
Synucleins: New Data on Misfolding, Aggregation and Role in Diseases


{kind=link}
{kind=link}
Abstract
:1. Introduction
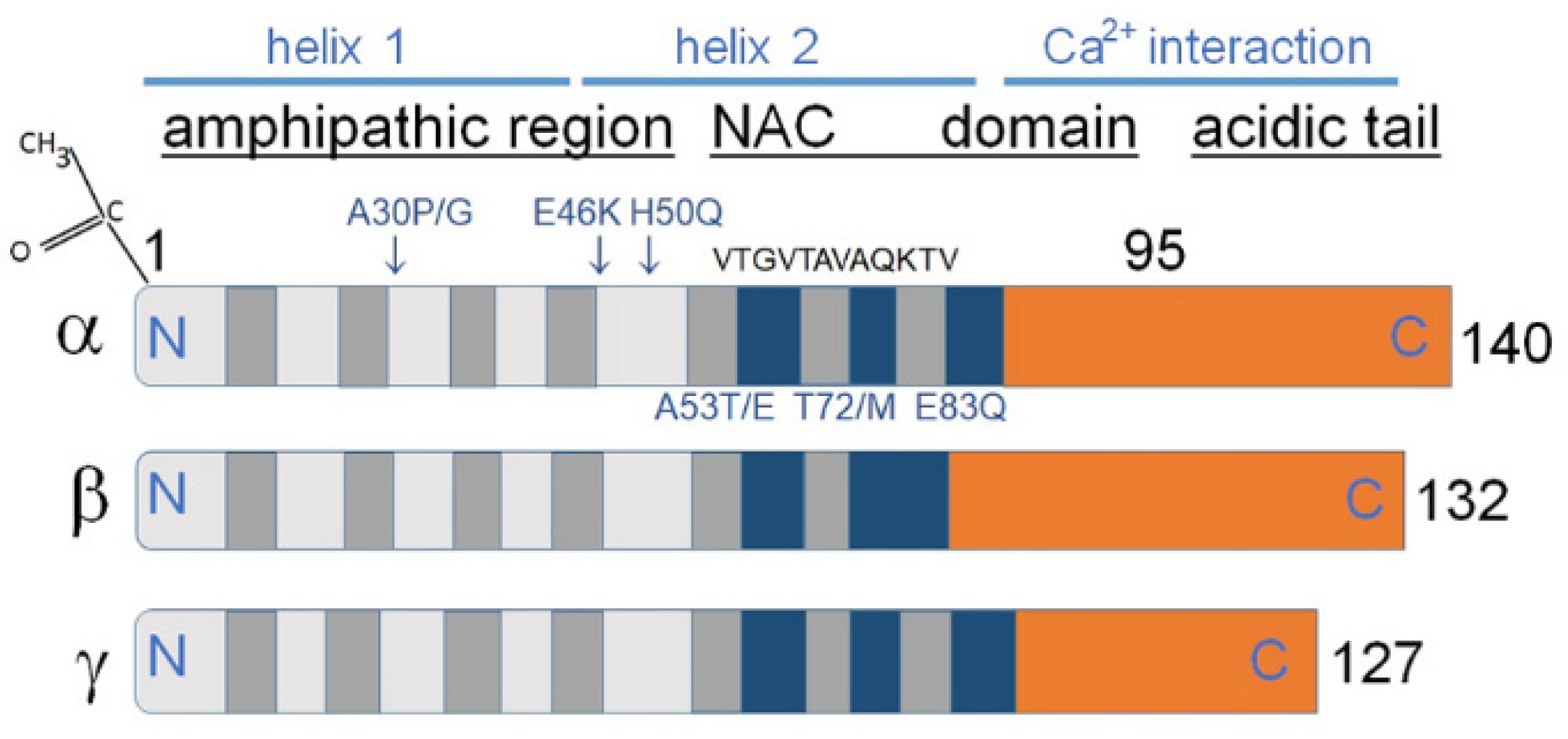
2. Three Members of The Synuclein Family
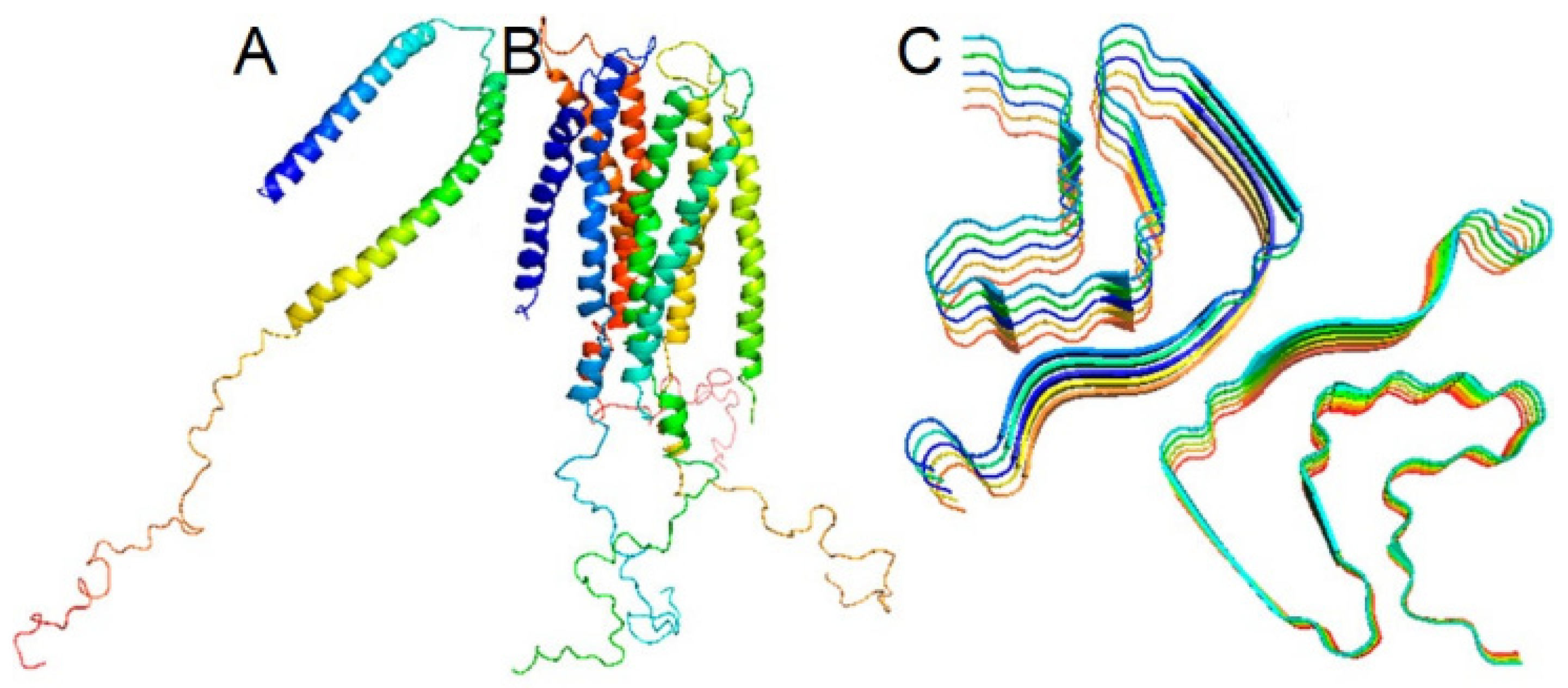
2.1. Common Structure of Members of the Synuclein Family
2.2. Synuclein’s Cellular Functions and Role in Pathology
3. α-Synuclein Misfolding, Aggregation, and Fibrillation
3.1. Post-Translational Modifications (PTMs) of α-Synuclein
3.2. Approaches to Reduce α-Synuclein Toxicity
3.3. The Quaternary Structure of α-Synuclein Fibrils Modulates α-Synuclein Pathology
4. Synuclein-Based Methods of Disease Diagnosis
5. Aggregation of β-Synuclein and γ-Synuclein and Their Role in Diseases
6. Inhibitors of α-Synuclein Aggregation and Fibrillation as Potential Tools for Therapy
7. Conclusions and Future Directions
7.1. Importance of Easily Accessible Samples for Diagnosis
7.2. Role of the Gut-Brain Axis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [Green Version]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Carnazza, K.E.; Komer, L.E.; Xie, Y.X.; Pineda, A.; Briano, J.A.; Gao, V.; Na, Y.; Ramlall, T.; Buchman, V.L.; Eliezer, D.; et al. Synaptic vesicle binding of α-synuclein is modulated by β- and γ-synucleins. Cell Rep. 2022, 39, 110675. [Google Scholar] [CrossRef]
- Yoshida, H.; Craxton, M.; Jakes, R.; Zibaee, S.; Tavaré, R.; Fraser, G.; Serpell, L.C.; Davletov, B.; Crowther, R.A.; Goedert, M. Synuclein proteins of the pufferfish Fugu rubripes: Sequences and functional characterization. Biochemistry 2006, 45, 2599–2607. [Google Scholar] [CrossRef]
- Toni, M.; Cioni, C. Fish Synucleins: An Update. Mar. Drugs 2015, 13, 6665–6686. [Google Scholar] [CrossRef] [PubMed]
- Deiana, A.; Forcelloni, S.; Porrello, A.; Giansanti, A. Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS ONE 2019, 14, e0217889. [Google Scholar] [CrossRef] [Green Version]
- Duperrier, S.; Bortolozzi, A.; Sgambato, V. Increased Expression of Alpha-, Beta-, and Gamma-Synucleins in Brainstem Regions of a Non-Human Primate Model of Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 8586. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-E.; Newman, A.J.; Imberdis, T.; Brontesi, L.; Tripathi, A.; Ramalingam, N.; Fanning, S.; Selkoe, D.; Dettmer, U. Excess membrane binding of monomeric alpha-, beta- and gamma-synuclein is invariably associated with inclusion formation and toxicity. Hum. Mol. Genet. 2021, 30, 2332–2346. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. Synucleinopathies and Tauopathies. In Basic Neurochemistry, 8th ed.; Principles of Molecular, Cellular, and Medical Neurobiology; Scott, T.B., George, J.S., Albers, R.W., Price, D.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; Chapter 47; pp. 829–843. [Google Scholar]
