
Involvement of Il-33 in the Pathogenesis and Prognosis of Major Respiratory Viral Infections: Future Perspectives for Personalized Therapy

 , , , and
, , , and
Abstract
:1. Introduction
2. IL-33: Synthesis, Receptor Effects and Pathogenetic Role in Respiratory Diseases
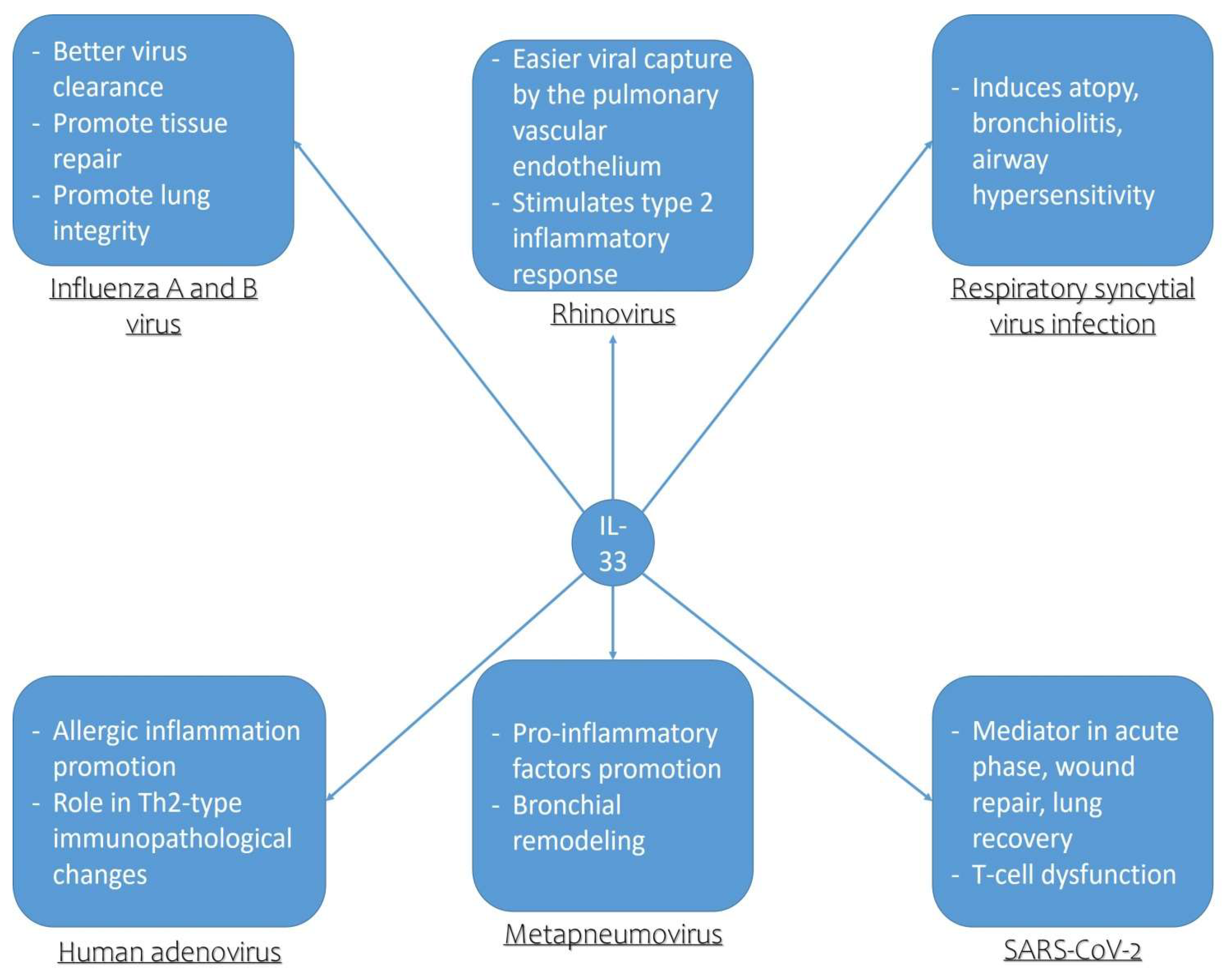
3. Role of IL-33 in Influenza A and B Virus Infection
4. Role of IL-33 in Rhinovirus Infection
5. Role of IL-33 in Respiratory Syncytial Virus Infection
6. Role of IL-33 in Human Adenovirus Infection
7. Role of IL-33 in Human Metapneumovirus Infection
8. Role of IL-33 in SARS-CoV-2 Infection
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allegra, A.; Di Gioacchino, M.; Tonacci, A.; Musolino, C.; Gangemi, S. Immunopathology of SARS-CoV-2 Infection: Immune Cells and Mediators, Prognostic Factors, and Immune-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 4782. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M. Innate and adaptive type 2 immunity in lung allergic inflammation. Immunol. Rev. 2017, 278, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Paladin, F.; Tonacci, A.; Isola, S.; Allegra, A.; Gangemi, S. The Potential Role of Cytokine Storm Pathway in the Clinical Course of Viral Respiratory Pandemic. Biomedicines 2021, 9, 1688. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, F.; Gangemi, S. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanetti, L.; Dijkhuis, A.; Dekker, T.; Sabogal Pineros, Y.S.; Ravi, A.; Dierdorp, B.S.; Erjefalt, J.S.; Mori, M.; Pavlidis, S.; Adcock, I.M.; et al. IL-33 drives influenza-induced asthma exacerbations by halting innate and adaptive antiviral immunity. J. Allergy Clin. Immunol. 2019, 143, 1355–1370.e1316. [Google Scholar] [CrossRef]
- Portugal, C.A.A.; de Araujo Castro, I.; Prates, M.C.M.; Gagliardi, T.B.; Martins, R.B.; de Jesus, B.L.S.; de Souza Cardoso, R.; da Silva, M.V.G.; Aragon, D.C.; Arruda Neto, E.; et al. IL-33 and ST2 as predictors of disease severity in children with viral acute lower respiratory infection. Cytokine 2020, 127, 154965. [Google Scholar] [CrossRef]
- Wu, Y.H.; Lai, A.C.; Chi, P.Y.; Thio, C.L.; Chen, W.Y.; Tsai, C.H.; Lee, Y.L.; Lukacs, N.W.; Chang, Y.J. Pulmonary IL-33 orchestrates innate immune cells to mediate respiratory syncytial virus-evoked airway hyperreactivity and eosinophilia. Allergy 2020, 75, 818–830. [Google Scholar] [CrossRef]
- Zizzo, G.; Cohen, P.L. Imperfect storm: Is interleukin-33 the Achilles heel of COVID-19? Lancet Rheumatol. 2020, 2, e779–e790. [Google Scholar] [CrossRef]
- Zeng, Z.; Hong, X.Y.; Li, Y.; Chen, W.; Ye, G.; Li, Y.; Luo, Y. Serum-soluble ST2 as a novel biomarker reflecting inflammatory status and illness severity in patients with COVID-19. Biomark. Med. 2020, 14, 1619–1629. [Google Scholar] [CrossRef]
- Liang, Y.; Ge, Y.; Sun, J. IL-33 in COVID-19: Friend or foe? Cell. Mol. Immunol. 2021, 18, 1602–1604. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Kotsiou, O.S.; Gourgoulianis, K.I.; Zarogiannis, S.G. IL-33/ST2 Axis in Organ Fibrosis. Front. Immunol. 2018, 9, 2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Li, C.; Wang, W.; Huang, Q.; Wang, J.; Tong, Z.; Huang, K.; Chen, Y.; Yuan, H.; Lv, Z.; et al. IL-33 induced airways inflammation is partially dependent on IL-9. Cell Immunol. 2020, 352, 104098. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, A.; Gangemi, S.; La Grutta, S.; Malizia, V.; Riccobono, L.; Colombo, P.; Cibella, F.; Profita, M. 25-Hydroxyvitamin D, IL-31, and IL-33 in children with allergic disease of the airways. Mediat. Inflamm. 2014, 2014, 520241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjota, M.; Williams, J.; Lu, T.; Clay, B.S.; Byrd, T.; Hrusch, C.L.; Decker, D.C.; De Araujo, C.A.; Bryce, P.J.; Sperling, A.I. IL-33-dependent induction of allergic lung inflammation by FcγRIII signaling. J. Clin. Investig. 2013, 123, 2287–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Goffic, R.; Arshad, M.I.; Rauch, M.; L’Helgoualc’h, A.; Delmas, B.; Piquet-Pellorce, C.; Samson, M. Infection with influenza virus induces IL-33 in murine lungs. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1125–1132. [Google Scholar] [CrossRef]
- Bian, J.R.; Nie, W.; Zang, Y.S.; Fang, Z.; Xiu, Q.Y.; Xu, X.X. Clinical aspects and cytokine response in adults with seasonal influenza infection. Int. J. Clin. Exp. Med. 2014, 7, 5593–5602. [Google Scholar]
- Yamaya, M.; Nomura, K.; Arakawa, K.; Sugawara, M.; Deng, X.; Lusamba Kalonji, N.; Nishimura, H.; Yamada, M.; Nagatomi, R.; Kawase, T. Clarithromycin decreases rhinovirus replication and cytokine production in nasal epithelial cells from subjects with bronchial asthma: Effects on IL-6, IL-8 and IL-33. Arch. Pharmacal Res. 2020, 43, 526–539. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Robinson, K.M.; Ramanan, K.; Clay, M.E.; McHugh, K.J.; Rich, H.E.; Alcorn, J.F. Novel protective mechanism for interleukin-33 at the mucosal barrier during influenza-associated bacterial superinfection. Mucosal Immunol. 2018, 11, 199–208. [Google Scholar] [CrossRef]
