
The Hallmarks of Glioblastoma: Heterogeneity, Intercellular Crosstalk and Molecular Signature of Invasiveness and Progression

 , , ,
, , ,  , , , ,
, , , ,  and
and 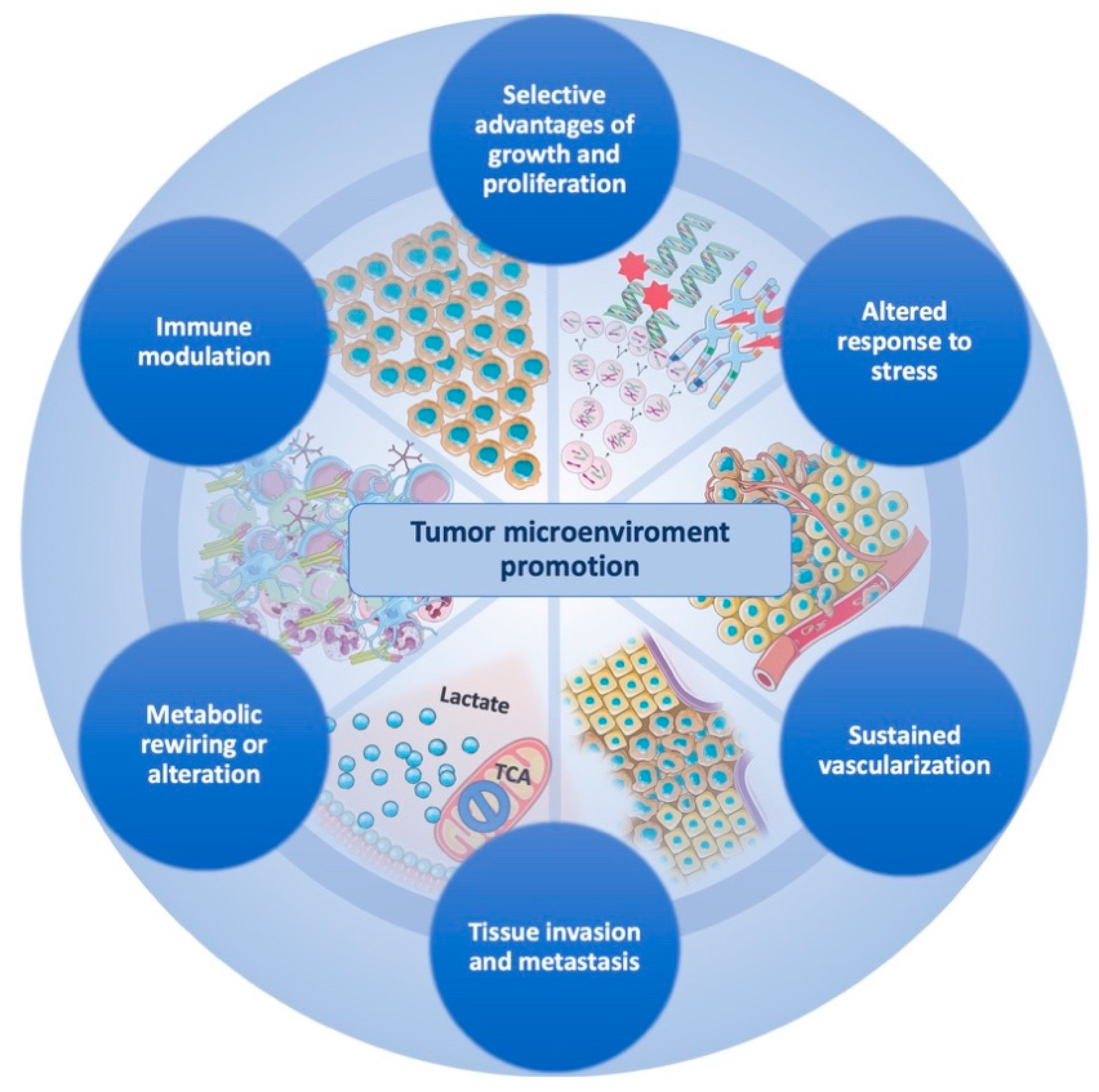{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
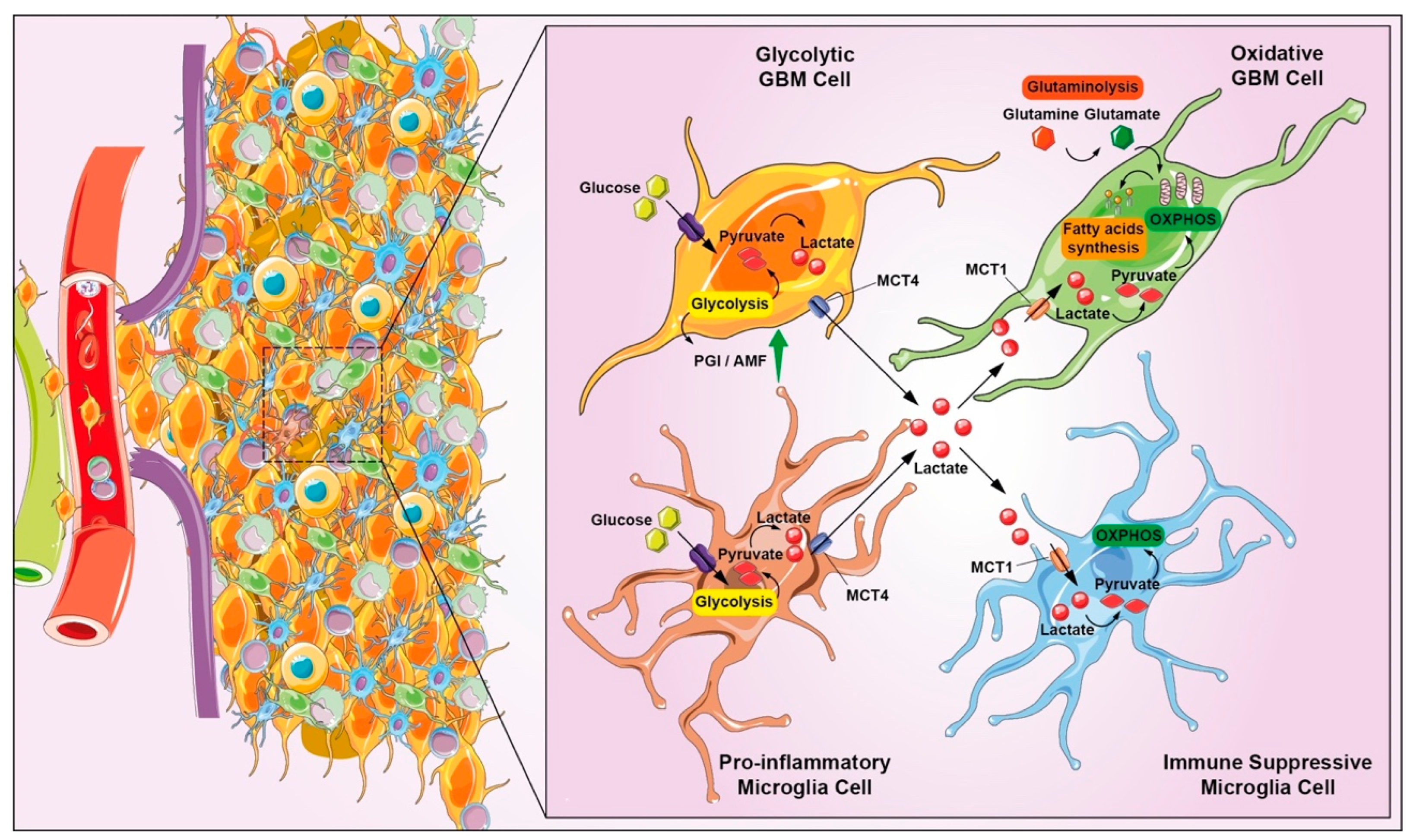{kind=link}
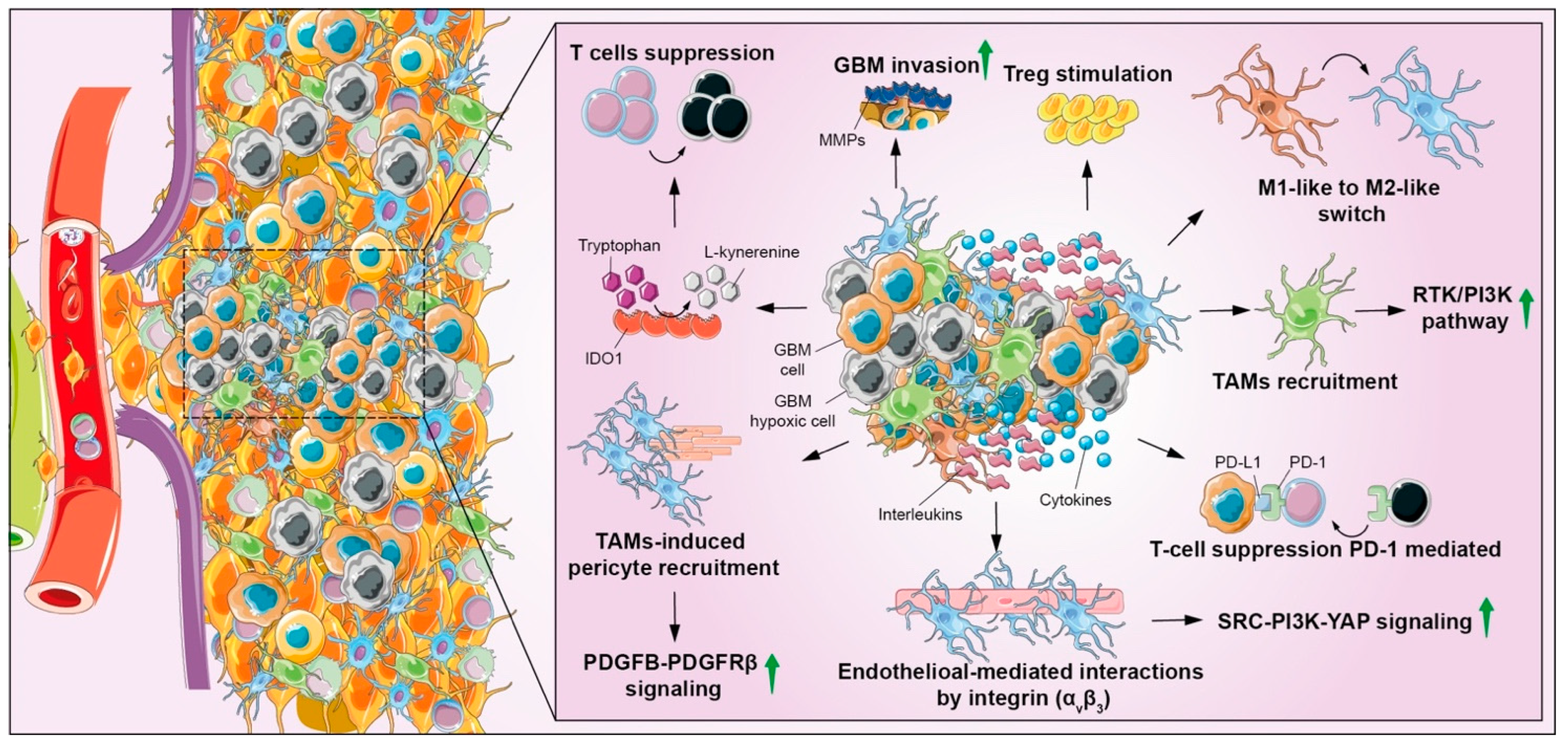{kind=link}
Abstract
:1. Introduction
2. Selective Advantages of Growth and Proliferation
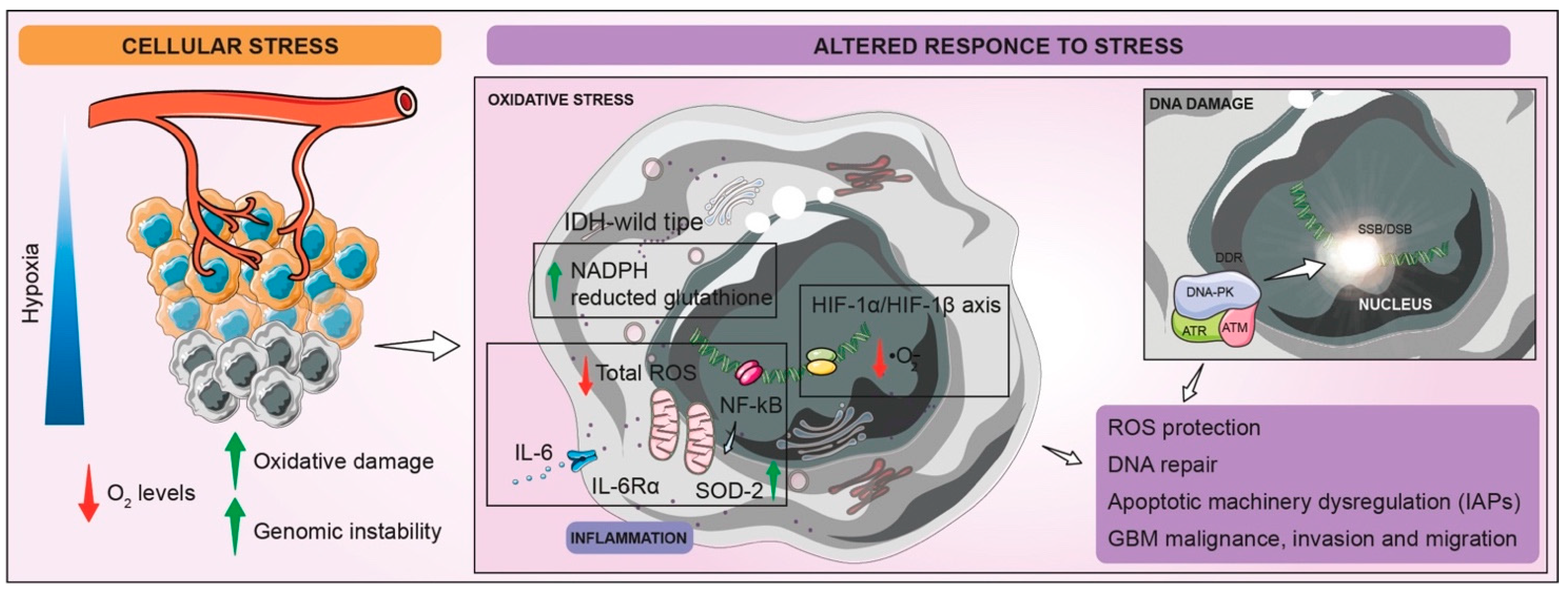
3. Altered Response to Stress
4. Sustained Vascularization
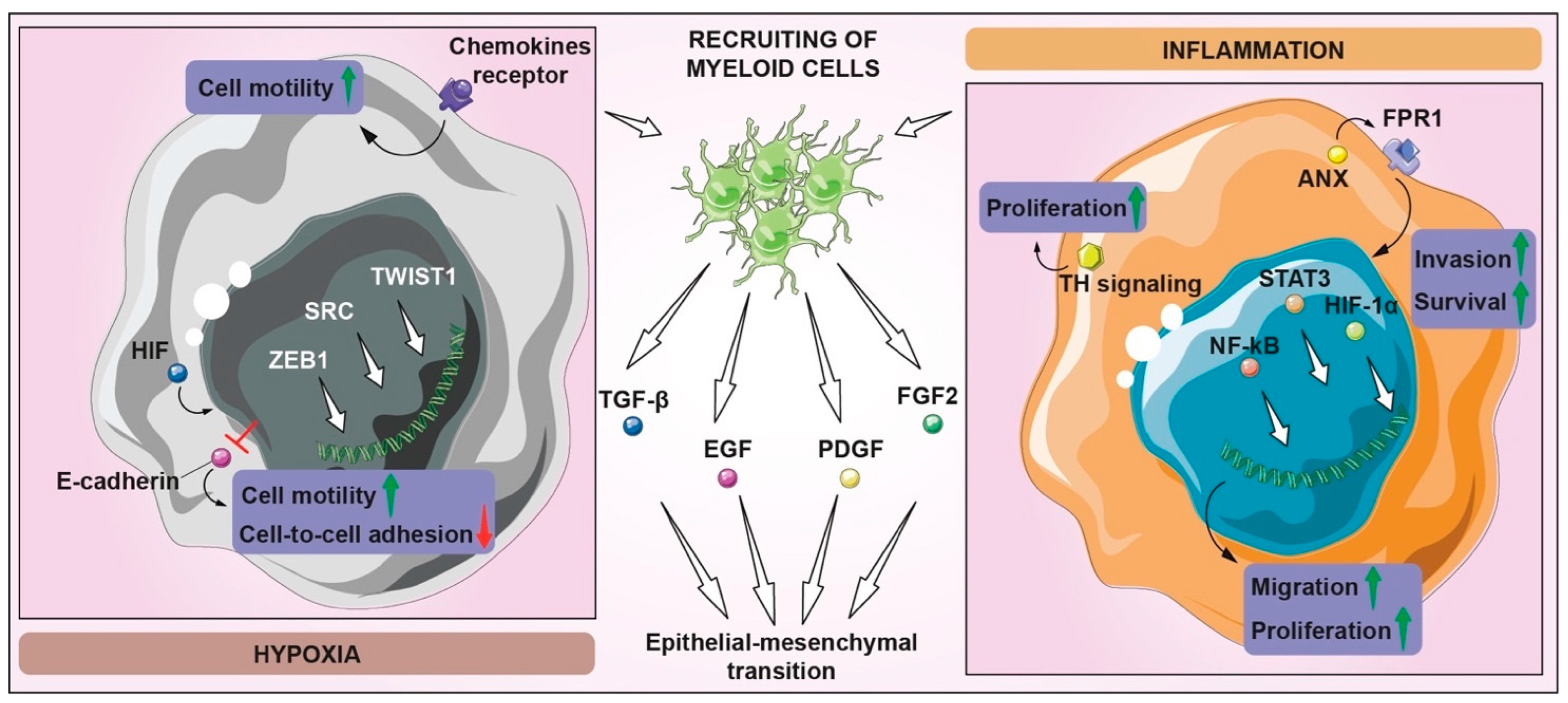
5. Tissue Invasion and Metastasis
6. Metabolic Rewiring and Adaptation
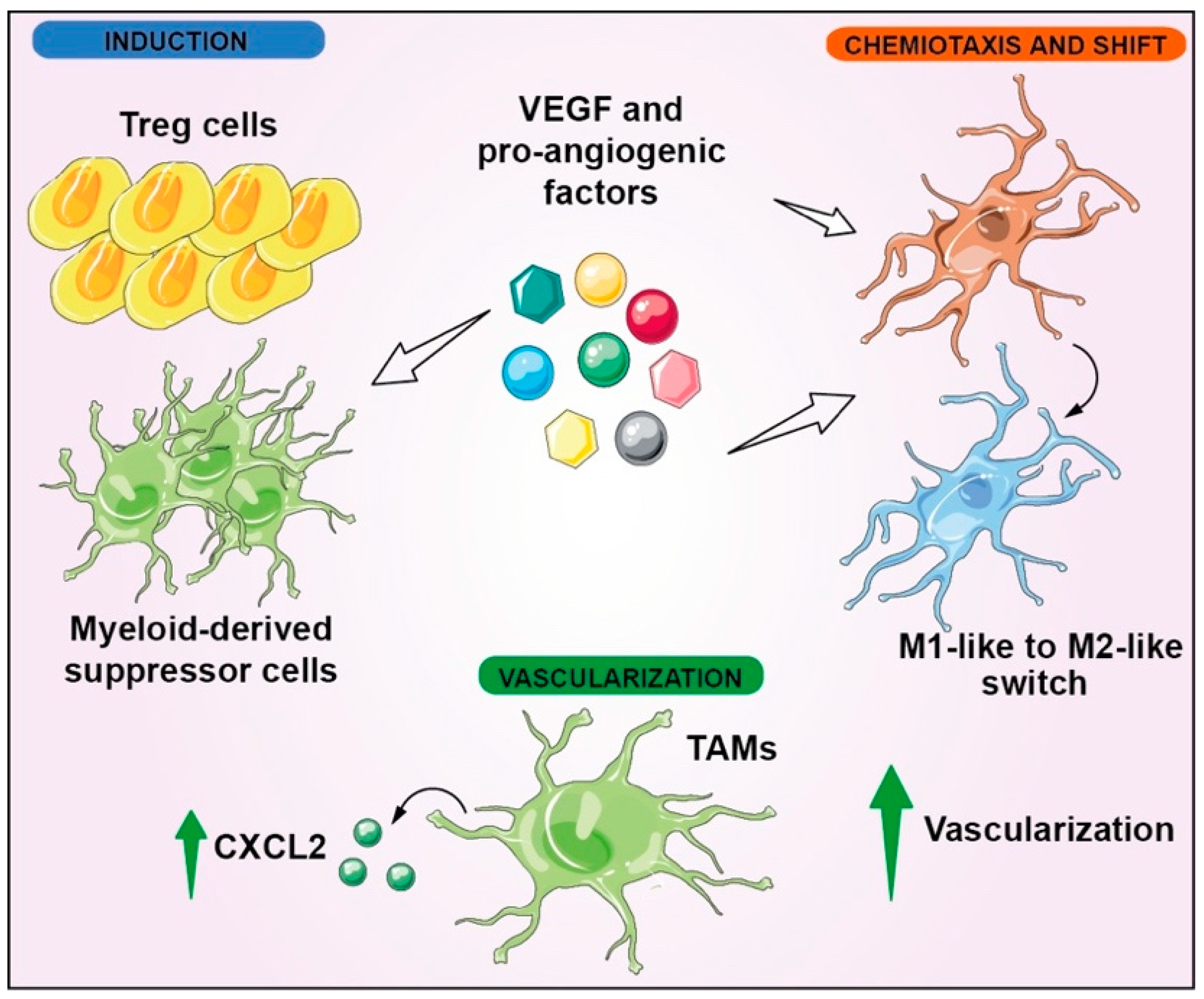
7. Immune Modulation
8. Interconnections and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeibouei, S.; Akbari, M.E.; Kalbasi, A.; Aref, A.R.; Ajoudanian, M.; Rezvani, A.; Zali, H. Personalized medicine in breast cancer: Pharmacogenomics approaches. Pharm. Pers. Med. 2019, 12, 59–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.W.H.; Law, B.M.H.; So, W.K.W.; Chow, K.M.; Waye, M.M.Y. Novel Strategies on Personalized Medicine for Breast Cancer Treatment: An Update. Int. J. Mol. Sci. 2017, 18, 2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Delgado-Lopez, P.D.; Corrales-Garcia, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, A.A.; Bartolotti, M.; Tosoni, A.; Franceschi, E. Nitrosoureas in the Management of Malignant Gliomas. Curr. Neurol. Neurosci. Rep. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Portnow, J.; Ahluwalia, M.; Baehring, J.; Brem, H.; Brem, S.; Butowski, N.; Campian, J.L.; Clark, S.W.; Fabiano, A.J.; et al. Central Nervous System Cancers, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Natl. Compr. Netw. 2020, 18, 1537–1570. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Rahman, M.; Sawyer, W.G.; Lindhorst, S.; Deleyrolle, L.P.; Harrison, J.K.; Karachi, A.; Dastmalchi, F.; Flores-Toro, J.; Mitchell, D.A.; Lim, M.; et al. Adult immuno-oncology: Using past failures to inform the future. Neuro Oncol. 2020, 22, 1249–1261. [Google Scholar] [CrossRef]
- Oliveira, A.I.; Anjo, S.I.; Vieira de Castro, J.; Serra, S.C.; Salgado, A.J.; Manadas, B.; Costa, B.M. Crosstalk between glial and glioblastoma cells triggers the "go-or-grow" phenotype of tumor cells. Cell Commun. Signal. 2017, 15, 37. [Google Scholar] [CrossRef] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Degl’Innocenti, A.; di Leo, N.; Ciofani, G. Genetic Hallmarks and Heterogeneity of Glioblastoma in the Single-Cell Omics Era. Adv. Ther. 2020, 3, 1900152. [Google Scholar] [CrossRef]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: Rationale and importance to inhibiting these pathways in human health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, H.R.; Camelo-Piragua, S.; Kurian, K.M. Prognostic and predictive biomarkers in adult and pediatric gliomas: Toward personalized treatment. Front. Oncol. 