- Bell, R.; Thrush, R.J.; Castellana-Cruz, M.; Oeller, M.; Staats, R.; Nene, A.; Flagmeier, P.; Xu, C.K.; Satapathy, S.; Galvagnion, C.; et al. N-Terminal Acetylation of α-Synuclein Slows down Its Aggregation Process and Alters the Morphology of the Resulting Aggregates. Biochemistry 2022, 61, 1743–1756. [Google Scholar] [CrossRef]
- Bell, R.; Castellana-Cruz, M.; Nene, A.; Thrush, R.J.; Xu, C.K.; Kumita, J.R.; Vendruscolo, M. Effects of N-terminal acetylation on the aggregation of disease-related α-synuclein variants. Mol. Biol. 2022, 10, 167825. [Google Scholar] [CrossRef]
- Newberry, R.W.; Leong, J.T.; Chow, E.D.; Kampmann, M.; DeGrado, W.F. Deep mutational scanning reveals the structural basis for α-synuclein activity. Nat. Chem. Biol. 2020, 16, 653–659. [Google Scholar] [CrossRef]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.-H.; Castillo, P.; Shinsky, N.; García-Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Edwards, R.H. The physiological role of α-synuclein and its relationship to Parkinson’s disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Barba, L.; Abu Rumeileh, S.; Bellomo, G.; Paoletti, F.P.; Halbgebauer, S.; Oeckl, P.; Steinacker, P.; Massa, F.; Gaetani, L.; Parnetti, L.; et al. Cerebrospinal fluid β-synuclein as a synaptic biomarker for preclinical Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2022, jnnp-2022-329124. [Google Scholar] [CrossRef]
- Halbgebauer, S.; Oeckl, P.; Steinacker, P.; Yilmazer-Hanke, D.; Anderl-Straub, S.; von Arnim, C.; Froelich, L.; Gomes, L.A.; Hausner, L.; Huss, A.; et al. Beta-Synuclein in cerebrospinal fluid as an early diagnostic marker of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2021, 92, 349–356. [Google Scholar] [CrossRef]
- Kokhan, V.; Kokhan, T.G.; Samsonova, A.N.; Fisenko, V.P.; Ustyugov, A.; Aliev, G.M. The Dopaminergic Dysfunction and Altered Working Memory Performance of Aging Mice Lacking Gamma-synuclein Gene. CNS Neurol. Disord.-Drug Targets 2018, 17, 604–607. [Google Scholar] [CrossRef]
- Surgucheva, I.; Sharov, V.S.; Surguchov, A. γ-Synuclein: Seeding of α-synuclein aggregation and transmission between cells. Biochemistry. Biochemistry 2012, 51, 4743–4754. [Google Scholar] [CrossRef]
- Ninkina, N.; Millership, S.J.; Peters, O.M.; Connor-Robson, N.; Chaprov, K.; Kopylov, A.T.; Montoya, A.; Kramer, H.; Withers, D.J.; Buchman, V.L. β-synuclein potentiates synaptic vesicle dopamine uptake and rescues dopaminergic neurons from MPTP-induced death in the absence of other synucleins. J. Biol. Chem. 2021, 297, 101375. [Google Scholar] [CrossRef]
- Al-Mazidi, S.; Al-Ayadhi, L.Y. Plasma levels of alpha and gamma synucleins in autism spectrum disorder: An indicator of severity. Med. Princ. Pract. 2021, 30, 160–167. [Google Scholar] [CrossRef]
