Acad. Sci. USA 1992, 89, 12160–12164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaskuła, K.; Sacharczuk, M.; Gaciong, Z.; Skiba, D.S. Cardiovascular Effects Mediated by HMMR and CD44. Mediat. Inflamm. 2021, 2021, e4977209. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Rizeq, B.; Saleh, H.A.; Rahman, M.M.; Zayed, H. Novel CD44-Downstream Signaling Pathways Mediating Breast Tumor Invasion. Int. J. Biol. Sci. 2018, 14, 1782–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Zhong, H.; Li, N.; Chen, K.; Chen, J.; Sun, J.; Xu, L.; Wang, J.; Zhang, M.; Liu, X.; et al. Osteopontin Activates Retinal Microglia Causing Retinal Ganglion Cells Loss via P38 MAPK Signaling Pathway in Glaucoma. FASEB J. 2021, 35, e21405. [Google Scholar] [CrossRef]
- Blasi, E.; Ardizzoni, A.; Colombari, B.; Neglia, R.; Baschieri, C.; Peppoloni, S.; Cinco, M. NF-KB Activation and P38 Phosphorilation in Microglial Cells Infected with Leptospira or Exposed to Partially Purified Leptospiral Lipoproteins. Microb. Pathog. 2007, 42, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK P38 Regulates Transcriptional Activity of NF-ΚB in Primary Human Astrocytes via Acetylation of P65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef] [Green Version]
- Guire, C.M.; Prinz, M.; Beyaert, R.; Loo, G. van Nuclear Factor Kappa B (NF-ΚB) in Multiple Sclerosis Pathology. Trends Mol. Med. 2013, 19, 604–613. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Singleton, P.A.; Zhu, H.; Zhou, B. Hyaluronan Promotes Signaling Interaction between CD44 and the Transforming Growth Factor β Receptor I in Metastatic Breast Tumor Cells *. J. Biol. Chem. 2002, 277, 39703–39712. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a Unique TGF-β–Dependent Molecular and Functional Signature in Microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Zöller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGFβ Signalling in Microglia Results in Impaired Homeostasis. Nat. Commun. 2018, 9, 4011. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Müller, P.F.; Wolf, Y.; Varol, D.; Yona, S.; Brendecke, S.M.; Kierdorf, K.; Staszewski, O.; Datta, M.; et al. A New Type of Microglia Gene Targeting Shows TAK1 to Be Pivotal in CNS Autoimmune Inflammation. Nat. Neurosci. 2013, 16, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Xu, M.; Chen, Z.J. Ubiquitin-Mediated Activation of TAK1 and IKK. Oncogene 2007, 26, 3214–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; Daems, C.; Staszewski, O.; Vande Walle, L.; Jordao, M.J.C.; et al. A20 Critically Controls Microglia Activation and Inhibits Inflammasome-Dependent Neuroinflammation. Nat. Commun. 2018, 9, 2036. [Google Scholar] [CrossRef] [Green Version]
- Wieghofer, P.; Knobeloch, K.-P.; Prinz, M. Genetic Targeting of Microglia. Glia 2015, 63, 1–22. [Google Scholar] [CrossRef]
- Dumas, A.A.; Borst, K.; Prinz, M. Current Tools to Interrogate Microglial Biology. Neuron 2021, 109, 2805–2819. [Google Scholar] [CrossRef]
- Masuda, T.; Amann, L.; Sankowski, R.; Staszewski, O.; Lenz, M.; D Errico, P.; Snaidero, N.; Costa Jordão, M.J.; Böttcher, C.; Kierdorf, K.; et al. Novel Hexb-Based Tools for Studying Microglia in the CNS. Nat. Immunol. 2020, 21, 802–815. [Google Scholar] [CrossRef]
- Kim, J.-S.; Kolesnikov, M.; Peled-Hajaj, S.; Scheyltjens, I.; Xia, Y.; Trzebanski, S.; Haimon, Z.; Shemer, A.; Lubart, A.; Van Hove, H.; et al. A Binary Cre Transgenic Approach Dissects Microglia and CNS Border-Associated Macrophages. Immunity 2021, 54, 176–190.e7. [Google Scholar] [CrossRef]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell Adhesion by Integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Milner, R.; Campbell, I.L. The Integrin Family of Cell Adhesion Molecules Has Multiple Functions within the CNS. J. Neurosci. Res. 2002, 69, 286–291. [Google Scholar] [CrossRef]
- Milner, R.; Campbell, I.L. The Extracellular Matrix and Cytokines Regulate Microglial Integrin Expression and Activation. J. Immunol. 2003, 170, 3850–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, D.; Ryu, J.K.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-Induced Perivascular Microglial Clustering Is Required for the Development of Axonal Damage in Neuroinflammation. Nat. Commun. 2012, 3, 1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harburger, D.S.; Calderwood, D.A. Integrin Signalling at a Glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Rong, Z.; Cheng, B.; Zhong, L.; Ye, X.; Li, X.; Jia, L.; Li, Y.; Shue, F.; Wang, N.; Cheng, Y.; et al. Activation of FAK/Rac1/Cdc42-GTPase Signaling Ameliorates Impaired Microglial Migration Response to Aβ42 in Triggering Receptor Expressed on Myeloid Cells 2 Loss-of-Function Murine Models. FASEB J. 2020, 34, 10984–10997. [Google Scholar] [CrossRef]