2014, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.W.; Hu, B.; Cheng, S.Y. Platelet-derived growth factor receptor alpha in glioma: A bad seed. Chin. J. Cancer 2011, 30, 590–602. [Google Scholar] [CrossRef]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e520. [Google Scholar] [CrossRef]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, R.C.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 2011, 3, 192–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahluwalia, M.S.; de Groot, J.; Liu, W.M.; Gladson, C.L. Targeting SRC in glioblastoma tumors and brain metastases: Rationale and preclinical studies. Cancer Lett. 2010, 298, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, E.; Koustas, E.; Goulielmaki, M.; Pintzas, A. BRAF vs. RAS oncogenes: Are mutations of the same pathway equal? Differential signalling and therapeutic implications. Oncotarget 2014, 5, 11752–11777. [Google Scholar] [CrossRef] [Green Version]
- Ronellenfitsch, M.W.; Zeiner, P.S.; Mittelbronn, M.; Urban, H.; Pietsch, T.; Reuter, D.; Senft, C.; Steinbach, J.P.; Westphal, M.; Harter, P.N. Akt and mTORC1 signaling as predictive biomarkers for the EGFR antibody nimotuzumab in glioblastoma. Acta Neuropathol. Commun. 2018, 6, 81. [Google Scholar] [CrossRef]
- Lo, H.W. Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Curr. Cancer Drug Targets 2010, 10, 840–848. [Google Scholar] [CrossRef]
- Holmen, S.L.; Williams, B.O. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res. 2005, 65, 8250–8255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, A.; Sarcar, B.; Miller, C.R.; Kim, S.H.; Nakano, I.; Forsyth, P.; Chinnaiyan, P. Ras-mediated modulation of pyruvate dehydrogenase activity regulates mitochondrial reserve capacity and contributes to glioblastoma tumorigenesis. Neuro-Oncology 2015, 17, 1220–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Searle, J.S. The rb pathway and cancer therapeutics. Curr. Drug Targets 2009, 10, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.L.; Ueki, K.; Jhung, S.L.; Koh, J.; Louis, D.N. Molecular genetic correlates of p16, cdk4, and pRb immunohistochemistry in glioblastomas. J. Neuropathol. Exp. Neurol. 1998, 57, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, W.A.; Araujo, M.D.; Anselmo, N.P.; de Oliveira, E.H.; Brito, J.R.; Burbano, R.R.; Harada, M.L.; Borges Bdo, N. Expression Analysis of Genes Involved in the RB/E2F Pathway in Astrocytic Tumors. PLoS ONE 2015, 10, e0137259. [Google Scholar] [CrossRef] [Green Version]
- Biasoli, D.; Kahn, S.A.; Cornelio, T.A.; Furtado, M.; Campanati, L.; Chneiweiss, H.; Moura-Neto, V.; Borges, H.L. Retinoblastoma protein regulates the crosstalk between autophagy and apoptosis, and favors glioblastoma resistance to etoposide. Cell Death Dis. 2013, 4, e767. [Google Scholar] [CrossRef] [Green Version]