- Rodríguez-Barrueco, R.; Latorre, J.; Devis-Jáuregui, L.; Lluch, A.; Bonifaci, N.; Llobet, F.J.; Olivan, M.; Coll-Iglesias, L.; Gassner, K.; Davis, M.L.; et al. A microRNA Cluster Controls Fat Cell Differentiation and Adipose Tissue Expansion By Regulating SNCG. Adv. Sci. 2022, 9, e2104759. [Google Scholar] [CrossRef]
- Tofaris, G.K. Initiation and progression of α-synuclein pathology in Parkinson’s disease. Cell. Mol. Life Sci. 2022, 79, 210. [Google Scholar] [CrossRef]
- Tran, C.H.; Saha, R.; Blanco, C.; Bagchi, D.; Chen, I.A. Modulation of α-Synuclein Aggregation In Vitro by a DNA Aptamer. Biochemistry 2022, 61, 1757–1765. [Google Scholar] [CrossRef]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef]
- Korneev, A.; Begun, A.; Liubimov, S.; Kachlishvili, K.; Molochkov, A.; Niemi, A.J.; Maisuradze, G.G. Exploring Structural Flexibility and Stability of α-Synuclein by the Landau-Ginzburg-Wilson Approach. J. Phys. Chem. B 2022, 126, 6878–6890. [Google Scholar] [CrossRef]
- Chiba-Falek, O. Structural variants in SNCA gene and the implication to synucleinopathies. Curr. Opin. Genet. Dev. 2017, 44, 110–116. [Google Scholar] [CrossRef]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [CrossRef] [Green Version]
- Antunes, A.S.L.M. Post-translational Modifications in Parkinson’s Disease. Adv. Exp. Med. Biol. 2022, 1382, 85–94. [Google Scholar] [CrossRef]
- Yoo, H.; Lee, J.; Kim, B.; Moon, H.; Jeong, H.; Lee, K.; Song, W.J.; Hur, J.K.; Oh, J.K.H.Y. Role of post-translational modifications on the alpha-synuclein aggregation-related pathogenesis of Parkinson’s disease. BMB Rep. 2022, 55, 323–335. [Google Scholar] [CrossRef]
- Stephens, A.D.; Zacharopoulou, M.; Moons, R.; Fusco, G.; Seetaloo, N.; Chiki, A.; Woodhams, P.J.; Mela, I.; Lashuel, H.A.; Phillips, J.J.; et al. Extent of N-terminus exposure of monomeric alpha-synuclein determines its aggregation propensity. Nat. Commun. 2020, 11, 2820. [Google Scholar] [CrossRef]
- Zhang, C.; Pei, Y.; Zhang, Z.; Xu, L.; Liu, X.; Jiang, L.; Pielak, G.J.; Zhou, X.; Liu, M.; Li, C. C-terminal truncation modulates α-Synuclein’s cytotoxicity and aggregation by promoting the interactions with membrane and chaperone. Commun. Biol. 2022, 5, 798. [Google Scholar] [CrossRef]
- Kalia, L.V. First trials test targeting of α-synuclein for Parkinson disease. Nat. Rev. Neurol. 2022, 18, 703–704. [Google Scholar] [CrossRef]
- Whone, A. Monoclonal Antibody Therapy in Parkinson’s Disease—The End? N. Engl. J. Med. 2022, 387, 466–467. [Google Scholar] [CrossRef]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Lang, A.E.; Siderowf, A.D.; Macklin, E.A.; Poewe, W.; Brooks, D.J.; Fernandez, H.H.; Rascol, O.; Giladi, N.; Stocchi, F.; Tanner, C.M.; et al. Trial of cinpanemab in early Parkinson’s disease. N. Engl. J. Med. 2022, 387, 408–420. [Google Scholar] [CrossRef]