- Kim, I.-D.; Lee, H.; Lee, Y.-C.J. and J.-K. Osteopontin Peptide Icosamer Containing RGD and SLAYGLR Motifs Enhances the Motility and Phagocytic Activity of Microglia. Exp. Neurobiol. 2017, 26, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J.; et al. Extracellular Signal-Regulated Kinase Regulates Microglial Immune Responses in Alzheimer’s Disease. J. Neurosci. Res. 2021, 99, 1704–1721. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Ajami, B.; Samusik, N.; Wieghofer, P.; Ho, P.P.; Crotti, A.; Bjornson, Z.; Prinz, M.; Fantl, W.J.; Nolan, G.P.; Steinman, L. Single-Cell Mass Cytometry Reveals Distinct Populations of Brain Myeloid Cells in Mouse Neuroinflammation and Neurodegeneration Models. Nat. Neurosci. 2018, 21, 541–551. [Google Scholar] [CrossRef]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single Cell RNA Sequencing of Human Microglia Uncovers a Subset Associated with Alzheimer’s Disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping Microglia States in the Human Brain through the Integration of High-Dimensional Techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-Cell Profiling of Myeloid Cells in Glioblastoma across Species and Disease Stage Reveals Macrophage Competition and Specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan Metabolism Drives Dynamic Immunosuppressive Myeloid States in IDH-Mutant Gliomas. Nat. Cancer 2021, 2, 723–740. [Google Scholar] [CrossRef]
- Wallin, M.T.; Culpepper, W.J.; Nichols, E.; Bhutta, Z.A.; Gebrehiwot, T.T.; Hay, S.I.; Khalil, I.A.; Krohn, K.J.; Liang, X.; Naghavi, M.; et al. Global, Regional, and National Burden of Multiple Sclerosis 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Schattling, B.; Steinbach, K.; Thies, E.; Kruse, M.; Menigoz, A.; Ufer, F.; Flockerzi, V.; Brück, W.; Pongs, O.; Vennekens, R.; et al. TRPM4 Cation Channel Mediates Axonal and Neuronal Degeneration in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Nat. Med. 2012, 18, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Carare, R.O.; Bechmann, I.; Flügel, A.; Laman, J.D.; Weller, R.O. Vascular, Glial, and Lymphatic Immune Gateways of the Central Nervous System. Acta Neuropathol. 2016, 132, 317–338. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, C.; van der Poel, M.; Fernández-Zapata, C.; Schlickeiser, S.; Leman, J.K.H.; Hsiao, C.-C.; Mizee, M.R.; Adelia; Vincenten, M.C.J.; Kunkel, D.; et al. Single-Cell Mass Cytometry Reveals Complex Myeloid Cell Composition in Active Lesions of Progressive Multiple Sclerosis. Acta Neuropathol. Commun. 2020, 8, 136. [Google Scholar] [CrossRef]
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.-H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, Ø.; Wang, X.; et al. Peli1 Promotes Microglia-Mediated CNS Inflammation by Regulating Traf3 Degradation. Nat. Med. 2013, 19, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Mundt, S.; Mrdjen, D.; Utz, S.G.; Greter, M.; Schreiner, B.; Becher, B. Conventional DCs Sample and Present Myelin Antigens in the Healthy CNS and Allow Parenchymal T Cell Entry to Initiate Neuroinflammation. Sci. Immunol. 2019, 4, eaau8380. [Google Scholar] [CrossRef]
- Jafari, M.; Schumacher, A.-M.; Snaidero, N.; Ullrich Gavilanes, E.M.; Neziraj, T.; Kocsis-Jutka, V.; Engels, D.; Jürgens, T.; Wagner, I.; Weidinger, J.D.F.; et al. Phagocyte-Mediated Synapse Removal in Cortical Neuroinflammation Is Promoted by Local Calcium Accumulation. Nat. Neurosci. 2021, 24, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, A.G.; Wishart, C.L.; Pamphlett, R.; Locatelli, G.; King, N.J.C. Microglia and Monocytes in Inflammatory CNS Disease: Integrating Phenotype and Function. Acta Neuropathol. 2021, 143, 179–224. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Buonsanti, C.; Mariani, M.; Cella, M.; Gilfillan, S.; Cross, A.H.; Colonna, M.; Panina-Bordignon, P. Blockade of TREM-2 Exacerbates Experimental Autoimmune Encephalomyelitis. Eur. J. Immunol. 2007, 37, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S.; Saitoh, S.; Miyajima, H.; Itokazu, T.; Yamashita, T. Microglia Suppress the Secondary Progression of Autoimmune Encephalomyelitis. Glia 2019, 67, 1694–1704. [Google Scholar] [CrossRef]
- Agah, E.; Zardoui, A.; Saghazadeh, A.; Ahmadi, M.; Tafakhori, A.; Rezaei, N. Osteopontin (OPN) as a CSF and Blood Biomarker for Multiple Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2018, 13, e0190252. [Google Scholar] [CrossRef]
- Miedema, A.; Gerrits, E.; Brouwer, N.; Jiang, Q.; Kracht, L.; Meijer, M.; Nutma, E.; Peferoen-Baert, R.; Pijnacker, A.T.E.; Wesseling, E.M.; et al. Brain Macrophages Acquire Distinct Transcriptomes in Multiple Sclerosis Lesions and Normal Appearing White Matter. Acta Neuropathol. Commun. 2022, 10, 8. [Google Scholar] [CrossRef]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A Randomized, Placebo-Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of Experimental Autoimmune Encephalomyelitis by Antibodies against Alpha 4 Beta 1 Integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef]
- Clemente, N.; Comi, C.; Raineri, D.; Cappellano, G.; Vecchio, D.; Orilieri, E.; Gigliotti, C.L.; Boggio, E.; Dianzani, C.; Sorosina, M.; et al. Role of Anti-Osteopontin Antibodies in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2017, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.F.; Davies, C.L.; Holloway, R.K.; Labrak, Y.; Ireland, G.; Carradori, D.; Dillenburg, A.; Borger, E.; Soong, D.; Richardson, J.C.; et al. Central Nervous System Regeneration Is Driven by Microglia Necroptosis and Repopulation. Nat. Neurosci. 2019, 22, 1046–1052. [Google Scholar] [CrossRef]