- Kim, C.W.; Yoo, H.J.; Park, J.H.; Oh, J.E.; Lee, H.K. Exogenous Interleukin-33 Contributes to Protective Immunity via Cytotoxic T-Cell Priming against Mucosal Influenza Viral Infection. Viruses 2019, 11, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, K.K.W.; Yip, C.C.Y.; Yuen, K.Y. Rhinovirus—From bench to bedside. J. Formos. Med. Assoc. 2017, 116, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.K.; Duan, W.; Doerner, A.M.; Traves, S.L.; Broide, D.H.; Proud, D.; Zuraw, B.L.; Croft, M. Rhinovirus infection interferes with induction of tolerance to aeroantigens through OX40 ligand, thymic stromal lymphopoietin, and IL-33. J. Allergy Clin. Immunol. 2016, 137, 278–288.e276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilvering, B.; Xue, L.; Pavord, I.D. IL-33-dependent Th2 response after rhinovirus infection in asthma: More information needed. Am. J. Respir. Crit. Care Med. 2015, 191, 237. [Google Scholar] [CrossRef]
- Jarjour, N.N.; Esnault, S. Interleukin-33: A potential link between rhinovirus infections and asthma exacerbation. Am. J. Respir. Crit. Care Med. 2014, 190, 1336–1337. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Rajput, C.; Hong, J.Y.; Lei, J.; Hinde, J.L.; Wu, Q.; Bentley, J.K.; Hershenson, M.B. The Innate Cytokines IL-25, IL-33, and TSLP Cooperate in the Induction of Type 2 Innate Lymphoid Cell Expansion and Mucous Metaplasia in Rhinovirus-Infected Immature Mice. J. Immunol. 2017, 199, 1308–1318. [Google Scholar] [CrossRef]
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.; Shamji, B.W.; Trujillo-Torralbo, M.B.; Footitt, J.; Jerico, D.-R.; Telcian, A.G.; Nikonova, A.; Zhu, J.; et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Jurak, L.M.; Xi, Y.; Landgraf, M.; Carroll, M.L.; Murray, L.; Upham, J.W. Interleukin 33 Selectively Augments Rhinovirus-Induced Type 2 Immune Responses in Asthmatic but not Healthy People. Front. Immunol. 2018, 9, 1895. [Google Scholar] [CrossRef]
- Ramu, S.; Calven, J.; Michaeloudes, C.; Menzel, M.; Akbarshahi, H.; Chung, K.F.; Uller, L. TLR3/TAK1 signalling regulates rhinovirus-induced interleukin-33 in bronchial smooth muscle cells. ERJ Open Res. 2020, 6, 147. [Google Scholar] [CrossRef]
- Bloodworth, M.H.; Rusznak, M.; Pfister, C.C.; Zhang, J.; Bastarache, L.; Calvillo, S.A.; Chappell, J.D.; Boyd, K.L.; Toki, S.; Newcomb, D.C.; et al. Glucagon-like peptide 1 receptor signaling attenuates respiratory syncytial virus-induced type 2 responses and immunopathology. J. Allergy Clin. Immunol. 2018, 142, 683–687.e612. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, A.; Gawrysiak, M.; Szewczyk, R.; Gulbas, I.; Likonska, A.; Michlewska, S.; Kowalski, M.L.; Chalubinski, M. IL-33 augments the effect of rhinovirus HRV16 on inflammatory activity of human lung vascular endothelium-possible implications for rhinoviral asthma exacerbations. Allergy 2021, 76, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Calven, J.; Akbarshahi, H.; Menzel, M.; Ayata, C.K.; Idzko, M.; Bjermer, L.; Uller, L. Rhinoviral stimuli, epithelial factors and ATP signalling contribute to bronchial smooth muscle production of IL-33. J. Transl. Med. 2015, 13, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, Y.; Gonzalez, L.; Noguera, L.; Gonzalez, P.A.; Riedel, C.A.; Bertrand, P.; Bueno, S.M. Cytokines in the Respiratory Airway as Biomarkers of Severity and Prognosis for Respiratory Syncytial Virus Infection: An Update. Front. Immunol. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Wu, J.; Liu, J.; Qi, F.; Liu, B. IL-33 Receptor (ST2) Signalling is Important for Regulation of Th2-Mediated Airway Inflammation in a Murine Model of Acute Respiratory Syncytial Virus Infection. Scand. J. Immunol. 2015, 81, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, P.; Lay, M.K.; Piedimonte, G.; Brockmann, P.E.; Palavecino, C.E.; Hernandez, J.; Leon, M.A.; Kalergis, A.M.; Bueno, S.M. Elevated IL-3 and IL-12p40 levels in the lower airway of infants with RSV-induced bronchiolitis correlate with recurrent wheezing. Cytokine 2015, 76, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.D.; Siefker, D.; Jones, T.L.; You, D.; Taylor, R.; DeVincenzo, J.; Cormier, S.A. Elevated Levels of Type 2 Respiratory Innate Lymphoid Cells in Human Infants with Severe Respiratory Syncytial Virus Bronchiolitis. Am. J. Respir. Crit. Care Med. 2019, 200, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Wang, D.; Liu, J.; Zeng, S.; Xu, L.; Hu, H.; Liu, B. Respiratory macrophages and dendritic cells mediate respiratory syncytial virus-induced IL-33 production in TLR3- or TLR7-dependent manner. Int. Immunopharmacol. 2015, 29, 408–415. [Google Scholar] [CrossRef]
- Zhang, L.; Wan, Y.; Ma, L.; Xu, K.; Cheng, B. Inhibition of NF-kappaB/IL-33/ST2 Axis Ameliorates Acute Bronchiolitis Induced by Respiratory Syncytial Virus. J. Immunol. Res. 2021, 2021, 6625551. [Google Scholar] [CrossRef]