- Kanu, O.O.; Hughes, B.; Di, C.; Lin, N.; Fu, J.; Bigner, D.D.; Yan, H.; Adamson, C. Glioblastoma Multiforme Oncogenomics and Signaling Pathways. Clin. Med. Oncol. 2009, 3, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Conti, A.; Guli, C.; La Torre, D.; Tomasello, C.; Angileri, F.F.; Aguennouz, M. Role of inflammation and oxidative stress mediators in gliomas. Cancers 2010, 2, 693–712. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audia, A.; Conroy, S.; Glass, R.; Bhat, K.P.L. The Impact of the Tumor Microenvironment on the Properties of Glioma Stem-Like Cells. Front. Oncol. 2017, 7, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padovan, M.; Eoli, M.; Pellerino, A.; Rizzato, S.; Caserta, C.; Simonelli, M.; Michiara, M.; Caccese, M.; Anghileri, E.; Cerretti, G.; et al. Depatuxizumab Mafodotin (Depatux-M) Plus Temozolomide in Recurrent Glioblastoma Patients: Real-World Experience from a Multicenter Study of Italian Association of Neuro-Oncology (AINO). Cancers 2021, 13, 2773. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; van den Bent, M.J.; Taphoorn, M.J.; Steuve, J.; Brandes, A.A.; Hamou, M.F.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.Y.; de Groot, J.F. Treatment of Glioblastoma. J. Oncol. Pract. 2017, 13, 629–638. [Google Scholar] [CrossRef]
- Zhou, M.; Niu, C.; Jia, L.; He, H. The value of MGMT promote methylation and IDH-1 mutation on diagnosis of pseudoprogression in patients with high-grade glioma: A meta-analysis. Medicine 2019, 98, e18194. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Shi, J.; Zuo, H.; Ni, L.; Xia, L.; Zhao, L.; Gong, M.; Nie, D.; Gong, P.; Cui, D.; Shi, W.; et al. An IDH1 mutation inhibits growth of glioma cells via GSH depletion and ROS generation. Neurol. Sci. 2014, 35, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Dattilo, S.; Petralia, A.; Parenti, R.; Pennisi, M.; Koverech, G.; Calabrese, V.; Graziano, A.; Monte, I.; Maiolino, L.; et al. Analytical approaches to the diagnosis and treatment of aging and aging-related disease: Redox status and proteomics. Free Radic. Res. 2015, 49, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C.; Seyfried, T.N. Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis. ASN Neuro 2018, 10, 1759091418818261. [Google Scholar] [CrossRef]
- Ferri, A.; Stagni, V.; Barila, D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2020, 7, 620677. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; De Laurenzi, V. Role of apoptosis in disease. Aging (Albany NY) 2012, 4, 330–349. [Google Scholar] [CrossRef]
- Trejo-Solis, C.; Serrano-Garcia, N.; Escamilla-Ramirez, A.; Castillo-Rodriguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Najera, A.; Cruz-Salgado, A.; et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrisi, F.; Alberghina, C.; Lo Furno, D.; Zappala, A.; Valable, S.; Li Volti, G.; Tibullo, D.; Vicario, N.; Parenti, R. Connexin 43 and Sonic Hedgehog Pathway Interplay in Glioblastoma Cell Proliferation and Migration. Biology 2021, 10, 767. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Rives, S.A.; Casique-Aguirre, D.; German-Castelan, L.; Velasco-Velazquez, M.A.; Gonzalez-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicario, N.; Bernstock, J.D.; Spitale, F.M.; Giallongo, C.; Giunta, M.A.S.; Li Volti, G.; Gulisano, M.; Leanza, G.; Tibullo, D.; Parenti, R.; et al. Clobetasol Modulates Adult Neural Stem Cell Growth via Canonical Hedgehog Pathway Activation. Int. J. Mol. Sci. 2019, 20, 1991. [Google Scholar] [CrossRef] [Green Version]