- Shinohara, M.L.; Kim, J.-H.; Garcia, V.A.; Cantor, H. Engagement of the Type I Interferon Receptor on Dendritic Cells Inhibits T Helper 17 Cell Development: Role of Intracellular Osteopontin. Immunity 2008, 29, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraju, R.; Bernasconi, L.; Losberger, C.; Graber, P.; Kadi, L.; Avellana-Adalid, V.; Picard-Riera, N.; Van Evercooren, A.B.; Cirillo, R.; Kosco-Vilbois, M.; et al. Osteopontin Is Upregulated during in Vivo Demyelination and Remyelination and Enhances Myelin Formation in Vitro. Mol. Cell. Neurosci. 2004, 25, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M. Changing Concepts of Alzheimer Disease. JAMA 2011, 305, 2458–2459. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Delatour, B.; Potier, M.-C. Classification and Basic Pathology of Alzheimer Disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef]
- Lowe, V.J.; Lundt, E.S.; Albertson, S.M.; Przybelski, S.A.; Senjem, M.L.; Parisi, J.E.; Kantarci, K.; Boeve, B.; Jones, D.T.; Knopman, D.; et al. Neuroimaging Correlates with Neuropathologic Schemes in Neurodegenerative Disease. Alzheimers Dement. 2019, 15, 927–939. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. The Role of Microglia in Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 79, 961–968. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and Neuron Numbers, but Not Amyloid Load, Predict Cognitive Status in Alzheimer’s Disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Mattiace, L.A.; Davies, P.; Yen, S.-H.; Dickson, D.W. Microglia in Cerebellar Plaques in Alzheimer’s Disease. Acta Neuropathol. 1990, 80, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid Appearance and Local Toxicity of Amyloid-β Plaques in a Mouse Model of Alzheimer’s Disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Braak, H.; Xue, Q.-S.; Bechmann, I. Dystrophic (Senescent) Rather than Activated Microglial Cells Are Associated with Tau Pathology and Likely Precede Neurodegeneration in Alzheimer’s Disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Tischer, J.; Krueger, M.; Mueller, W.; Staszewski, O.; Prinz, M.; Streit, W.J.; Bechmann, I. Inhomogeneous Distribution of Iba-1 Characterizes Microglial Pathology in Alzheimer’s Disease. Glia 2016, 64, 1562–1572. [Google Scholar] [CrossRef]
- Young, A.M.H.; Kumasaka, N.; Calvert, F.; Hammond, T.R.; Knights, A.; Panousis, N.; Park, J.S.; Schwartzentruber, J.; Liu, J.; Kundu, K.; et al. A Map of Transcriptional Heterogeneity and Regulatory Variation in Human Microglia. Nat. Genet. 2021, 53, 861–868. [Google Scholar] [CrossRef]
- de Lopes, K.P.; Snijders, G.J.L.; Humphrey, J.; Allan, A.; Sneeboer, M.A.M.; Navarro, E.; Schilder, B.M.; Vialle, R.A.; Parks, M.; Missall, R.; et al. Genetic Analysis of the Human Microglial Transcriptome across Brain Regions, Aging and Disease Pathologies. Nat. Genet. 2022, 54, 4–17. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-Avidity Binding to Beta-Amyloid and Increased Frequency of Type 4 Allele in Late-Onset Familial Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [Green Version]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E Immunoreactivity in Cerebral Amyloid Deposits and Neurofibrillary Tangles in Alzheimer’s Disease and Kuru Plaque Amyloid in Creutzfeldt-Jakob Disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Song, W.M.; Colonna, M. The Identity and Function of Microglia in Neurodegeneration. Nat. Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Ulland, T.K.; Mahan, T.E.; Nyström, S.; Nilsson, K.P.; Song, W.M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; et al. ApoE Facilitates the Microglial Response to Amyloid Plaque Pathology. J. Exp. Med. 2018, 215, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 Function Impedes Tau Seeding in Neuritic Plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and Microglia Mediate Early Synapse Loss in Alzheimer Mouse Models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia Contribute to the Propagation of Aβ into Unaffected Brain Tissue. Nat. Neurosci. 2021, 25, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Wung, J.K.; Perry, G.; Kowalski, A.; Harris, P.L.R.; Bishop, G.M.; Trivedi, M.A.; Johnson, S.C.; Smith, M.A.; Denhardt, D.T.; Atwood, C.S. Increased Expression of the Remodeling- and Tumorigenic-Associated Factor Osteopontin in Pyramidal Neurons of the Alzheimer’s Disease Brain. Curr. Alzheimer Res. 2007, 4, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Comi, C.; Carecchio, M.; Chiocchetti, A.; Nicola, S.; Galimberti, D.; Fenoglio, C.; Cappellano, G.; Monaco, F.; Scarpini, E.; Dianzani, U. Osteopontin Is Increased in the Cerebrospinal Fluid of Patients with Alzheimer’s Disease and Its Levels Correlate with Cognitive Decline. J. Alzheimers Dis. 2010, 19, 1143–1148. [Google Scholar] [CrossRef]
- Shen, X.; Qiu, Y.; Wight, A.E.; Kim, H.-J.; Cantor, H. Definition of a Mouse Microglial Subset That Regulates Neuronal Development and Proinflammatory Responses in the Brain. Proc. Natl. Acad. Sci. USA 2022, 119, e2116241119. [Google Scholar] [CrossRef]
- Pinner, E.; Gruper, Y.; Ben Zimra, M.; Kristt, D.; Laudon, M.; Naor, D.; Zisapel, N. CD44 Splice Variants as Potential Players in Alzheimer’s Disease Pathology. J. Alzheimer’s Dis. 2017, 58, 1137–1149. [Google Scholar] [CrossRef]