- Werder, R.B.; Zhang, V.; Lynch, J.P.; Snape, N.; Upham, J.W.; Spann, K.; Phipps, S. Chronic IL-33 expression predisposes to virus-induced asthma exacerbations by increasing type 2 inflammation and dampening antiviral immunity. J. Allergy Clin. Immunol. 2018, 141, 1607–1619.e1609. [Google Scholar] [CrossRef] [Green Version]
- Hammond, C.; Kurten, M.; Kennedy, J.L. Rhinovirus and asthma: A storied history of incompatibility. Curr. Allergy Asthma Rep. 2015, 15, 502. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.A. Adenovirus infections in solid organ transplant recipients. Curr. Opin. Organ Transplant. 2009, 14, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.P., 3rd; Kajon, A.E. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Punga, T.; Pettersson, U. Adenovirus in the omics era—A multipronged strategy. FEBS Lett. 2020, 594, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Ballegeer, M.; Saelens, X. Cell-Mediated Responses to Human Metapneumovirus Infection. Viruses 2020, 12, 542. [Google Scholar] [CrossRef]
- Kenmoe, S.; Vernet, M.A.; Penlap Beng, V.; Vabret, A.; Njouom, R. Phylogenetic variability of Human Metapneumovirus in patients with acute respiratory infections in Cameroon, 2011–2014. J. Infect. Public Health 2020, 13, 606–612. [Google Scholar] [CrossRef]
- Lay, M.K.; Cespedes, P.F.; Palavecino, C.E.; Leon, M.A.; Diaz, R.A.; Salazar, F.J.; Mendez, G.P.; Bueno, S.M.; Kalergis, A.M. Human metapneumovirus infection activates the TSLP pathway that drives excessive pulmonary inflammation and viral replication in mice. Eur. J. Immunol. 2015, 45, 1680–1695. [Google Scholar] [CrossRef]
- Burke, H.; Freeman, A.; Cellura, D.C.; Stuart, B.L.; Brendish, N.J.; Poole, S.; Borca, F.; Phan, H.T.T.; Sheard, N.; Williams, S.; et al. Inflammatory phenotyping predicts clinical outcome in COVID-19. Respir. Res. 2020, 21, 245. [Google Scholar] [CrossRef]
- Markovic, S.S.; Jovanovic, M.; Gajovic, N.; Jurisevic, M.; Arsenijevic, N.; Jovanovic, M.; Jovanovic, M.; Mijailovic, Z.; Lukic, S.; Zornic, N.; et al. IL 33 Correlates With COVID-19 Severity, Radiographic and Clinical Finding. Front. Med. 2021, 8, 749569. [Google Scholar] [CrossRef]
- Soto, J.A.; Galvez, N.M.S.; Benavente, F.M.; Pizarro-Ortega, M.S.; Lay, M.K.; Riedel, C.; Bueno, S.M.; Gonzalez, P.A.; Kalergis, A.M. Human Metapneumovirus: Mechanisms and Molecular Targets Used by the Virus to Avoid the Immune System. Front. Immunol. 2018, 9, 2466. [Google Scholar] [CrossRef] [Green Version]
- Kayamuro, H.; Yoshioka, Y.; Abe, Y.; Arita, S.; Katayama, K.; Nomura, T.; Yoshikawa, T.; Kubota-Koketsu, R.; Ikuta, K.; Okamoto, S.; et al. Interleukin-1 family cytokines as mucosal vaccine adjuvants for induction of protective immunity against influenza virus. J. Virol. 2010, 84, 12703–12712. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.J.; Dash, P.; Crawford, J.C.; Allen, E.K.; Zamora, A.E.; Boyd, D.F.; Duan, S.; Bajracharya, R.; Awad, W.A.; Apiwattanakul, N.; et al. Lung gammadelta T Cells Mediate Protective Responses during Neonatal Influenza Infection that Are Associated with Type 2 Immunity. Immunity 2018, 49, 531–544.e536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, J.L.; Turner, R.B.; Braciale, T.; Heymann, P.W.; Borish, L. Pathogenesis of rhinovirus infection. Curr. Opin. Virol. 2012, 2, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenes, J.A., Jr.; Srivastava, V.; ReddyVari, H.; Kotnala, S.; Mishra, R.; Farazuddin, M.; Li, W.; Sajjan, U.S. Rhinovirus-induces progression of lung disease in a mouse model of COPD via IL-33/ST2 signaling axis. Clin. Sci. 2019, 133, 983–996. [Google Scholar] [CrossRef]
- Ganesan, S.; Pham, D.; Jing, Y.; Farazuddin, M.; Hudy, M.H.; Unger, B.; Comstock, A.T.; Proud, D.; Lauring, A.S.; Sajjan, U.S. TLR2 Activation Limits Rhinovirus-Stimulated CXCL-10 by Attenuating IRAK-1-Dependent IL-33 Receptor Signaling in Human Bronchial Epithelial Cells. J. Immunol. 2016, 197, 2409–2420. [Google Scholar] [CrossRef] [Green Version]
- Norlander, A.E.; Peebles, R.S., Jr. Innate Type 2 Responses to Respiratory Syncytial Virus Infection. Viruses 2020, 12, 521. [Google Scholar] [CrossRef]
- Garcia-Garcia, M.L.; Calvo, C.; Moreira, A.; Canas, J.A.; Pozo, F.; Sastre, B.; Quevedo, S.; Casas, I.; Del Pozo, V. Thymic stromal lymphopoietin, IL-33, and periostin in hospitalized infants with viral bronchiolitis. Medicine 2017, 96, e6787. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Bai, S.; Wang, D.; Xu, L.; Hu, H.; Zeng, S.; Chai, R.; Liu, B. Macrophages produce IL-33 by activating MAPK signaling pathway during RSV infection. Mol. Immunol. 2017, 87, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, J.; Qi, F.; Zeng, S.; Xu, L.; Hu, H.; Wang, D.; Liu, B. Natural helper cells contribute to pulmonary eosinophilia by producing IL-13 via IL-33/ST2 pathway in a murine model of respiratory syncytial virus infection. Int. Immunopharmacol. 2015, 28, 337–343. [Google Scholar] [CrossRef]