- Mallat, Z.; Tedgui, A. Apoptosis in the vasculature: Mechanisms and functional importance. Br. J. Pharmacol. 2000, 130, 947–962. [Google Scholar] [CrossRef] [Green Version]
- Zagzag, D.; Amirnovin, R.; Greco, M.A.; Yee, H.; Holash, J.; Wiegand, S.J.; Zabski, S.; Yancopoulos, G.D.; Grumet, M. Vascular apoptosis and involution in gliomas precede neovascularization: A novel concept for glioma growth and angiogenesis. Lab. Investig. 2000, 80, 837–849. [Google Scholar] [CrossRef] [Green Version]
- Jawhari, S.; Ratinaud, M.H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone ‘menage-a-trois’. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M.; Khuchua, Z.; et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef]
- Guo, J.C.; Wei, Q.S.; Dong, L.; Fang, S.S.; Li, F.; Zhao, Y. Prognostic Value of an Autophagy-Related Five-Gene Signature for Lower-Grade Glioma Patients. Front. Oncol. 2021, 11, 644443. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, R.; Li, X.; Dong, N.; Wu, D.; Hou, L.; Yin, K.; Zhao, C. An Autophagy-Related Gene Signature Associated With Clinical Prognosis and Immune Microenvironment in Gliomas. Front. Oncol. 2020, 10, 571189. [Google Scholar] [CrossRef]
- Beckta, J.M.; Bindra, R.S.; Chalmers, A.J. Targeting DNA repair in gliomas. Curr. Opin. Neurol. 2019, 32, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, S.; Wagemakers, M.; Walenkamp, A.M.; Kruyt, F.A.; den Dunnen, W.F. Novel insights into vascularization patterns and angiogenic factors in glioblastoma subclasses. J. Neurooncol. 2017, 131, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ali, S.; Clarke, J.; Cha, S. Bevacizumab in Recurrent Glioma: Patterns of Treatment Failure and Implications. Brain Tumor Res. Treat. 2017, 5, 1–9. [Google Scholar] [CrossRef]
- Huveldt, D.; Lewis-Tuffin, L.J.; Carlson, B.L.; Schroeder, M.A.; Rodriguez, F.; Giannini, C.; Galanis, E.; Sarkaria, J.N.; Anastasiadis, P.Z. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS ONE 2013, 8, e56505. [Google Scholar] [CrossRef] [Green Version]
- Kuang, R.; Jahangiri, A.; Mascharak, S.; Nguyen, A.; Chandra, A.; Flanigan, P.M.; Yagnik, G.; Wagner, J.R.; De Lay, M.; Carrera, D.; et al. GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight 2017, 2, e88815. [Google Scholar] [CrossRef]
- Vallee, A.; Lecarpentier, Y.; Vallee, J.N. The Key Role of the WNT/beta-Catenin Pathway in Metabolic Reprogramming in Cancers under Normoxic Conditions. Cancers 2021, 13, 5557. [Google Scholar] [CrossRef]