- Greenwood, E.K.; Brown, D.R. Senescent Microglia: The Key to the Ageing Brain? Int. J. Mol. Sci. 2021, 22, 4402. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic Microglia in the Aging Human Brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Stark, B.A.; Johnson, C.O.; Roth, G.A.; Bisignano, C.; Abady, G.G.; Abbasifard, M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abedi, V.; et al. Global, Regional, and National Burden of Stroke and Its Risk Factors, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic Stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Granger, D.N. Leukocyte Recruitment and Ischemic Brain Injury. Neuromolecular. Med. 2010, 12, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil Cerebrovascular Transmigration Triggers Rapid Neurotoxicity through Release of Proteases Associated with Decondensed DNA. J. Immunol. 2012, 189, 381–392. [Google Scholar] [CrossRef]
- Planas, A.M.; Gómez-Choco, M.; Urra, X.; Gorina, R.; Caballero, M.; Chamorro, Á. Brain-Derived Antigens in Lymphoid Tissue of Patients with Acute Stroke. J. Immunol. 2012, 188, 2156–2163. [Google Scholar] [CrossRef]
- Ortega, S.B.; Noorbhai, I.; Poinsatte, K.; Kong, X.; Anderson, A.; Monson, N.L.; Stowe, A.M. Stroke Induces a Rapid Adaptive Autoimmune Response to Novel Neuronal Antigens. Discov. Med. 2015, 19, 381–392. [Google Scholar]
- Rupalla, K.; Allegrini, P.R.; Sauer, D.; Wiessner, C. Time Course of Microglia Activation and Apoptosis in Various Brain Regions after Permanent Focal Cerebral Ischemia in Mice. Acta Neuropathol. 1998, 96, 172–178. [Google Scholar] [CrossRef]
- Iadecola, C.; Buckwalter, M.S.; Anrather, J. Immune Responses to Stroke: Mechanisms, Modulation, and Therapeutic Potential. J. Clin. Investig. 2020, 130, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia Protect against Brain Injury and Their Selective Elimination Dysregulates Neuronal Network Activity after Stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.-N.; Shi, S.X.-Y.; Li, Z.; Li, M.; Wood, K.; Gonzales, R.J.; Liu, Q. Depletion of Microglia Exacerbates Postischemic Inflammation and Brain Injury. J. Cereb. Blood Flow Metab. 2017, 37, 2224–2236. [Google Scholar] [CrossRef] [Green Version]
- Otxoa-de-Amezaga, A.; Miró-Mur, F.; Pedragosa, J.; Gallizioli, M.; Justicia, C.; Gaja-Capdevila, N.; Ruíz-Jaen, F.; Salas-Perdomo, A.; Bosch, A.; Calvo, M.; et al. Microglial Cell Loss after Ischemic Stroke Favors Brain Neutrophil Accumulation. Acta Neuropathol. 2019, 137, 321–341. [Google Scholar] [CrossRef] [Green Version]
- Jackson, L.; Dumanli, S.; Johnson, M.H.; Fagan, S.C.; Ergul, A. Microglia Knockdown Reduces Inflammation and Preserves Cognition in Diabetic Animals after Experimental Stroke. J. Neuroinflammation 2020, 17, 137. [Google Scholar] [CrossRef] [PubMed]
- Ellison, J.A.; Velier, J.J.; Spera, P.; Jonak, Z.L.; Wang, X.; Barone, F.C.; Feuerstein, G.Z. Osteopontin and Its Integrin Receptor Alpha(v)Beta3 Are Upregulated during Formation of the Glial Scar after Focal Stroke. Stroke 1998, 29, 1698–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meller, R.; Stevens, S.L.; Minami, M.; Cameron, J.A.; King, S.; Rosenzweig, H.; Doyle, K.; Lessov, N.S.; Simon, R.P.; Stenzel-Poore, M.P. Neuroprotection by Osteopontin in Stroke. J. Cereb. Blood Flow Metab. 2005, 25, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, M.; Zickler, P.; Denhardt, D.T.; Hartung, H.-P.; Jander, S. Increased Thalamic Neurodegeneration Following Ischaemic Cortical Stroke in Osteopontin-Deficient Mice. Brain 2006, 129, 1426–1437. [Google Scholar] [CrossRef]
- Shin, Y.-J.; Kim, H.L.; Choi, J.-S.; Choi, J.-Y.; Cha, J.-H.; Lee, M.-Y. Osteopontin: Correlation with Phagocytosis by Brain Macrophages in a Rat Model of Stroke. Glia 2011, 59, 413–423. [Google Scholar] [CrossRef]
- Gliem, M.; Krammes, K.; Liaw, L.; van Rooijen, N.; Hartung, H.-P.; Jander, S. Macrophage-Derived Osteopontin Induces Reactive Astrocyte Polarization and Promotes Re-Establishment of the Blood Brain Barrier after Ischemic Stroke. Glia 2015, 63, 2198–2207. [Google Scholar] [CrossRef]
- Ladwig, A.; Walter, H.L.; Hucklenbroich, J.; Willuweit, A.; Langen, K.-J.; Fink, G.R.; Rueger, M.A.; Schroeter, M. Osteopontin Augments M2 Microglia Response and Separates M1- and M2-Polarized Microglial Activation in Permanent Focal Cerebral Ischemia. Mediat. Inflamm. 2017, 2017, 7189421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davaanyam, D.; Kim, I.-D.; Lee, J.-K. Intranasal Delivery of RGD-Containing Osteopontin Heptamer Peptide Confers Neuroprotection in the Ischemic Brain and Augments Microglia M2 Polarization. Int. J. Mol. Sci. 2021, 22, 9999. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T Cells Are Key Cerebroprotective Immunomodulators in Acute Experimental Stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sun, Z.; Su, W.; Xu, F.; Xie, D.; Zhang, Q.; Dai, X.; Iyer, K.; Hitchens, T.K.; Foley, L.M.; et al. Treg Cell-Derived Osteopontin Promotes Microglia-Mediated White Matter Repair after Ischemic Stroke. Immunity 2021, 54, 1527–1542.e8. [Google Scholar] [CrossRef] [PubMed]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected Increase in Amyotrophic Lateral Sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-Wide Association Analyses Identify New Risk Variants and the Genetic Architecture of Amyotrophic Lateral Sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Strong, M.J.; Kesavapany, S.; Pant, H.C. The Pathobiology of Amyotrophic Lateral Sclerosis: A Proteinopathy? J. Neuropathol. Exp. Neurol. 2005, 64, 649–664. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Canto, M.C.D.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.