
gldc Is Essential for Renal Progenitor Patterning during Kidney Development




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Zebrafish Husbandry
2.2. Morpholino Knockdown
2.3. capped RNA (cRNA) Synthesis and Rescue Studies
2.4. Whole-Mount In Situ Hybridization (WISH)
2.5. Dextran FITC Injections
2.6. Acridine Orange (AO) Assay
2.7. Alcian Blue Stain
2.8. Dextran-Rhodamine Brain Ventricle Injections
2.9. Glycine Treatment
2.10. Image Acquisition and Statistical Analysis
3. Results
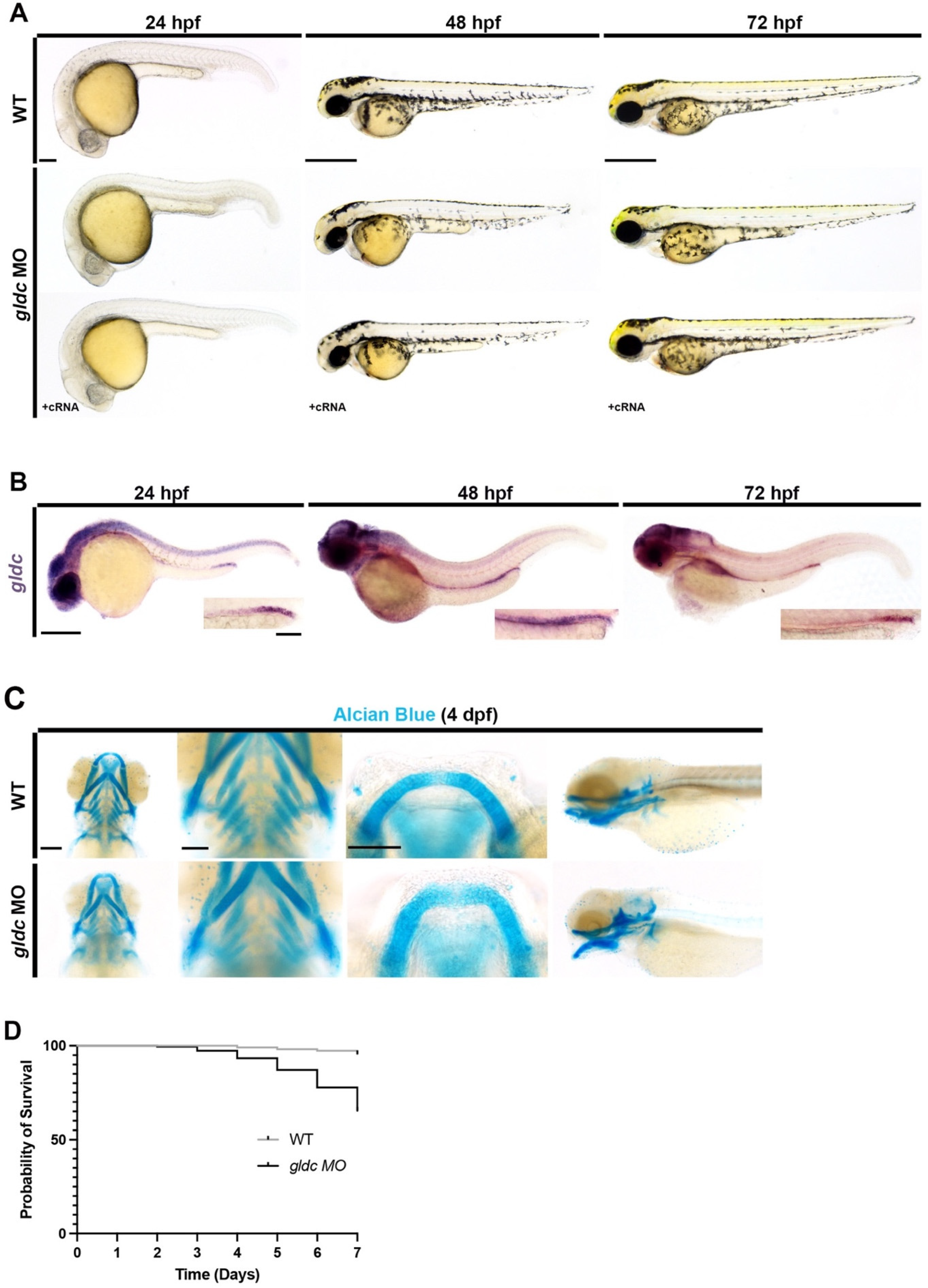
3.1. A Severe gldc Deficient Model Exhibits Phenotypes Consistent with Impaired Kidney Function