- Saravia, J.; You, D.; Shrestha, B.; Jaligama, S.; Siefker, D.; Lee, G.I.; Harding, J.N.; Jones, T.L.; Rovnaghi, C.; Bagga, B.; et al. Respiratory Syncytial Virus Disease Is Mediated by Age-Variable IL-33. PLoS Pathog. 2015, 11, e1005217. [Google Scholar] [CrossRef] [Green Version]
- Stier, M.T.; Bloodworth, M.H.; Toki, S.; Newcomb, D.C.; Goleniewska, K.; Boyd, K.L.; Quitalig, M.; Hotard, A.L.; Moore, M.L.; Hartert, T.V.; et al. Respiratory syncytial virus infection activates IL-13-producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2016, 138, 814–824.e811. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Chai, R.; Qi, F.; Bai, S.; Cui, Y.; Teng, Y.; Liu, B. Natural helper cells mediate respiratory syncytial virus-induced airway inflammation by producing type 2 cytokines in an IL-33-dependent manner. Immunotherapy 2017, 9, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.P., 3rd; Fishbein, M.; Echavarria, M. Adenovirus. Semin. Respir. Crit. Care Med. 2011, 32, 494–511. [Google Scholar] [CrossRef] [PubMed]
- Wold, W.S.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.J.; Byrnes, A.P. Interaction of adenovirus with antibodies, complement, and coagulation factors. FEBS Lett. 2019, 593, 3449–3460. [Google Scholar] [CrossRef]
- Stav-Noraas, T.E.; Edelmann, R.J.; Poulsen, L.C.; Sundnes, O.; Phung, D.; Kuchler, A.M.; Muller, F.; Kamen, A.A.; Haraldsen, G.; Kaarbo, M.; et al. Endothelial IL-33 Expression Is Augmented by Adenoviral Activation of the DNA Damage Machinery. J. Immunol. 2017, 198, 3318–3325. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Li, X.Y.; Liu, T.; Yuan, B.H.; Zhang, B.B.; Hu, S.L.; Gu, H.B.; Jin, X.B.; Zhu, J.Y. Adenovirus-mediated delivery of soluble ST2 attenuates ovalbumin-induced allergic asthma in mice. Clin. Exp. Immunol. 2012, 170, 1–9. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, Y.; Li, L.; Ye, X.; Wang, J.; Wang, Q.; Li, P.; Li, N.; Zheng, X.; Gao, X.; et al. Immunization with an adenovirus-vectored TB vaccine containing Ag85A-Mtb32 effectively alleviates allergic asthma. J. Mol. Med. 2018, 96, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Uche, I.K.; Guerrero-Plata, A. Interferon-Mediated Response to Human Metapneumovirus Infection. Viruses 2018, 10, 505. [Google Scholar] [CrossRef] [Green Version]
- McMichael, T.M.; Zhang, Y.; Kenney, A.D.; Zhang, L.; Zani, A.; Lu, M.; Chemudupati, M.; Li, J.; Yount, J.S. IFITM3 Restricts Human Metapneumovirus Infection. J. Infect. Dis. 2018, 218, 1582–1591. [Google Scholar] [CrossRef] [Green Version]
- Howard, L.M.; Edwards, K.M.; Zhu, Y.; Grijalva, C.G.; Self, W.H.; Jain, S.; Ampofo, K.; Pavia, A.T.; Arnold, S.R.; McCullers, J.A.; et al. Clinical Features of Human Metapneumovirus-Associated Community-acquired Pneumonia Hospitalizations. Clin. Infect. Dis. 2021, 72, 108–117. [Google Scholar] [CrossRef]
- Chow, J.Y.; Wong, C.K.; Cheung, P.F.; Lam, C.W. Intracellular signaling mechanisms regulating the activation of human eosinophils by the novel Th2 cytokine IL-33: Implications for allergic inflammation. Cell Mol. Immunol. 2010, 7, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munitz, A.; Edry-Botzer, L.; Itan, M.; Tur-Kaspa, R.; Dicker, D.; Marcoviciu, D.; Goren, M.G.; Mor, M.; Lev, S.; Gottesman, T.; et al. Rapid seroconversion and persistent functional IgG antibodies in severe COVID-19 patients correlates with an IL-12p70 and IL-33 signature. Sci. Rep. 2021, 11, 3461. [Google Scholar] [CrossRef] [PubMed]
- Stanczak, M.A.; Sanin, D.E.; Apostolova, P.; Nerz, G.; Lampaki, D.; Hofmann, M.; Steinmann, D.; Krohn-Grimberghe, M.; Thimme, R.; Mittler, G.; et al. IL-33 expression in response to SARS-CoV-2 correlates with seropositivity in COVID-19 convalescent individuals. Nat. Commun. 2021, 12, 2133. [Google Scholar] [CrossRef] [PubMed]
- Cristinziano, L.; Poto, R.; Criscuolo, G.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Loffredo, S.; De Paulis, A.; Marone, G.; Spadaro, G.; et al. IL-33 and Superantigenic Activation of Human Lung Mast Cells Induce the Release of Angiogenic and Lymphangiogenic Factors. Cells 2021, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Humbert, M.; Hanania, N.A.; Zhang, N.; Holgate, S.; Buhl, R.; Bröker, B.M. Staphylococcus aureus and its IgE-inducing enterotoxins in asthma: Current knowledge. Eur. Respir. J. 2020, 55, 1901592. [Google Scholar] [CrossRef] [PubMed]
- Abdurrahman, G.; Schmiedeke, F.; Bachert, C.; Bröker, B.M.; Holtfreter, S. Allergy—A New Role for T Cell Superantigens of Staphylococcus aureus? Toxins 2020, 12, 176. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Nie, J.; Wang, H.; Zhao, Q.; Xiong, Y.; Deng, L.; Song, S.; Ma, Z.; Mo, P.; Zhang, Y. Characteristics of Peripheral Lymphocyte Subset Alteration in COVID-19 Pneumonia. J. Infect. Dis. 2020, 221, 1762–1769. [Google Scholar] [CrossRef] [Green Version]