-X.; et al. Motor Neuron Degeneration in Mice That Express a Human Cu,Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Vocadlo, D.; Krieger, C. Mislocalization of TDP-43 in the G93A Mutant SOD1 Transgenic Mouse Model of ALS. Neurosci. Lett. 2009, 458, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Brioschi, S.; d’Errico, P.; Amann, L.S.; Janova, H.; Wojcik, S.M.; Meyer-Luehmann, M.; Rajendran, L.; Wieghofer, P.; Paolicelli, R.C.; Biber, K. Detection of Synaptic Proteins in Microglia by Flow Cytometry. Front. Mol. Neurosci. 2020, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.-Y.; et al. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 2017, 95, 297–308.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.F.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H.; et al. Motor Neurons in Cu/Zn Superoxide Dismutase-Deficient Mice Develop Normally but Exhibit Enhanced Cell Death after Axonal Injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef]
- Troost, D.; van den OORD, J.J.; Jong, J.M.B.V.D. Immunohistochemical Characterization of the Inflammatory Infiltrate in Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 1990, 16, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of Widespread Cerebral Microglial Activation in Amyotrophic Lateral Sclerosis: An [11C](R)-PK11195 Positron Emission Tomography Study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-Type Microglia Extend Survival in PU.1 Knockout Mice with Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef] [Green Version]
- Gowing, G.; Philips, T.; Van Wijmeersch, B.; Audet, J.-N.; Dewil, M.; Van Den Bosch, L.; Billiau, A.D.; Robberecht, W.; Julien, J.-P. Ablation of Proliferating Microglia Does Not Affect Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis Caused by Mutant Superoxide Dismutase. J. Neurosci. 2008, 28, 10234–10244. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M.V. Local Self-Renewal Can Sustain CNS Microglia Maintenance and Function throughout Adult Life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Chiot, A.; Zaïdi, S.; Iltis, C.; Ribon, M.; Berriat, F.; Schiaffino, L.; Jolly, A.; de la Grange, P.; Mallat, M.; Bohl, D.; et al. Modifying Macrophages at the Periphery Has the Capacity to Change Microglial Reactivity and to Extend ALS Survival. Nat. Neurosci. 2020, 23, 1339–1351. [Google Scholar] [CrossRef] [PubMed]
- Von Neuhoff, N.; Oumeraci, T.; Wolf, T.; Kollewe, K.; Bewerunge, P.; Neumann, B.; Brors, B.; Bufler, J.; Wurster, U.; Schlegelberger, B.; et al. Monitoring CSF Proteome Alterations in Amyotrophic Lateral Sclerosis: Obstacles and Perspectives in Translating a Novel Marker Panel to the Clinic. PLoS ONE 2012, 7, e44401. [Google Scholar] [CrossRef] [PubMed]
- Varghese, A.M.; Sharma, A.; Mishra, P.; Vijayalakshmi, K.; Harsha, H.C.; Sathyaprabha, T.N.; Bharath, S.M.; Nalini, A.; Alladi, P.A.; Raju, T.R. Chitotriosidase—A Putative Biomarker for Sporadic Amyotrophic Lateral Sclerosis. Clin. Proteom. 2013, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Silva, K.; Hope-Lucas, C.; White, T.; Hairston, T.-K.; Rameau, T.; Brown, A. Cortical Neurons Are a Prominent Source of the Proinflammatory Cytokine Osteopontin in HIV-Associated Neurocognitive Disorders. J. Neurovirol. 2015, 21, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Murayama, S.; Takao, M.; Isa, T.; Higo, N. Expression of Secreted Phosphoprotein 1 (Osteopontin) in Human Sensorimotor Cortex and Spinal Cord: Changes in Patients with Amyotrophic Lateral Sclerosis. Brain Res. 2017, 1655, 168–175. [Google Scholar] [CrossRef]
- Chiu, I.M.; Morimoto, E.T.A.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A Neurodegeneration-Specific Gene-Expression Signature of Acutely Isolated Microglia from an Amyotrophic Lateral Sclerosis Mouse Model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Imagama, S.; Hirano, K.; Ohgomori, T.; Natori, T.; Kobayashi, K.; Muramoto, A.; Ishiguro, N.; Kadomatsu, K. CD44 Expression in Astrocytes and Microglia Is Associated with ALS Progression in a Mouse Model. Neurosci. Lett. 2012, 520, 115–120. [Google Scholar] [CrossRef]
- Morisaki, Y.; Niikura, M.; Watanabe, M.; Onishi, K.; Tanabe, S.; Moriwaki, Y.; Okuda, T.; Ohara, S.; Murayama, S.; Takao, M.; et al. Selective Expression of Osteopontin in ALS-Resistant Motor Neurons Is a Critical Determinant of Late Phase Neurodegeneration Mediated by Matrix Metalloproteinase-9. Sci. Rep. 2016, 6, 27354. [Google Scholar] [CrossRef] [Green Version]
- Nikodemova, M.; Small, A.L.; Smith, S.M.C.; Mitchell, G.S.; Watters, J.J. Spinal but Not Cortical Microglia Acquire an Atypical Phenotype with High VEGF, Galectin-3 and Osteopontin, and Blunted Inflammatory Responses in ALS Rats. Neurobiol. Dis. 2014, 69, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Hunter, M.; Spiller, K.J.; Dominique, M.A.; Xu, H.; Hunter, F.W.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglial Transcriptome Analysis in the RNLS8 Mouse Model of TDP-43 Proteinopathy Reveals Discrete Expression Profiles Associated with Neurodegenerative Progression and Recovery. Acta Neuropathol. Commun. 2021, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global Prevalence of Age-Related Macular Degeneration and Disease Burden Projection for 2020 and 2040: A Systematic Review and Meta-Analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-Related Macular Degeneration. Nat. Rev. Dis. Primers 2021, 7, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Tenbrock, L.; Wolf, J.; Boneva, S.; Schlecht, A.; Agostini, H.; Wieghofer, P.; Schlunck, G.; Lange, C. Subretinal Fibrosis in Neovascular Age-Related Macular Degeneration: Current Concepts, Therapeutic Avenues, and Future Perspectives. Cell Tissue Res. 2022, 387, 361–375. [Google Scholar] [CrossRef]