3.2. gldc Deficient Zebrafish Display Phenotypes Consistent with Gldc Deficient Mammals
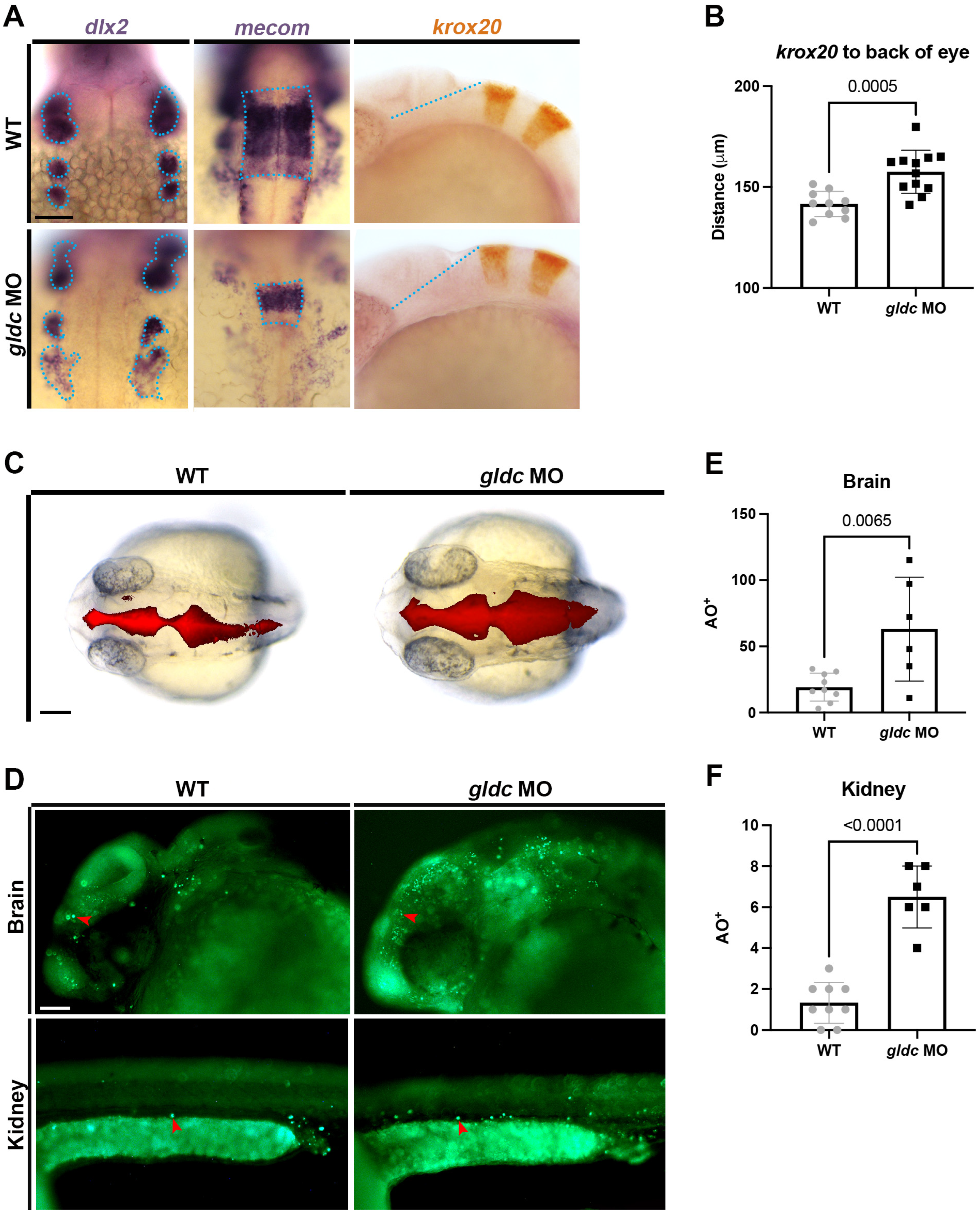
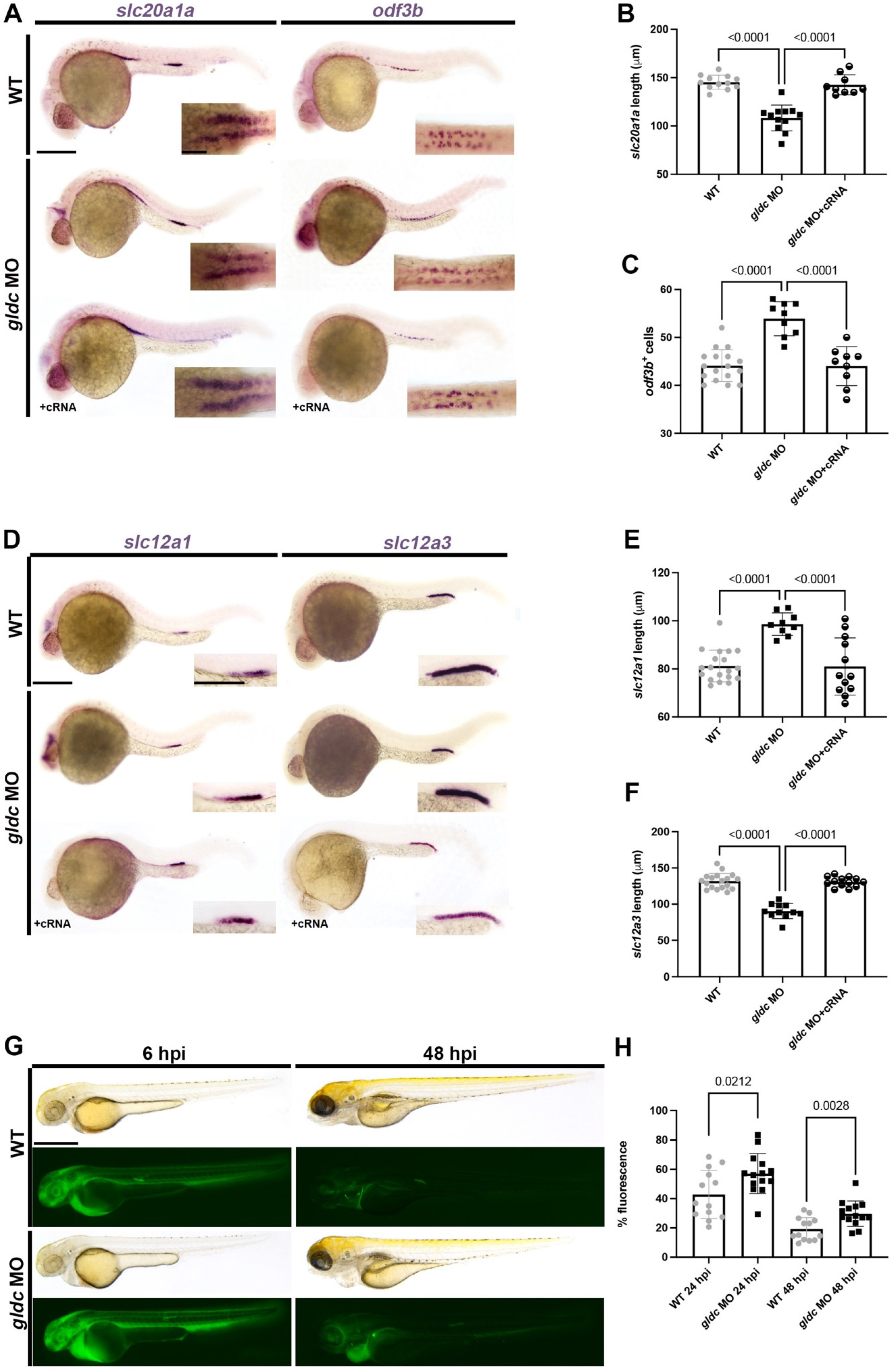
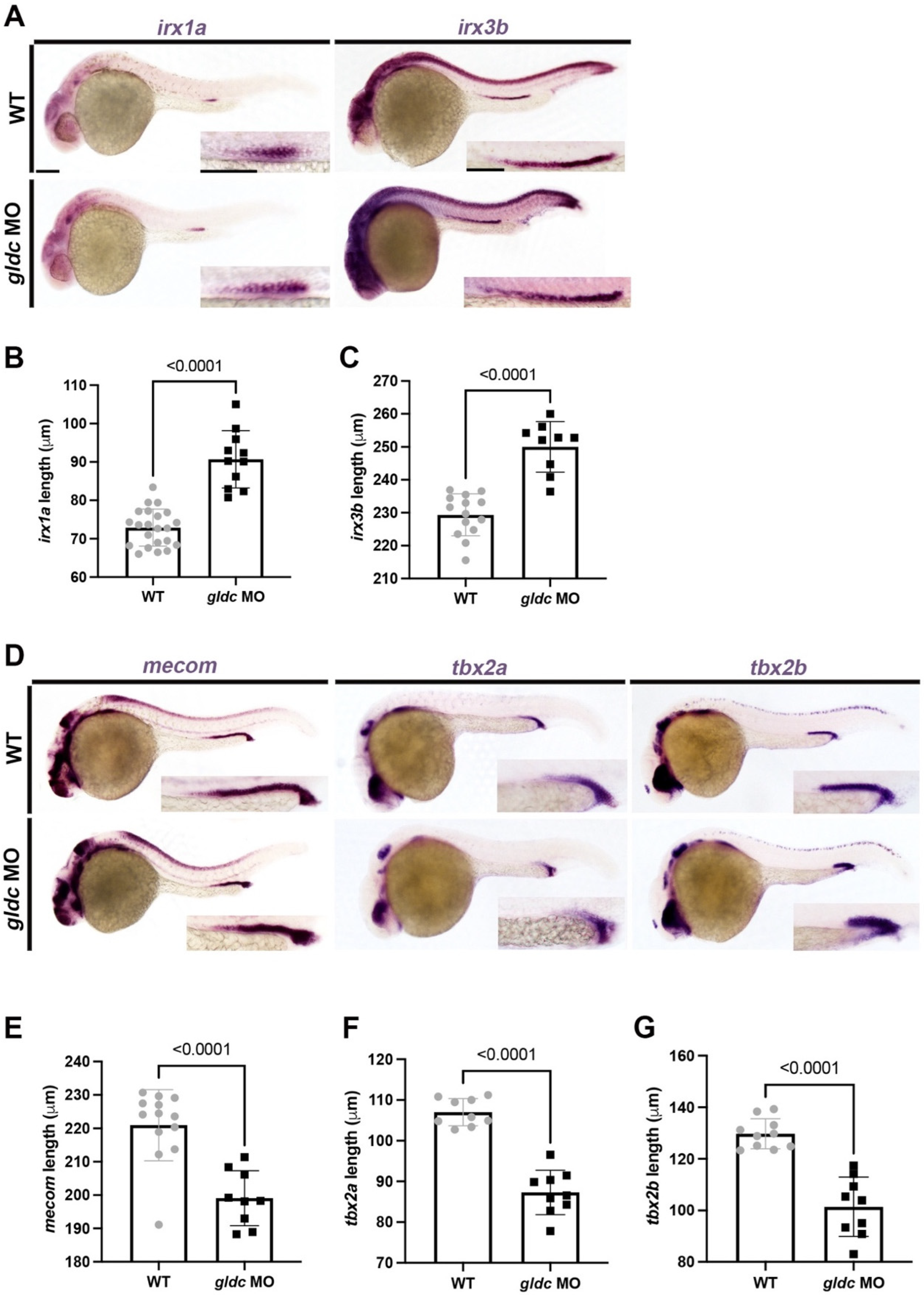
3.3. gldc Is Necessary for Proper Pronephros Segment Patterning
3.4. Loss of gldc Affects DE and DL Precursor Populations
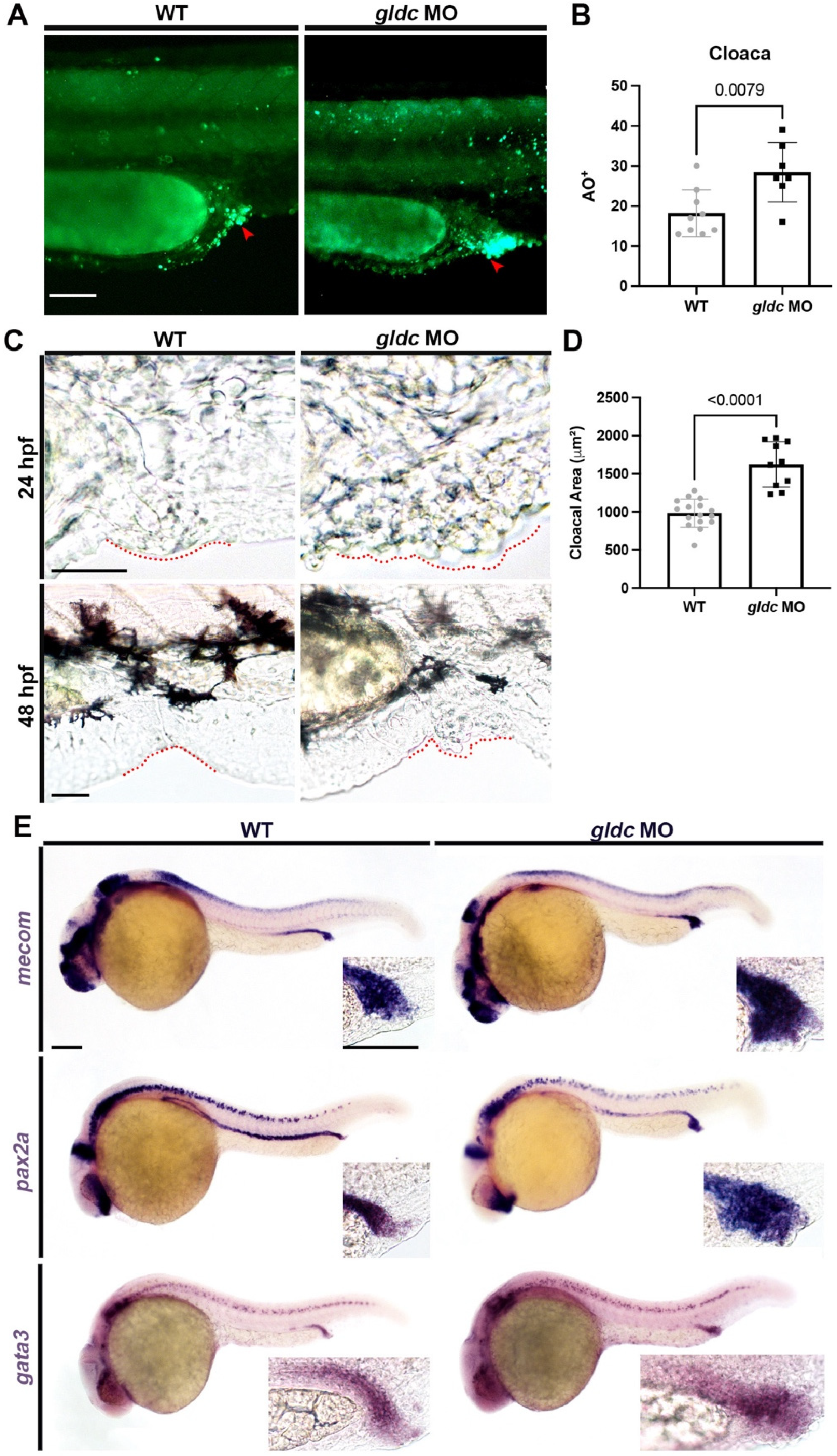
3.5. gldc Morphant Cloacae Exhibit Increased Levels of Apoptosis and Structural Abnormalities
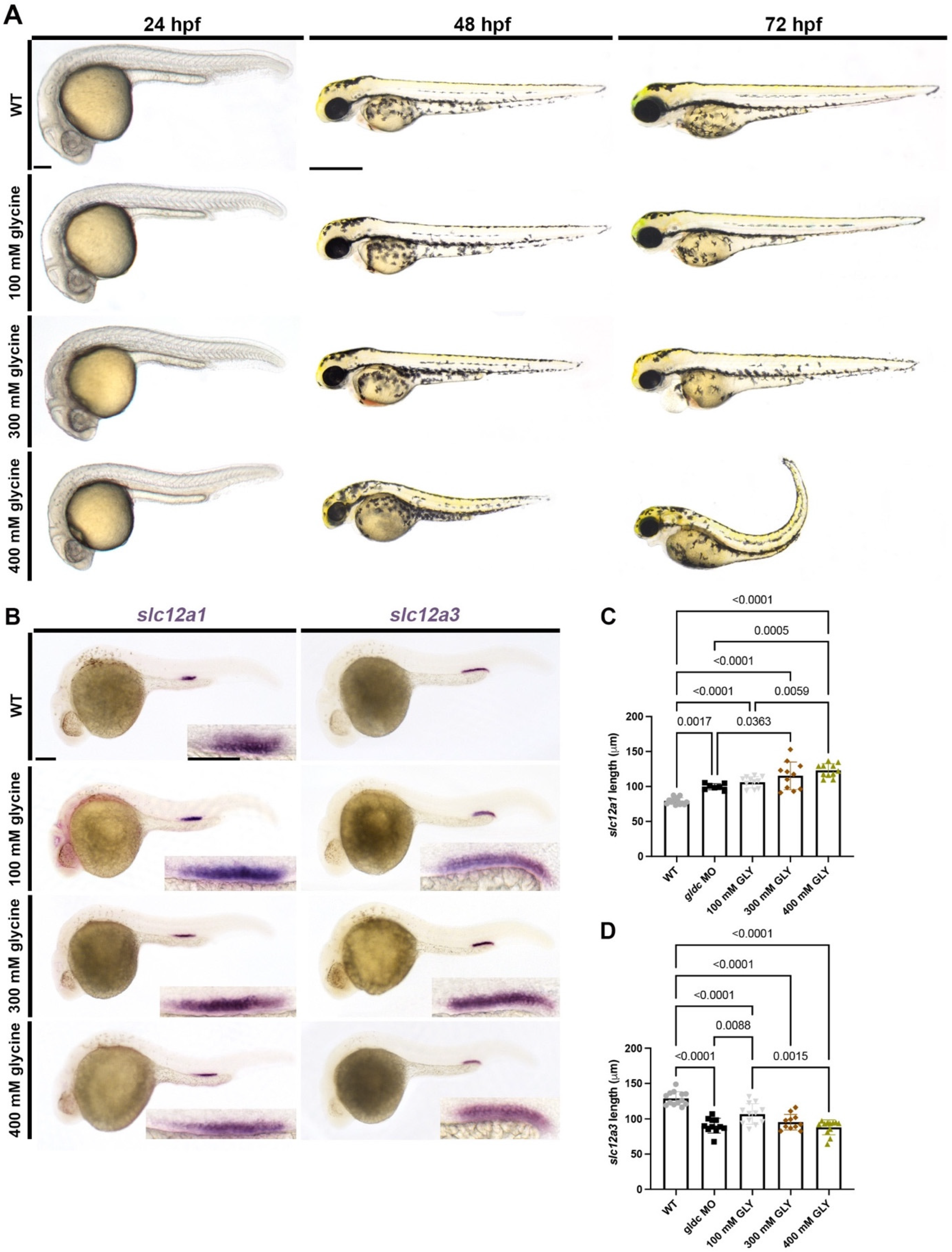