- Gaurav, R.; Anderson, D.R.; Radio, S.J.; Bailey, K.L.; England, B.R.; Mikuls, T.R.; Thiele, G.M.; Strah, H.M.; Romberger, D.J.; Wyatt, T.A.; et al. IL-33 Depletion in COVID-19 Lungs. Chest 2021, 160, 1656–1659. [Google Scholar] [CrossRef]
- Jeican, I.I.; Gheban, D.; Barbu-Tudoran, L.; Inisca, P.; Albu, C.; Ilies, M.; Albu, S.; Vica, M.L.; Matei, H.V.; Tripon, S.; et al. Respiratory Nasal Mucosa in Chronic Rhinosinusitis with Nasal Polyps versus COVID-19: Histopathology, Electron Microscopy Analysis and Assessing of Tissue Interleukin-33. J. Clin. Med. 2021, 10, 4110. [Google Scholar] [CrossRef]
- Kumar, N.P.; Banurekha, V.V.; Kumar, G.; Nancy, A.; Padmapriyadarsini, C.; Mary, A.S.; Devi, K.R.U.; Murhekar, M.; Babu, S. Prime-Boost Vaccination With Covaxin/BBV152 Induces Heightened Systemic Cytokine and Chemokine Responses. Front. Immunol. 2021, 12, 752397. [Google Scholar] [CrossRef]
- Gabryelska, A.; Kuna, P.; Antczak, A.; Bialasiewicz, P.; Panek, M. IL-33 Mediated Inflammation in Chronic Respiratory Diseases-Understanding the Role of the Member of IL-1 Superfamily. Front. Immunol. 2019, 10, 692. [Google Scholar] [CrossRef] [PubMed]
- Farne, H.A.; Wilson, A.; Powell, C.; Bax, L.; Milan, S.J. Anti-IL5 therapies for asthma. Cochrane Database Syst. Rev. 2017, 9, CD010834. [Google Scholar] [CrossRef]
- Tan, R.; Liew, M.F.; Lim, H.F.; Leung, B.P.; Wong, W.S.F. Promises and challenges of biologics for severe asthma. Biochem. Pharmacol. 2020, 179, 114012. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Pelaia, G.; Longhini, F.; Crimi, C.; Calabrese, C.; Gallelli, L.; Sciacqua, A.; Vatrella, A. Monoclonal Antibodies Targeting Alarmins: A New Perspective for Biological Therapies of Severe Asthma. Biomedicines 2021, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Porsbjerg, C.M.; Sverrild, A.; Lloyd, C.M.; Menzies-Gow, A.N.; Bel, E.H. Anti-alarmins in asthma: Targeting the airway epithelium with next-generation biologics. Eur. Respir. J. 2020, 56, 2000260. [Google Scholar] [CrossRef]
- Warren, K.J.; Poole, J.A.; Sweeter, J.M.; DeVasure, J.M.; Dickinson, J.D.; Peebles, R.S., Jr.; Wyatt, T.A. Neutralization of IL-33 modifies the type 2 and type 3 inflammatory signature of viral induced asthma exacerbation. Respir. Res. 2021, 22, 206. [Google Scholar] [CrossRef]


{kind=link}
| Author | Year | Type of Study | Objective | Outcome |
|---|---|---|---|---|
| Ronan Le Goffic et al. [11] | 2011 | Research study | Evaluation of IL-33 expression and release in lungs of influenza A virus-infected mice in vivo and in murine respiratory epithelial cells | Significant increase in mRNA expression of IL-33 in the virus-infected mice at day 3, compared with non-infected control mice. A significant correlation was found between IL-33 mRNA induction and mRNA levels of TNF-a, IFN-g, and IL-6, but not with IL-1b. The protein expression of IL-33 in virus-infected lungs and in BAL was significantly higher than in controls, especially at day 3. We found a significant increase in IL-33 mRNA expression in IAV-infected transformed murine respiratory epithelial cell line, MLE-15, and human pulmonary epithelial cell line A549, compared with non-infected cells. |
| Jia-Rong Bian et al. [12] | 2014 | Research study | Evaluation of serum levels of pro-inflammatory cytokines in adult patients with seasonal influenza infection | Higher serum concentration of IL-6, IL-33, and TNF-α at admission, compared to controls. Higher levels of IL-33 were found in influenza A-infected patients. |
| Kayamuro H et al. [13] | 2010 | Research study | Evaluation of specific antibody response and specific cellular toxicity in intranasally immunized mice with hemagglutinin and various cytokines | IL-1 family cytokines were able to induce the highest IgG and IgA production. IL-33 and IL-18 were able to elicit both Th1- and Th2-type cytokine responses and high-avidity CD8+ cytotoxic lymphocytes. |
| Xi-zhi J. Guo et al. [14] | 2018 | Research study | Evaluation of γδ T cells and production of IL-33 in mouse models of influenza infection | IL-33 induces a local type-2 immune response with increased accumulation of type-2 innate lymphoid cells and T regulatory cells in the lung, which promotes tissue repair and lung integrity after influenza infection. |
| Monticelli et al. [15] | 2011 | Research study | Evaluation of the role of innate lymphoid cells after influenza virus infection | Innate lymphoid cells, induced by IL-33, are of crucial importance in promoting airway epithelial integrity and lung tissue homeostasis through production of the epidermal growth factor family member amphiregulin. |
| Robinson et al. [16] | 2018 | Research study | Evaluation of the role of IL-33 in mucosal anti-bacterial host defense in influenza infection and bacterial superinfection | Reduction in IL-33 correlates with a negative outcome after influenza infection and bacterial superinfection. Its restoration is correlated with an improvement in bacterial clearance, not related to innate lymphoid cells or type-2 macrophages but to neutrophil recruitment. |