- Meng, Y.; Pople, C.B.; Huang, Y.; Jones, R.M.; Ottoy, J.; Goubran, M.; Oliveira, L.M.; Davidson, B.; Lawrence, L.S.; Lau, A.Z.; et al. Putaminal Recombinant Glucocerebrosidase Delivery with Magnetic Resonance—Guided Focused Ultrasound in Parkinson’s Disease: A Phase I Study. Mov. Disord. 2022, 37, 2134–2139. [Google Scholar] [CrossRef]
- Shin, S.-M.; Choi, D.-K.; Jung, K.; Bae, J.; Kim, J.-S.; Park, S.-W.; Song, K.-H.; Kim, Y.-S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017, 8, 15090. [Google Scholar] [CrossRef] [Green Version]
- Frieg, B.; Geraets, J.A.; Strohäker, T.; Dienemann, C.; Mavroeidi, P.; Jung, B.C.; Kim, W.S.; Lee, S.-J.; Xilouri, M.; Zweckstetter, M.; et al. Quaternary structure of patient-homogenate amplified α-synuclein fibrils modulates seeding of endogenous α-synuclein. Commun. Biol. 2022, 5, 1040. [Google Scholar] [CrossRef]
- Murray, K.A.; Hu, C.J.; Griner, S.L.; Pan, H.; Bowler, J.T.; Abskharon, R.; Rosenberg, G.M.; Cheng, X.; Seidler, P.M.; Eisenberg, D.S. De novo designed protein inhibitors of amyloid aggregation and seeding. Proc. Natl. Acad. Sci. USA 2022, 119, e2206240119. [Google Scholar] [CrossRef]
- Bagre, G.; Srivastava, T.; Mahasivam, S.; Sinha, M.; Bansal, V.; Ramanathan, R.; Priya, S.; Sharma, S.K. Differential interactions of α-synuclein conformers affect refolding and activity of proteins. J. Biochem. 2022, mvac095. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Ninkina, N.; Peters, O.; Millership, S.; Salem, H.; Van Der Putten, H.; Buchman, V.L. Gamma-synucleinopathy: Neurodegeneration associated with overexpression of the mouse protein. Hum. Mol. Genet. 2009, 18, 1779–1794. [Google Scholar] [CrossRef] [Green Version]
- Surgucheva, I.; Newell, K.L.; Burns, J.; Surguchov, A. New α- and γ-synuclein immunopathological lesions in human brain. Acta Neuropathol. Commun. 2014, 2, 132. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [Green Version]
- Morten, M.J.; Sirvio, L.; Rupawala, H.; Hayes, E.M.; Franco, A.; Radulescu, C.; Ying, L.; Barnes, S.J.; Muga, A.; Yu, Y. Quantitative super-resolution imaging of pathological aggregates reveals distinct toxicity profiles in different synucleinopathies. Proc. Natl. Acad. Sci. USA 2022, 119, e2205591119. [Google Scholar] [CrossRef]
- Sekiya, H.; Tsuji, A.; Hashimoto, Y.; Takata, M.; Koga, S.; Nishida, K.; Futamura, N.; Kawamoto, M.; Kohara, N.; Dickson, D.W.; et al. Discrepancy between distribution of alpha-synuclein oligomers and Lewy-related pathology in Parkinson’s disease. Acta Neuropathol. Commun. 2022, 10, 133. [Google Scholar] [CrossRef]