- Brandenburg, S.; Muller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef]
- De Palma, M.; Murdoch, C.; Venneri, M.A.; Naldini, L.; Lewis, C.E. Tie2-expressing monocytes: Regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 2007, 28, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Forstreuter, F.; Lucius, R.; Mentlein, R. Vascular endothelial growth factor induces chemotaxis and proliferation of microglial cells. J. Neuroimmunol. 2002, 132, 93–98. [Google Scholar] [CrossRef]
- Souberan, A.; Brustlein, S.; Gouarne, C.; Chasson, L.; Tchoghandjian, A.; Malissen, M.; Rougon, G. Effects of VEGF blockade on the dynamics of the inflammatory landscape in glioblastoma-bearing mice. J. Neuroinflammation 2019, 16, 191. [Google Scholar] [CrossRef] [PubMed]
- Popescu, A.M.; Purcaru, S.O.; Alexandru, O.; Dricu, A. New perspectives in glioblastoma antiangiogenic therapy. Contemp. Oncol. 2016, 20, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Lee, E.S.; You, H.J.; Lee, J.W.; Park, J.W.; Chun, Y.S. Ras-dependent induction of HIF-1alpha785 via the Raf/MEK/ERK pathway: A novel mechanism of Ras-mediated tumor promotion. Oncogene 2004, 23, 9427–9431. [Google Scholar] [CrossRef] [Green Version]
- Domenech, M.; Hernandez, A.; Plaja, A.; Martinez-Balibrea, E.; Balana, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef]
- Kim, W.; Yoo, H.; Shin, S.H.; Gwak, H.S.; Lee, S.H. Extraneural Metastases of Glioblastoma without Simultaneous Central Nervous System Recurrence. Brain Tumor Res. Treat. 2014, 2, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Cunha, M.; Maldaun, M.V.C. Metastasis from glioblastoma multiforme: A meta-analysis. Rev. Assoc. Med. Bras. 2019, 65, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrisi, F.; Vicario, N.; Spitale, F.M.; Cammarata, F.P.; Minafra, L.; Salvatorelli, L.; Russo, G.; Cuttone, G.; Valable, S.; Gulino, R.; et al. The Role of Hypoxia and SRC Tyrosine Kinase in Glioblastoma Invasiveness and Radioresistance. Cancers 2020, 12, 2860. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrisi, F.; Minafra, L.; Cammarata, F.P.; Savoca, G.; Calvaruso, M.; Vicario, N.; Maccari, L.; Peres, E.A.; Ozcelik, H.; Bernaudin, M.; et al. SRC Tyrosine Kinase Inhibitor and X-rays Combined Effect on Glioblastoma Cell Lines. Int. J. Mol. Sci. 2020, 21, 3917. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Calabrese, G.; Zappala, A.; Parenti, C.; Forte, S.; Graziano, A.C.E.; Vanella, L.; Pellitteri, R.; Cardile, V.; Parenti, R. Inhibition of Cx43 mediates protective effects on hypoxic/reoxygenated human neuroblastoma cells. J. Cell Mol. Med. 2017, 21, 2563–2572. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhao, J.; Chen, K.; Bian, X.; Wang, C.; Shi, Y.; Wang, J.M. G protein-coupled receptor FPR1 as a pharmacologic target in inflammation and human glioblastoma. Int. Immunopharmacol. 