- Brown, D.M.; Kaiser, P.K.; Michels, M.; Soubrane, G.; Heier, J.S.; Kim, R.Y.; Sy, J.P.; Schneider, S. ANCHOR Study Group Ranibizumab versus Verteporfin for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2006, 355, 1432–1444. [Google Scholar] [CrossRef] [Green Version]
- Daniel, E.; Toth, C.A.; Grunwald, J.E.; Jaffe, G.J.; Martin, D.F.; Fine, S.L.; Huang, J.; Ying, G.; Hagstrom, S.A.; Winter, K.; et al. Risk of Scar in the Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2014, 121, 656–666. [Google Scholar] [CrossRef] [Green Version]
- Wecker, T.; Ehlken, C.; Bühler, A.; Lange, C.; Agostini, H.; Böhringer, D.; Stahl, A. Five-Year Visual Acuity Outcomes and Injection Patterns in Patients with pro-Re-Nata Treatments for AMD, DME, RVO and Myopic CNV. Br. J. Ophthalmol. 2017, 101, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of Three Key Innate Immune Pathways for the Most Common Retinal Degenerative Diseases. EMBO Mol. Med. 2018, 10, e8259. [Google Scholar] [CrossRef]
- Cipriani, V.; Matharu, B.K.; Khan, J.C.; Shahid, H.; Stanton, C.M.; Hayward, C.; Wright, A.F.; Bunce, C.; Clayton, D.G.; Moore, A.T.; et al. Genetic Variation in Complement Regulators and Susceptibility to Age-Related Macular Degeneration. Immunobiology 2012, 217, 158–161. [Google Scholar] [CrossRef] [Green Version]
- Lorés-Motta, L.; Paun, C.C.; Corominas, J.; Pauper, M.; Geerlings, M.J.; Altay, L.; Schick, T.; Daha, M.R.; Fauser, S.; Hoyng, C.B.; et al. Genome-Wide Association Study Reveals Variants in CFH and CFHR4 Associated with Systemic Complement Activation: Implications in Age-Related Macular Degeneration. Ophthalmology 2018, 125, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Lipo, E.; Cashman, S.M.; Kumar-Singh, R. Aurintricarboxylic Acid Inhibits Complement Activation, Membrane Attack Complex, and Choroidal Neovascularization in a Model of Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7107–7114. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated Microglia in Human Retinitis Pigmentosa, Late-Onset Retinal Degeneration, and Age-Related Macular Degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Combadière, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodéro, M.; Pézard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. CX3CR1-Dependent Subretinal Microglia Cell Accumulation Is Associated with Cardinal Features of Age-Related Macular Degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef] [Green Version]
- Schlecht, A.; Boneva, S.; Gruber, M.; Zhang, P.; Horres, R.; Bucher, F.; Auw-Haedrich, C.; Hansen, L.; Stahl, A.; Hilgendorf, I.; et al. Transcriptomic Characterization of Human Choroidal Neovascular Membranes Identifies Calprotectin as a Novel Biomarker for Patients with Age-Related Macular Degeneration. Am. J. Pathol. 2020, 190, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Lambert, V.; Lecomte, J.; Hansen, S.; Blacher, S.; Gonzalez, M.-L.A.; Struman, I.; Sounni, N.E.; Rozet, E.; de Tullio, P.; Foidart, J.M.; et al. Laser-Induced Choroidal Neovascularization Model to Study Age-Related Macular Degeneration in Mice. Nat. Protoc. 2013, 8, 2197–2211. [Google Scholar] [CrossRef] [PubMed]
- Monsorno, K.; Buckinx, A.; Paolicelli, R.C. Microglial Metabolic Flexibility: Emerging Roles for Lactate. Trends Endocrinol. Metab. 2022, 33, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Schlecht, A.; Wolf, J.; Boneva, S.; Laich, Y.; Koch, J.; Ludwig, F.; Boeck, M.; Thien, A.; Härdtner, C.; et al. The Role of Interferon Regulatory Factor 8 for Retinal Tissue Homeostasis and Development of Choroidal Neovascularisation. J. Neuroinflammation 2021, 18, 215. [Google Scholar] [CrossRef]
- Wolf, A.; Herb, M.; Schramm, M.; Langmann, T. The TSPO-NOX1 Axis Controls Phagocyte-Triggered Pathological Angiogenesis in the Eye. Nat. Commun. 2020, 11, 2709. [Google Scholar] [CrossRef]
- Lückoff, A.; Caramoy, A.; Scholz, R.; Prinz, M.; Kalinke, U.; Langmann, T. Interferon-Beta Signaling in Retinal Mononuclear Phagocytes Attenuates Pathological Neovascularization. EMBO Mol. Med. 2016, 8, 670–678. [Google Scholar] [CrossRef]
- Dai, J.; Peng, L.; Fan, K.; Wang, H.; Wei, R.; Ji, G.; Cai, J.; Lu, B.; Li, B.; Zhang, D.; et al. Osteopontin Induces Angiogenesis through Activation of PI3K/AKT and ERK1/2 in Endothelial Cells. Oncogene 2009, 28, 3412–3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Fujita, S.; Ogata, N.; Matsuoka, M.; Okada, Y.; Kon, S.; Uede, T.; Saika, S. Endogenous Osteopontin Involvement in Laser-Induced Choroidal Neovascularization in Mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9310–9315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdaleno, F.; Ge, X.; Fey, H.; Lu, Y.; Gaskell, H.; Blajszczak, C.C.; Aloman, C.; Fiel, M.I.; Nieto, N. Osteopontin Deletion Drives Hematopoietic Stem Cell Mobilization to the Liver and Increases Hepatic Iron Contributing to Alcoholic Liver Disease. Hepatol. Commun. 2018, 2, 84–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-Related Macular Degeneration: Genetics and Biology Coming Together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Lin, S.; Copland, D.A.; Dick, A.D.; Liu, J. Cellular Senescence in the Aging Retina and Developments of Senotherapies for Age-Related Macular Degeneration. J. Neuroinflammation 2021, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Boneva, S.; Rosmus, D.-D.; Agostini, H.; Schlunck, G.; Wieghofer, P.; Schlecht, A.; Lange, C. In-Depth Molecular Profiling Specifies Human Retinal Microglia Identity. Front. Immunol. 2022, 13, 863158. [Google Scholar] [CrossRef]