- Espay, A.J. Movement disorders research in 2021: Cracking the paradigm. Lancet Neurol. 2022, 21, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Rouvière, L.; Giorla, E.; Farrugia, C.; El Waly, B.; Poindron, P.; Callizot, N. Alpha-Synuclein: The Spark That Flames Dopaminergic Neurons, In Vitro and In Vivo Evidence. Int. J. Mol. Sci. 2022, 23, 9864. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Martens, Y.A.; Meneses, A.; Ryu, D.H.; Lu, W.; Raulin, A.C.; Li, F.; Zhao, J.; Chen, Y.; Jin, Y.; et al. LRP1 is a neuronal receptor for α-synuclein uptake and spread. Mol. Neurodegener. 2022, 17, 57. [Google Scholar] [CrossRef]
- Ozdilek, B.; Agirbasli, M. Soluble LRP-1 in Parkinson’s disease: Clues for paradoxical effects. Int. J. Neurosci. 2022, 1–12. [Google Scholar] [CrossRef]
- Iba, M.; McDevitt, R.A.; Kim, C.; Roy, R.; Sarantopoulou, D.; Tommer, E.; Siegars, B.; Sallin, M.; Kwon, S.; Sen, J.M.; et al. Aging exacerbates the brain inflammatory micro-environment contributing to α-synuclein pathology and functional deficits in a mouse model of DLB/PD. Mol. Neurodegener. 2022, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Gracia, P.; Polanco, D.; Tarancón-Díez, J.; Serra, I.; Bracci, M.; Oroz, J.; Laurents, D.V.; García, I.; Cremades, N. Molecular mechanism for the synchronized electrostatic coacervation and co-aggregation of alpha-synuclein and tau. Nat. Commun. 2022, 13, 4586. [Google Scholar] [CrossRef] [PubMed]
- Takada, F.; Kasahara, T.; Otake, K.; Maru, T.; Miwa, M.; Muto, K.; Sasaki, M.; Hirozane, Y.; Yoshikawa, M.; Yamaguchi, J. Identification of α-Synuclein Proaggregator: Rapid Synthesis and Streamlining RT-QuIC Assays in Parkinson’s Disease. ACS Med. Chem. Lett. 2022, 13, 1421–1426. [Google Scholar] [CrossRef]
- Yoo, J.M.; Lin, Y.; Heo, Y.; Lee, Y.-H. Polymorphism in alpha-synuclein oligomers and its implications in toxicity under disease conditions. Front. Mol. Biosci. 2022, 9, 959425. [Google Scholar] [CrossRef]
- Masliah, E.; Hashimoto, M. Development of New Treatments for Parkinson’s Disease in Transgenic Animal Models: A Role for β-Synuclein. NeuroToxicology 2002, 23, 461–468. [Google Scholar] [CrossRef]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Mallory, M.; Masliah, E. beta-Synuclein inhibits alpha-synuclein aggregation: A possible role as an anti-parkinsonian factor. Neuron 2001, 32, 213–223. [Google Scholar] [CrossRef]
- Zhang, F.; Wu, Z.; Long, F.; Tan, J.; Gong, N.; Li, X.; Lin, C. The Roles of ATP13A2 Gene Mutations Leading to Abnormal Aggregation of α-Synuclein in Parkinson’s Disease. Front. Cell. Neurosci. 2022, 16, 927682. [Google Scholar] [CrossRef]
- Bellomo, G.; De Luca, C.M.G.; Paoletti, F.P.; Gaetani, L.; Moda, F.; Parnetti, L. α-Synuclein Seed Amplification Assays for Diagnosing Synucleinopathies: The Way Forward. Neurology 2022, 99, 195–205. [Google Scholar] [CrossRef]
- Majbour, N.; Aasly, J.; Abdi, I.; Ghanem, S.; Erskine, D.; van de Berg, W.; El-Agnaf, O. Disease-Associated α-Synuclein Aggregates as Biomarkers of Parkinson Disease Clinical Stage. Neurology 2022, 99, e2417–e2427. [Google Scholar] [CrossRef]