3.6. Exogenous Glycine Treatment Recapitulates the Live Morphology and Segment Patterning of gldc Deficient Embryos in a Dose-Dependent Manner
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coughlin, C.R.; Swanson, M.A.; Kronquist, K.; Acquaviva, C.; Hutchin, T.; Rodríguez-Pombo, P.; Väisänen, M.-L.; Spector, E.; Creadon-Swindell, G.; Brás-Goldberg, A.M.; et al. The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT. Anesthesia Analg. 2017, 19, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Johnston, M.V. Nonketotic hyperglycinemia. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw Hill: New York, NY, USA, 2019. [Google Scholar]
- van Hove, J.L.K.; Coughlin, C., II; Swanson, M.; Hennermann, J.B.; Adam, M.P.; Everman, D.B.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; et al. (Eds.) Nonketotic Hyperglycinemia. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Toone, J.R.; Applegarth, D.A.; Coulter-Mackie, M.; James, E.R. Recurrent Mutations in P- and T-Proteins of the Glycine Cleavage Complex and a Novel T-Protein Mutation (N145I): A Strategy for the Molecular Investigation of Patients with Nonketotic Hyperglycinemia (NKH). Mol. Genet. Metab. 2001, 72, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Chuchra, P.; Paprocka, J. Nonketotic Hyperglycinemia: Insight into Current Therapies. J. Clin. Med. 2022, 11, 3027. [Google Scholar] [CrossRef] [PubMed]
- Farris, J.; Calhoun, B.; Alam, S.; Lee, S.; Haldar, K. Large scale analyses of genotype-phenotype relationships of glycine decarboxylase mutations and neurological disease severity. PLoS Comput. Biol. 2020, 16, e1007871. [Google Scholar] [CrossRef]
- Swanson, M.A.; Coughlin, C.R.; Scharer, G.H.; Szerlong, H.J.; Bjoraker, K.J.; Spector, E.B.; Creadon-Swindell, G.; Mahieu, V.; Matthijs, G.; Hennermann, J.B.; et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann. Neurol. 