2012, 14, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.E.; Lumibao, J.; Leary, S.; Sarkaria, J.N.; Steelman, A.J.; Gaskins, H.R.; Harley, B.A.C. Crosstalk between microglia and patient-derived glioblastoma cells inhibit invasion in a three-dimensional gelatin hydrogel model. J. Neuroinflamm. 2020, 17, 346. [Google Scholar] [CrossRef]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wei, Z.; Dong, B.; Lian, Z.; Xu, Y. Silencing of phosphoglucose isomerase/autocrine motility factor decreases U87 human glioblastoma cell migration. Int. J. Mol. Med. 2016, 37, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.; Fingleton, B. Non-canonical roles for metabolic enzymes and intermediates in malignant progression and metastasis. Clin. Exp. Metastasis 2019, 36, 211–224. [Google Scholar] [CrossRef]
- Garcia, J.H.; Jain, S.; Aghi, M.K. Metabolic Drivers of Invasion in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 683276. [Google Scholar] [CrossRef] [PubMed]
- Duraj, T.; Garcia-Romero, N.; Carrion-Navarro, J.; Madurga, R.; Mendivil, A.O.; Prat-Acin, R.; Garcia-Canamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology 2017, 19, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [Green Version]
- Poteet, E.; Choudhury, G.R.; Winters, A.; Li, W.; Ryou, M.G.; Liu, R.; Tang, L.; Ghorpade, A.; Wen, Y.; Yuan, F.; et al. Reversing the Warburg effect as a treatment for glioblastoma. J. Biol. Chem. 2013, 288, 9153–9164. [Google Scholar] [CrossRef] [Green Version]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Crane, C.A.; Austgen, K.; Haberthur, K.; Hofmann, C.; Moyes, K.W.; Avanesyan, L.; Fong, L.; Campbell, M.J.; Cooper, S.; Oakes, S.A.; et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc. Natl. Acad. Sci. USA 2014, 111, 12823–12828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirasani, S.R.; Leukel, P.; Gottfried, E.; Hochrein, J.; Stadler, K.; Neumann, B.; Oefner, P.J.; Gronwald, W.; Bogdahn, U.; Hau, P.; et al. Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. Int. J. Cancer 2013, 132, 843–853. [Google Scholar] [CrossRef]
- Virtuoso, A.; Giovannoni, R.; De Luca, C.; Gargano, F.; Cerasuolo, M.; Maggio, N.; Lavitrano, M.; Papa, M. The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence. Int. J. Mol. Sci. 2021, 22, 3301. [Google Scholar] [CrossRef]
- Han, C.J.; Zheng, J.Y.; Sun, L.; Yang, H.C.; Cao, Z.Q.; Zhang, X.H.; Zheng, L.T.; Zhen, X.C. The oncometabolite 2-hydroxyglutarate inhibits microglial activation via the AMPK/mTOR/NF-kappaB pathway. Acta Pharmacol. Sin. 2019, 40, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strahle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, W.J.; Deshane, J.S.; Leavenworth, J.W.; Oliva, C.R.; Griguer, C.E. Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 2019, 3, 47–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejera, D.; Kushnirsky, M.; Gultekin, S.H.; Lu, M.; Steelman, L.; de la Fuente, M.I. Ivosidenib, an IDH1 inhibitor, in a patient with recurrent, IDH1-mutant glioblastoma: A case report from a Phase I study. CNS Oncol. 2020, 9, CNS62. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, S.; Golbourn, B.; Huang, X.; Remke, M.; Younger, S.; Cairns, R.A.; Chalil, A.; Smith, C.A.; Krumholtz, S.L.; Mackenzie, D.; et al. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in Glioblastoma. Cancer Res. 2016, 76, 4708–4719. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas-an emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef]