- Emin, D.; Zhang, Y.P.; Lobanova, E.; Miller, A.; Li, X.; Xia, Z.; Dakin, H.; Sideris, D.I.; Lam, J.Y.L.; Ranasinghe, R.T.; et al. Small soluble α-synuclein aggregates are the toxic species in Parkinson’s disease. Nat. Commun. 2022, 13, 5512. [Google Scholar] [CrossRef]
- Ducas, V.C.; Rhoades, E. Investigation of Intramolecular Dynamics and Conformations of α-, β- and γ-Synuclein. PLoS ONE 2014, 9, e86983. [Google Scholar] [CrossRef] [Green Version]
- Janowska, M.K.; Baum, J. The loss of inhibitory C-terminal conformations in disease associated P123H β-Synuclein. Protein Sci. 2016, 25, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Janowska, M.K.; Wu, K.-P.; Baum, J. Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci. Rep. 2015, 5, 15164. [Google Scholar] [CrossRef] [Green Version]
- Biere, A.L.; Wood, S.J.; Wypych, J.; Steavenson, S.; Jiang, Y.; Anafi, D.; Jacobsen, F.W.; Jarosinski, M.A.; Wu, G.-M.; Louis, J.-C.; et al. Parkinson’s disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J. Biol. Chem. 2000, 275, 34574–34579. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, J.; Carver, J.A. β-Synuclein: An Enigmatic Protein with Diverse Functionality. Biomolecules 2022, 12, 142. [Google Scholar] [CrossRef]
- Ohtake, H.; Limprasert, P.; Fan, Y.; Onodera, O.; Kakita, A.; Takahashi, H.; Bonner, L.T.; Tsuang, D.W.; Murray, I.V.; Lee, V.M.-Y.; et al. Beta-synuclein gene alterations in dementia with Lewy bodies. Neurology 2004, 63, 805–811. [Google Scholar] [CrossRef]
- Wei, J.; Fujita, M.; Nakai, M.; Waragai, M.; Watabe, K.; Akatsu, H.; Rockenstein, E.; Masliah, E.; Hashimoto, M. Enhanced lysosomal pathology caused by β-Synuclein mutants linked to dementia with Lewy bodies. J. Biol. Chem. 2007, 282, 28904–28914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of α-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.K.; Singh, P.; Roy, S.; Bhat, R. Comparative Analysis of the Conformation, Aggregation, Interaction, and Fibril Morphologies of Human α-, β-, and γ-Synuclein Proteins. Biochemistry 2018, 57, 3830–3848. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Millership, S.; Shelkovnikova, T.A.; Soto, I.; Keeling, L.; Hann, A.; Marsh-Armstrong, N.; Buchman, V.L.; Ninkina, N. Selective pattern of motor system damage in gamma-synuclein transgenic mice mirrors the respective pathology in amyotrophic lateral sclerosis. Neurobiol. Dis. 2012, 48, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Peters, O.M.; Shelkovnikova, T.; Highley, J.R.; Cooper-Knock, J.; Hortobágyi, T.; Troakes, C.; Ninkina, N.; Buchman, V.L. Gamma-synuclein pathology in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2015, 2, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.K.; Yang, X.; Atieh, T.B.; Olson, M.P.; Khare, S.D.; Baum, J. Multi-Pronged Interactions Underlie Inhibition of α-Synuclein Aggregation by β-Synuclein. J. Mol. Biol. 2018, 430, 2360–2371. [Google Scholar] [CrossRef]