- Schlecht, A.; Thien, A.; Wolf, J.; Prinz, G.; Agostini, H.; Schlunck, G.; Wieghofer, P.; Boneva, S.; Lange, C. Immunosenescence in Choroidal Neovascularization (CNV)-Transcriptional Profiling of Naïve and CNV-Associated Retinal Myeloid Cells during Aging. Int. J. Mol. Sci. 2021, 22, 13318. [Google Scholar] [CrossRef]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic Retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Yau, J.W.Y.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.-J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global Prevalence and Major Risk Factors of Diabetic Retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.Y.; Cheung, C.M.G.; Larsen, M.; Sharma, S.; Simó, R. Diabetic Retinopathy. Nat. Rev. Dis. Primers 2016, 2, 16012. [Google Scholar] [CrossRef]
- Klein, B.E.; Moss, S.E.; Klein, R. Effect of Pregnancy on Progression of Diabetic Retinopathy. Diabetes Care 1990, 13, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, Q.; Gillies, M.C.; Wong, T.Y. Management of Diabetic RetinopathyA Systematic Review. JAMA 2007, 298, 902–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simó, R.; Hernández, C. Neurodegeneration in the Diabetic Eye: New Insights and Therapeutic Perspectives. Trends Endocrinol. Metab. 2014, 25, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Murata, T.; Tsujikawa, A.; Kirchhof, B.; Bursell, S.E.; Adamis, A.P. Leukocyte-Mediated Endothelial Cell Injury and Death in the Diabetic Retina. Am. J. Pathol. 2001, 158, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Lange, C.A.K.; Stavrakas, P.; Luhmann, U.F.O.; de Silva, D.J.; Ali, R.R.; Gregor, Z.J.; Bainbridge, J.W.B. Intraocular Oxygen Distribution in Advanced Proliferative Diabetic Retinopathy. Am. J. Ophthalmol. 2011, 152, 406–412.e3. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, M.; Forrester, J.V. Para-Inflammation in the Aging Retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef]
- Simo, R.; Carrasco, E.; Garcia-Ramirez, M.; Hernandez, C. Angiogenic and Antiangiogenic Factors in Proliferative Diabetic Retinopathy. Curr. Diabetes Rev. 2006, 2, 71–98. [Google Scholar] [CrossRef]
- Liyanage, S.E.; Fantin, A.; Villacampa, P.; Lange, C.A.; Denti, L.; Cristante, E.; Smith, A.J.; Ali, R.R.; Luhmann, U.F.; Bainbridge, J.W.; et al. Myeloid-Derived Vascular Endothelial Growth Factor and Hypoxia-Inducible Factor Are Dispensable for Ocular Neovascularization—Brief Report. Arter. Thromb. Vasc. Biol. 2016, 36, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Boeck, M.; Thien, A.; Wolf, J.; Hagemeyer, N.; Laich, Y.; Yusuf, D.; Backofen, R.; Zhang, P.; Boneva, S.; Stahl, A.; et al. Temporospatial Distribution and Transcriptional Profile of Retinal Microglia in the Oxygen-Induced Retinopathy Mouse Model. Glia 2020, 68, 1859–1873. [Google Scholar] [CrossRef]
- Boneva, S.K.; Wolf, J.; Hajdú, R.I.; Prinz, G.; Salié, H.; Schlecht, A.; Killmer, S.; Laich, Y.; Faatz, H.; Lommatzsch, A.; et al. In-Depth Molecular Characterization of Neovascular Membranes Suggests a Role for Hyalocyte-to-Myofibroblast Transdifferentiation in Proliferative Diabetic Retinopathy. Front. Immunol. 2021, 12, 757607. [Google Scholar] [CrossRef]
- Zeng, H.; Green, W.R.; Tso, M.O.M. Microglial Activation in Human Diabetic Retinopathy. Arch. Ophthalmol. 2008, 126, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakoczy, E.P.; Rahman, I.S.A.; Binz, N.; Li, C.-R.; Vagaja, N.N.; de Pinho, M.; Lai, C.-M. Characterization of a Mouse Model of Hyperglycemia and Retinal Neovascularization. Am. J. Pathol. 2010, 177, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, I.; De Groef, L.; Boeckx, B.; Modave, E.; Hu, T.-T.; Beets, K.; Etienne, I.; Van Bergen, T.; Lambrechts, D.; Moons, L.; et al. Single-Cell Transcriptome Analysis of the Akimba Mouse Retina Reveals Cell-Type-Specific Insights into the Pathobiology of Diabetic Retinopathy. Diabetologia 2020, 63, 2235–2248. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Jiang, C.; Xue, B.; Zhu, S.; Wang, X. High Glucose Stimulates TNFα and MCP-1 Expression in Rat Microglia via ROS and NF-ΚB Pathways. Acta Pharm. Sin. 2011, 32, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinuthia, U.M.; Wolf, A.; Langmann, T. Microglia and Inflammatory Responses in Diabetic Retinopathy. Front. Immunol. 2020, 11, 564077. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.H.; Yun, J.-H.; Cho, C.S.; Kim, J.H.; Kim, J.H.; Cho, C.-H. Interaction between Microglia and Retinal Pigment Epithelial Cells Determines the Integrity of Outer Blood-Retinal Barrier in Diabetic Retinopathy. Glia 2019, 67, 321–331. [Google Scholar] [CrossRef]