| Chae Won Kim et al. [17] | 2019 | Research study | Evaluation of antiviral protection against influenza virus infection by exogenous IL-33 | Exogenous administration of IL-33 was related to a better outcome in mice infected with influenza IL-33 virus. IL-33 increased the number of innate lymphoid cells, eosinophils, and dendritic cells and CD8+ T-cell activity. |
| García-García ML, Calvo C et al. [18]. | 2017 | Experimental study | Investigate whether infants exhibit enhanced nasal airway secretion of TSLP, IL-33, and periostin during natural respiratory viral bronchiolitis | Bronchiolitis caused by common respiratory viruses is associated with elevated nasal levels of TSLP, IL-33, and periostin, factors known to be important in the development of Th2-response. |
| Mehta AK, Duan W et al. [19] | 2016 | Experimental study | Examine whether rhinovirus infection of the respiratory tract can block airway tolerance by modulating Treg cells | Infection of the respiratory epithelium with rhinovirus can antagonize tolerance to inhaled antigen through combined induction of TSLP, IL-33, and OX40 ligand, and this can lead to susceptibility to asthmatic lung inflammation. |
| Jarjour NN, Esnault S [20] | 2014 | Review | Demonstrate the mechanism of exacerbation in human asthma during rhinovirus infection | IL-33 is likely a major cause of viral-induced asthma exacerbations and a potential therapeutic target in asthma. |
| Jackson DJ, Makrinioti H et al. [21] | 2014 | Experimental study | Assess whether rhinovirus induces a type-2 inflammatory response in asthma in vivo and define a role for IL-33 in this pathway | IL-33 and type-2 cytokines are induced during a rhinovirus-induced asthma exacerbation in vivo. |
| Han M, Rajput C et al. [22] | 2017 | Experimental study | IL-33 and TSLP expression is also induced by RV infection in immature mice and required for maximum ILC2 expansion and mucous metaplasia | The generation of mucous metaplasia in immature, RV-infected mice involves a complex interplay between the innate cytokines IL-25, IL-33, and TSLP. |
| Jurak LM, Xi Y et al. [23] | 2018 | Case-control study | Investigate the effects of IL-33 on rhinovirus (RV)-induced immune responses by circulating leukocytes from people with allergic asthma, and how this response may differ from non-allergic controls | RV infections and IL-33 might interact in asthmatic individuals to exacerbate type-2 immune responses and allergic airway inflammation. |
| Calvén J, Akbarshahi H et al. [24] | 2015 | Experimental study | Investigate effects of epithelial-derived media and viral stimuli on IL-33 expression in human BSMCs | RV infection of BSMCs and activation of TLR3 and RIG-I-like receptors cause expression and production of IL-33. |
| Ramu S, Calvén J et al. [25] | 2020 | Case-control study | Compare levels of RV-induced IL-33 in BSMCs from healthy and asthmatic subjects | RV infection cause higher levels of IL-33 and increased pro-inflammatory and type-2 cytokine release in BSMCs from patients with non-severe asthma. |
| Ganesan S, Pham D et al. [26] | 2016 | Experimental study | Examine the role of TLR2 and IRAK-1 in RV-induced IFN-β, IFN-λ1, and CXCL-10, which require signaling by viral RNA | RV stimulates CXCL-10 expression via the IL-33/ST2 signaling axis, and TLR2 signaling limits RV-induced CXCL-10 via IRAK-1 depletion at least in airway epithelial cells. |
| Gimenes JA Jr, Srivastava V et al. [27] | 2019 | Experimental study | Examine the mechanisms underlying the RV-induced persistent inflammation and progression of emphysema in mice with COPD phenotype | RV may stimulate expression of CXCL-10 and IFN-γ via activation of the ST2/IL-33 signaling axis, which in turn promotes accumulation of CD11b+/CD11c+ macrophages and CD8+ T cells. |
| Gajewski A, Gawrysiak M et al. [28] | 2019 | Experimental study | Analyze the effect of IL-33, the cytokine widely distributed in large amounts in airways of asthmatic individuals, on the HRV-induced inflammatory response in the human lung vascular endothelium. | In asthmatics, IL-33 may facilitate higher viral load in the lung vascular endothelium, while IL-33-orchestrated cytokine milieu may enhance innate inflammatory responses without any concomitant increase in antiviral innate and adaptive mechanisms. |
| Werder RB, Zhang V et al. [29]. | 2018 | Experimental study | Determine whether anti-IL-33 therapy is effective during disease progression, established disease, or viral exacerbation using a preclinical model of chronic asthma and in vitro human primary airway epithelial cells (AECs) | The latter phenotype was replicated in rhinovirus-infected human AECs, suggesting that anti-IL-33 therapy has the additional benefit of enhancing host defense |