- Yang, X.; Williams, J.K.; Yan, R.; Mouradian, M.M.; Baum, J. Increased Dynamics of α-Synuclein Fibrils by β-Synuclein Leads to Reduced Seeding and Cytotoxicity. Sci. Rep. 2019, 9, 17579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, J.; Nübling, G.; Giese, A.; Janzen, A.; Oertel, W. Neuroprotektive Therapien bei idiopathischen, genetischen und atypischen Parkinson-Syndromen mit α-Synuklein—Pathologie: Neuroprotective treatment of idiopathic, genetic and atypical Parkinson’s disease with alpha-synuclein-Pathology. Der Nervenarzt 2021, 92, 1249–1259. [Google Scholar] [CrossRef]
- Kurnik, M.; Sahin, C.; Andersen, C.B.; Lorenzen, N.; Giehm, L.; Mohammad-Beigi, H.; Jessen, C.M.; Pedersen, J.S.; Christiansen, G.; Petersen, S.V.; et al. Potent α-Synuclein Aggregation Inhibitors, Identified by High-Throughput Screening, Mainly Target the Monomeric State. Cell Chem. Biol. 2018, 25, 1389–1402.e9. [Google Scholar] [CrossRef]
- Kline, E.M.; Houser, M.C.; Herrick, M.K.; Seibler, P.; Klein, C.; West, A.; Tansey, M.G. Genetic and Environmental Factors in Parkinson’s Disease Converge on Immune Function and Inflammation. Mov. Disord. 2021, 36, 25–36. [Google Scholar] [CrossRef]
- Eteläinen, T.S.; Kilpeläinen, T.P.; Ignatius, A.; Auno, S.; De Lorenzo, F.; Uhari-Väänänen, J.K.; Julku, U.H.; Myöhänen, T.T. Removal of proteinase K resistant αSyn species does not correlate with cell survival in a virus vector-based Parkinson’s disease mouse model. Neuropharmacology 2022, 218, 109213. [Google Scholar] [CrossRef] [PubMed]
- Hmila, I.; Sudhakaran, I.P.; Ghanem, S.S.; Vaikath, N.N.; Poggiolini, I.; Abdesselem, H.; El-Agnaf, O.M.A. Inhibition of α-Synuclein Seeding-Dependent Aggregation by ssDNA Aptamers Specific to C-Terminally Truncated α-Synuclein Fibrils. ACS Chem. Neurosci. 2022, 13, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A.; Bernal, L.; Surguchev, A.A. Phytochemicals as regulators of genes involved in synucleinopathies. Biomolecules 2021, 11, 624. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Quan, Z.; Wong, W.; Guo, J.; Zhang, R.; Yang, Q.; Dai, R.; McGeer, P.L.; Qing, H. Epigallocatechin Gallate (EGCG) Inhibits Alpha-Synuclein Aggregation: A Potential Agent for Parkinson’s Disease. Neurochem. Res. 2016, 41, 2788–2796. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chen, J.; Liu, Y. Curcumin Interacts with α-Synuclein Condensates To Inhibit Amyloid Aggregation under Phase Separation. ACS Omega 2022, 7, 30281–30290. [Google Scholar] [CrossRef]