- Couto, M.; Coelho-Santos, V.; Santos, L.; Fontes-Ribeiro, C.; Silva, A.P.; Gomes, C.M.F. The interplay between glioblastoma and microglia cells leads to endothelial cell monolayer dysfunction via the interleukin-6-induced JAK2/STAT3 pathway. J. Cell Physiol. 2019, 234, 19750–19760. [Google Scholar] [CrossRef]
- Daubon, T.; Hemadou, A.; Romero Garmendia, I.; Saleh, M. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 2020, 11, 585616. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [Green Version]
- Lehtipuro, S.; Nykter, M.; Granberg, K.J. Modes of immunosuppression in glioblastoma microenvironment. Oncotarget 2019, 10, 920–921. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Vicario, N.; Rong, L.; Valdes, P.A.; Choi, B.D.; Chen, J.A.; DiToro, D.; Osorio, D.S.; Kachurak, K.; Gessler, F.; et al. A novel in situ multiplex immunofluorescence panel for the assessment of tumor immunopathology and response to virotherapy in pediatric glioblastoma reveals a role for checkpoint protein inhibition. Oncoimmunology 2019, 8, e1678921. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.Y.; Hu, D.X.; Chen, W.Q.; Chen, R.Q.; Qian, S.R.; Li, C.Y.; Li, Y.J.; Xiong, X.X.; Liu, D.; Pan, F.; et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Gyorffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Gregoire, H.; Roncali, L.; Rousseau, A.; Cherel, M.; Delneste, Y.; Jeannin, P.; Hindre, F.; Garcion, E. Targeting Tumor Associated Macrophages to Overcome Conventional Treatment Resistance in Glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Morales, R.T.; Qian, W.; Wang, H.; Gagner, J.P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef]
- Zhu, C.; Chrifi, I.; Mustafa, D.; van der Weiden, M.; Leenen, P.J.M.; Duncker, D.J.; Kros, J.M.; Cheng, C. CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Oncogene 2017, 36, 5356–5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achyut, B.R.; Angara, K.; Jain, M.; Borin, T.F.; Rashid, M.H.; Iskander, A.S.M.; Ara, R.; Kolhe, R.; Howard, S.; Venugopal, N.; et al. Canonical NFkappaB signaling in myeloid cells is required for the glioblastoma growth. Sci. Rep. 2017, 7, 13754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Long, W.; Xing, C.; Chu, J.; Luo, M.; Wang, H.Y.; Liu, Q.; Wang, R.F. Cancer Stem Cells and Immunosuppressive Microenvironment in Glioma. Front. Immunol. 2018, 9, 2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, M.L.; Garcia-Bernal, D.; Martinez, S.; Valdor, R. Autophagy in the Immunosuppressive Perivascular Microenvironment of Glioblastoma. Cancers 2019, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [Green Version]