| Han Xu et al. [30] | 2017 | Research Study | Evaluation of the role of natural helper cells in influenza virus-induced airway hyper-responsiveness | Blockage of IL-33 reduces natural helper cell recruitment in lungs, thus suggesting IL-33 is necessary for activating Th2-type response. |
| Wu Yi-Hsiu et al. [7] | 2019 | Research study | Evaluation of the individual roles of IL-33-activated innate immune cells, including ILC2s and ST2+ myeloid cells, in RSV infection-triggered pathophysiology. | IL-33 is crucial for the activation of ILC2s and the development of airway hyperreactivity and airway inflammation. IL-33 through lung myeloid cells mediates cellular infiltration but not airway hyperreactivity. |
| Liwen Zhang et al. [31] | 2021 | Research study | Investigation of the role of NF-κB/IL-33/ST2 axis on RSV-induced acute bronchiolitis | IL-33 level was significantly elevated in infants with RSV acute bronchiolitis. The NF-κB/IL-33/ST2 axis is important in the establishment of the Th2 environment after RSV infection. The use of an anti-IL-33 antibody blocks that mechanism, thus suggesting the crucial role of IL-33, especially produced by macrophages. |
| Allison E. Norlander and R. Stokes Peebles, Jr. [32] | 2020 | Review | Review of the impact of L-33, IL-25, thymic stromal lymphopoietin (TSLP), and high mobility group box 1 after RSV infection | ILC2 activation leads to the production of type-2 cytokines and the induction of a type-2 response during RSV infection. |
| Carolina Augusta Arantes Portugal et al. [6] | 2020 | Research study | Assess the role of IL-33-ST2 axis in acute lower respiratory infection by RSV | IL-33 and ST2 in nasopharyngeal aspirates on admission were associated with higher risk for mechanical ventilation. |
| Jing Liu et al. [33] | 2015 | Research study | Evaluation of IL-13-IL-33-ST2 axis and natural helper cells in the development of RSV-induced airway inflammation | RSV infection induces an increase in the number of IL-13-producing natural helper cells in an IL-33-dependent pathway. |
| Jordy Saravia et al. [34] | 2015 | Research Study | Evaluation of the pathogenic mechanisms responsible for RSV-induced immunopathophysiology | Infection with RSV induced rapid IL-33 expression and an increase in ILC2 numbers in the lungs of neonatal mice, in contrast with adult mice. Blocking IL-33 during infection was sufficient to inhibit RSV airway hyperresponsiveness, Th2 inflammation, eosinophilia, and mucus hyperproduction, whereas administration of IL-33 to adult mice during RSV infection was sufficient to induce RSV disease. Elevated IL-33 and IL-13 were observed in nasal aspirates from infants hospitalized with RSV. |
| Feifei Qi et al. [35] | 2015 | Research study | Evaluation of cellular source of IL-33, particularly the types of IL-33-producing cells in innate immune cells during RSV infection | IL-33 plays a key role in RSV-induced airway inflammation. Alveolar macrophages and dendritic cells are a cellular source of IL-33. RSV infection increases expression of IL-33 in pulmonary dendritic cells but not in interstitial macrophages. Macrophages and dendritic cells mediate the production of IL-33 through interaction with TLR3 or TLR7. |
| Feifei Qi et al. [36] | 2017 | Research study | Evaluation of specific signaling pathways for activation of macrophages during RSV infection | RSV infection can promote both the expression of mRNAs for MAPK molecules and the levels of MAPK proteins in lung macrophages. This mechanism may participate in the process of RSV-induced IL-33 secretion by macrophages, demonstrated by an attenuation of IL-33 production when mice were treated with a special MAPK inhibitor before RSV infection. |
| Stier MT et al. [37] | 2016 | Research study | Determination of the capacity of RSV infection to stimulate group 2 innate lymphoid cells (ILC2s) and the associated mechanism in a murine model | RSV-infected IL-33 knock-out mice presented a reduced lung concentration of IL-13, thus highlighting an important role for IL-33 in ILC2 activation. |
| Zeng S. et al. [38] | 2015 | Research study | Evaluation and understanding of the function of IL-33/ST2 signaling pathway during respiratory syncytial virus (RSV) infection | Following intranasal infection with RSV, BALB/c mice showed a marked increase in the production of IL-33, with elevated expression of ST2 mRNA as well as a massive infiltration of CD45+ST2+ cells in the lungs, suggesting that during the early phase of RSV infection, IL-33 target cells, which express ST2 on cell surface, may play a critical role for the development of RSV-induced airway inflammation. Indeed, blocking ST2 signaling using anti-ST2 monoclonal antibody diminished not only RSV-induced eosinophil recruitment, but also the amounts of Th2-associated cytokines, particularly IL-13, and Th17-type cytokine IL-17A in the lungs of infected mice. |