- Mills, S.A.; Jobling, A.I.; Dixon, M.A.; Bui, B.V.; Vessey, K.A.; Phipps, J.A.; Greferath, U.; Venables, G.; Wong, V.H.Y.; Wong, C.H.Y.; et al. Fractalkine-Induced Microglial Vasoregulation Occurs within the Retina and Is Altered Early in Diabetic Retinopathy. Proc. Natl. Acad. Sci. USA 2021, 118, e2112561118. [Google Scholar] [CrossRef]
- Vujosevic, S.; Bini, S.; Midena, G.; Berton, M.; Pilotto, E.; Midena, E. Hyperreflective Intraretinal Spots in Diabetics without and with Nonproliferative Diabetic Retinopathy: An in Vivo Study Using Spectral Domain OCT. J. Diabetes Res. 2013, 2013, 491835. [Google Scholar] [CrossRef] [Green Version]
- Ong, J.X.; Nesper, P.L.; Fawzi, A.A.; Wang, J.M.; Lavine, J.A. Macrophage-Like Cell Density Is Increased in Proliferative Diabetic Retinopathy Characterized by Optical Coherence Tomography Angiography. Investig. Ophthalmol. Vis. Sci. 2021, 62, 2. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Y.-S.; Li, X.-Q.; Hou, H.-Y.; Su, J.-B.; Yao, L.-B.; Zhang, J. Macrophages Promote Vasculogenesis of Retinal Neovascularization in an Oxygen-Induced Reti.inopathy Model in Mice. Cell Tissue Res. 2016, 364, 599–610. [Google Scholar] [CrossRef]
- Ritter, M.R.; Banin, E.; Moreno, S.K.; Aguilar, E.; Dorrell, M.I.; Friedlander, M. Myeloid Progenitors Differentiate into Microglia and Promote Vascular Repair in a Model of Ischemic Retinopathy. J. Clin. Investig. 2006, 116, 3266–3276. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, J.; Ma, Q.; Zhang, X.; Yang, Q.; Wang, L.; Cao, Y.; Xu, Z.; Tawfik, A.; Sun, Y.; et al. Glycolysis Links Reciprocal Activation of Myeloid Cells and Endothelial Cells in the Retinal Angiogenic Niche. Sci. Transl. Med. 2020, 12, eaay1371. [Google Scholar] [CrossRef] [PubMed]
- Kase, S.; Yokoi, M.; Saito, W.; Furudate, N.; Ohgami, K.; Kitamura, M.; Kitaichi, N.; Yoshida, K.; Kase, M.; Ohno, S.; et al. Increased Osteopontin Levels in the Vitreous of Patients with Diabetic Retinopathy. Ophthalmic Res. 2007, 39, 143–147. [Google Scholar] [CrossRef]
- Zhang, X.; Chee, W.K.; Liu, S.; Tavintharan, S.; Sum, C.F.; Lim, S.C.; Kumari, N. Association of Plasma Osteopontin with Diabetic Retinopathy in Asians with Type 2 Diabetes. Mol. Vis. 2018, 24, 165–173. [Google Scholar] [PubMed]
- Duan, P.; Chen, S.; Zeng, Y.; Xu, H.; Liu, Y. Osteopontin Upregulates Col IV Expression by Repressing MiR-29a in Human Retinal Capillary Endothelial Cells. Mol. Ther. Nucleic Acids 2020, 20, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Garzon, R.; Sun, H.; Ladner, K.J.; Singh, R.; Dahlman, J.; Cheng, A.; Hall, B.M.; Qualman, S.J.; Chandler, D.S.; et al. NF-KappaB-YY1-MiR-29 Regulatory Circuitry in Skeletal Myogenesis and Rhabdomyosarcoma. Cancer Cell 2008, 14, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlecht, A.; Wolf, J.; Boneva, S.; Prinz, G.; Braunger, B.M.; Wieghofer, P.; Agostini, H.; Schlunck, G.; Lange, C. Transcriptional and Distributional Profiling of Microglia in Retinal Angiomatous Proliferation. Int. J. Mol. Sci. 2022, 23, 3443. [Google Scholar] [CrossRef]
- Hu, Z.; Mao, X.; Chen, M.; Wu, X.; Zhu, T.; Liu, Y.; Zhang, Z.; Fan, W.; Xie, P.; Yuan, S.; et al. Single-Cell Transcriptomics Reveals Novel Role of Microglia in Fibrovascular Membrane of Proliferative Diabetic Retinopathy. Diabetes 2022, 71, 762–773. [Google Scholar] [CrossRef]
- Boneva, S.K.; Wolf, J.; Rosmus, D.-D.; Schlecht, A.; Prinz, G.; Laich, Y.; Boeck, M.; Zhang, P.; Hilgendorf, I.; Stahl, A.; et al. Transcriptional Profiling Uncovers Human Hyalocytes as a Unique Innate Immune Cell Population. Front. Immunol. 2020, 11, 567274. [Google Scholar] [CrossRef]
- Wolf, J.; Boneva, S.; Schlecht, A.; Lapp, T.; Auw-Haedrich, C.; Lagrèze, W.; Agostini, H.; Reinhard, T.; Schlunck, G.; Lange, C. The Human Eye Transcriptome Atlas: A Searchable Comparative Transcriptome Database for Healthy and Diseased Human Eye Tissue. Genomics 2022, 114, 110286. [Google Scholar] [CrossRef]
- Wolf, J.; Boneva, S.; Rosmus, D.-D.; Agostini, H.; Schlunck, G.; Wieghofer, P.; Schlecht, A.; Lange, C. Deciphering the Molecular Signature of Human Hyalocytes in Relation to Other Innate Immune Cell Populations. Investig. Ophthalmol. Vis. Sci. 2022, 63, 9. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Missotten, L.; Geboes, K. Expression of High-Mobility Groups Box-1/Receptor for Advanced Glycation End Products/Osteopontin/Early Growth Response-1 Pathway in Proliferative Vitreoretinal Epiretinal Membranes. Mol. Vis. 2011, 17, 508–518. [Google Scholar]
- Rodriques, S.G.; Chen, L.M.; Liu, S.; Zhong, E.D.; Scherrer, J.R.; Boyden, E.S.; Chen, F. RNA Timestamps Identify the Age of Single Molecules in RNA Sequencing. Nat. Biotechnol. 2021, 39, 320–325. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosmus, D.-D.; Lange, C.; Ludwig, F.; Ajami, B.; Wieghofer, P. The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives. Biomedicines 2022, 10, 840. https://doi.org/10.3390/biomedicines10040840
Rosmus D-D, Lange C, Ludwig F, Ajami B, Wieghofer P. The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives. Biomedicines. 2022; 10(4):840. https://doi.org/10.3390/biomedicines10040840
Chicago/Turabian StyleRosmus, Dennis-Dominik, Clemens Lange, Franziska Ludwig, Bahareh Ajami, and Peter Wieghofer. 2022. "The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives" Biomedicines 10, no. 4: 840. https://doi.org/10.3390/biomedicines10040840