- Aum, D.J.; Kim, D.H.; Beaumont, T.L.; Leuthardt, E.C.; Dunn, G.P.; Kim, A.H. Molecular and cellular heterogeneity: The hallmark of glioblastoma. Neurosurg. Focus 2014, 37, E11. [Google Scholar] [CrossRef] [Green Version]
- Noroxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef]
- Valtorta, S.; Salvatore, D.; Rainone, P.; Belloli, S.; Bertoli, G.; Moresco, R.M. Molecular and Cellular Complexity of Glioma. Focus on Tumour Microenvironment and the Use of Molecular and Imaging Biomarkers to Overcome Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 5631. [Google Scholar] [CrossRef]
- Walsh, J.J.; Parent, M.; Akif, A.; Adam, L.C.; Maritim, S.; Mishra, S.K.; Khan, M.H.; Coman, D.; Hyder, F. Imaging Hallmarks of the Tumor Microenvironment in Glioblastoma Progression. Front. Oncol. 2021, 11, 692650. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.J.; Chen, W.W.; Zhang, X. Glioblastoma multiforme: Effect of hypoxia and hypoxia inducible factors on therapeutic approaches. Oncol. Lett. 2016, 12, 2283–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercurio, L.; Ajmone-Cat, M.A.; Cecchetti, S.; Ricci, A.; Bozzuto, G.; Molinari, A.; Manni, I.; Pollo, B.; Scala, S.; Carpinelli, G.; et al. Targeting CXCR4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J. Exp. Clin. Cancer Res. 2016, 35, 55. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Maxwell, R.; Xia, Y.; Cardarelli, P.; Oyasu, M.; Belcaid, Z.; Kim, E.; Hung, A.; Luksik, A.S.; Garzon-Muvdi, T.; et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J. Neurooncol. 2019, 143, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: Shaping the phenotype of cancer stem-like cells. Neuro Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torrisi, F.; Alberghina, C.; D’Aprile, S.; Pavone, A.M.; Longhitano, L.; Giallongo, S.; Tibullo, D.; Di Rosa, M.; Zappalà, A.; Cammarata, F.P.; et al. The Hallmarks of Glioblastoma: Heterogeneity, Intercellular Crosstalk and Molecular Signature of Invasiveness and Progression. Biomedicines 2022, 10, 806. https://doi.org/10.3390/biomedicines10040806
Torrisi F, Alberghina C, D’Aprile S, Pavone AM, Longhitano L, Giallongo S, Tibullo D, Di Rosa M, Zappalà A, Cammarata FP, et al. The Hallmarks of Glioblastoma: Heterogeneity, Intercellular Crosstalk and Molecular Signature of Invasiveness and Progression. Biomedicines. 2022; 10(4):806. https://doi.org/10.3390/biomedicines10040806
Chicago/Turabian StyleTorrisi, Filippo, Cristiana Alberghina, Simona D’Aprile, Anna M. Pavone, Lucia Longhitano, Sebastiano Giallongo, Daniele Tibullo, Michelino Di Rosa, Agata Zappalà, Francesco P. Cammarata, and et al. 2022. "The Hallmarks of Glioblastoma: Heterogeneity, Intercellular Crosstalk and Molecular Signature of Invasiveness and Progression" Biomedicines 10, no. 4: 806. https://doi.org/10.3390/biomedicines10040806