| Bertrand P. et al. [39] | 2015 | Research study | Evaluation of possible mechanisms that connect RSV bronchiolitis to asthma and recurrent wheezing | Patients with family history of atopy presented a high level of IL-33 in nasopharyngeal aspirates. |
| García-García ML et al. [18] | 2017 | Research study | Assessment of the role of thymic stromal lymphopoietin, IL-33, and periostin in viral bronchiolitis | Infants with bronchiolitis had higher levels of TSLP (p = 0.02), IL-33 (p < 0.001) and periostin (p = 0.003) than healthy controls. TSLP and IL-33 were more common in coinfections, mainly RSV and rhinovirus, than in single-infections (p < 0.05). |
| Vu et al. [40] | 2019 | Research study | Investigate the role of mucosal innate immune responses to RSV and respiratory viral load in infants hospitalized with the natural disease | Levels of IL-4, IL-13, IL-33, and IL-1β were significantly higher in nasal aspirates of patients with severe disease compared with those of patients with moderate disease. The authors highlighted the prevalence of type-2 responses to RSV infection in infants and suggested an important role of ILC2 in shaping the immune response early during RSV infection. |
| Stav-Noraas TE et al. [41] | 2017 | Experimental study | Investigate whether endothelial IL-33 expression is augmented by adenoviral activation of the DNA damage machinery | Adenoviral transduction stimulates IL-33 expression in endothelial cells in a way that depends on the DNA-binding protein MRE11 and the antiviral factor IRF1 but not on downstream DNA damage response signaling |
| Zhang Y et al. [42] | 2018 | Experimental study | Evaluate the protective effects of Ad5-gsgAM in an ovalbumin (OVA)-induced asthmatic mouse model | Modulating the IL-33/ST2 axis via adenovirus-vectored mycobacterial antigen vaccination may provide clinical benefits in allergic inflammatory airways disease |
| Yin H et al. [43] | 2012 | Experimental study | Determine whether high levels of local soluble ST2 can ameliorate ovalbumin (OVA)-induced allergic airway inflammation | Single intranasal delivery of Ad-sST2-Fc to OVA-sensitized mice reduces significantly the production of Th2 cytokines, bronchoalveolar lavage eosinophil infiltrates and histopathological changes in the lung. Moreover, the protective effect is related to blocking IL-33/ST2L signaling. |
| Stanczak M.A. [44] | 2021 | Observational study | To analyze the seroprevalence and immune responses in subjects exposed to SARS-CoV-2 |
|
| Liang Y. [10] | 2021 | Original article, in vitro study | To test whether SARS- CoV-2 infection induces IL-33 expression in epithelial cells | IL-33 transcript levels significantly increased in cell lines at 72 h post-infection. |
| Burke H. [45] | 2020 | Observational study | To measure serum IL-6, IL-8, TNF, IL-1β, GM-CSF, IL-10, IL-33 and IFN-γ using a multiplex cytokine assay, in 100 hospitalized patients with COVID-19 | Increased IL-33 levels were associated with adverse outcomes. |
| Munitz A. [46] | 2021 | Original article | To investigate a correlation of IL-33 and IgG seroconversion with disease severity |
|
| Gaurav R. [47] | 2021 | Observational-post mortem | To characterize IL-33 expression in the lungs of patients with fulminant COVID-19, compared with other inflammatory lung diseases |
|
| Jeican I. [48] | 2021 | Original article | To perform a comparative morphological characterization of the respiratory nasal mucosa in CRSwNP versus COVID-19 and tissue IL-33 | The tissue IL-33 concentration in CRSwNP was higher than in COVID-19. |
| Markovic S. [49] | 2021 | Observational study | To analyze the correlation of IL-33 and other innate immunity cytokines with disease severity |
|
| Zeng Z. [9] | 2020 | Original article | To study the role of soluble ST2 in COVID-19 and its relationship with inflammatory status and disease severity |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murdaca, G.; Paladin, F.; Tonacci, A.; Borro, M.; Greco, M.; Gerosa, A.; Isola, S.; Allegra, A.; Gangemi, S. Involvement of Il-33 in the Pathogenesis and Prognosis of Major Respiratory Viral Infections: Future Perspectives for Personalized Therapy. Biomedicines 2022, 10, 715. https://doi.org/10.3390/biomedicines10030715
Murdaca G, Paladin F, Tonacci A, Borro M, Greco M, Gerosa A, Isola S, Allegra A, Gangemi S. Involvement of Il-33 in the Pathogenesis and Prognosis of Major Respiratory Viral Infections: Future Perspectives for Personalized Therapy. Biomedicines. 2022; 10(3):715. https://doi.org/10.3390/biomedicines10030715
Chicago/Turabian StyleMurdaca, Giuseppe, Francesca Paladin, Alessandro Tonacci, Matteo Borro, Monica Greco, Alessandra Gerosa, Stefania Isola, Alessandro Allegra, and Sebastiano Gangemi. 2022. "Involvement of Il-33 in the Pathogenesis and Prognosis of Major Respiratory Viral Infections: Future Perspectives for Personalized Therapy" Biomedicines 10, no. 3: 715. https://doi.org/10.3390/biomedicines10030715