- Al-Yousef, N.; Shinwari, Z.; Al-Shahrani, B.; Al-Showimi, M.; Al-Moghrabi, N. Curcumin induces re expression of BRCA1 and suppression of γ synuclein by modulating DNA promoter methylation in breast cancer cell lines. Oncol. Rep. 2020, 43, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, S.; Li, Q.; Lang, W.; Li, W.; Jiang, X.; Wan, Z.; Chen, J.; Wang, H. Epigallocatechin-3-gallate: A phytochemical as a promising drug candidate for the treatment of Parkinson’s disease. Front. Pharmacol. 2022, 13, 977521. [Google Scholar] [CrossRef]
- Gezen-Ak, D.; Yurttaş, Z.; Çamoǧlu, T.; Dursun, E. Could Amyloid-β 1–42 or α-Synuclein Interact Directly with Mitochondrial DNA? A Hypothesis. ACS Chem. ACS Chem. Neurosci. 2022, 13, 2803–2812. [Google Scholar] [CrossRef]
- Toba, S.; Jin, M.; Yamada, M.; Kumamoto, K.; Matsumoto, S.; Yasunaga, T.; Fukunaga, Y.; Miyazawa, A.; Fujita, S.; Itoh, K.; et al. Alpha-synuclein facilitates to form short unconventional microtubules that have a unique function in the axonal transport. Sci. Rep. 2017, 7, 16386. [Google Scholar] [CrossRef] [Green Version]
- Hansson, O. Biomarkers for neurodegenerative diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef]
- Surguchov, A. Biomarkers in Parkinson’s Disease. In Neurodegenerative Diseases Biomarkers; Peplow, P.V., Martinez, B., Gennarelli, T.A., Eds.; Humana: New York, NY, USA, 2022; Volume 173, pp. 155–180. [Google Scholar]
- Manne, S.; Kondru, N.; Jin, H.; Serrano, G.E.; Anantharam, V.; Kanthasamy, A.; Adler, C.H.; Beach, T.G.; Kanthasamy, A.G. Blinded RT-QuIC Analysis of α-Synuclein Biomarker in Skin Tissue From Parkinson’s Disease Patients. Mov. Disord. 2020, 35, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sci. 2022, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease—The gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.H.; Lim, S.Y.; Lang, A.E. The microbiome-gut-brain axis in Parkinson disease—From basic research to the clinic. Nat. Rev. Neurol. 2022, 18, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.-L.; Chang, E.E.-S.; Leung, C.-T.; Liu, H.; Malki, Y.; Pang, S.Y.-Y.; Choi, Z.Y.-K.; Liang, Y.; Lai, W.S.; Ruan, Y.; et al. Long-term inhibition of mutant LRRK2 hyper-kinase activity reduced mouse brain α-synuclein oligomers without adverse effects. NPJ Park. Dis. 2022, 8, 115. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surguchov, A.; Surguchev, A. Synucleins: New Data on Misfolding, Aggregation and Role in Diseases. Biomedicines 2022, 10, 3241. https://doi.org/10.3390/biomedicines10123241
Surguchov A, Surguchev A. Synucleins: New Data on Misfolding, Aggregation and Role in Diseases. Biomedicines. 2022; 10(12):3241. https://doi.org/10.3390/biomedicines10123241
Chicago/Turabian StyleSurguchov, Andrei, and Alexei Surguchev. 2022. "Synucleins: New Data on Misfolding, Aggregation and Role in Diseases" Biomedicines 10, no. 12: 3241. https://doi.org/10.3390/biomedicines10123241