2015, 78, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Hübschmann, O.K.; Juliá-Palacios, N.A.; Olivella, M.; Guder, P.; Zafeiriou, D.I.; Horvath, G.; Kulhánek, J.; Pearson, T.S.; Kuster, A.; Cortès-Saladelafont, E.; et al. Integrative Approach to Predict Severity in Nonketotic Hyperglycinemia. Ann. Neurol. 2022, 92, 292–303. [Google Scholar] [CrossRef]
- Pai, Y.J.; Leung, K.-Y.; Savery, D.; Hutchin, T.; Prunty, H.; Heales, S.; Brosnan, M.E.; Brosnan, J.T.; Copp, A.; Greene, N.D. Glycine decarboxylase deficiency causes neural tube defects and features of non-ketotic hyperglycinemia in mice. Nat. Commun. 2015, 6, 6388. [Google Scholar] [CrossRef] [Green Version]
- Mademont-Soler, I.; Casellas-Vidal, D.; Trujillo, A.; Espuña-Capote, N.; Maroto, A.; García-González, M.D.M.; Ruiz, M.D.; Diego-Álvarez, D.; Queralt, X.; Perapoch, J.; et al. GLYT1 encephalopathy: Further delineation of disease phenotype and discussion of pathophysiological mechanisms. Am. J. Med. Genet. Part A 2021, 185, 476–485. [Google Scholar] [CrossRef]
- Press, G.A.; Barshop, B.A.; Haas, R.H.; Nyhan, W.L.; Glass, R.F.; Hesselink, J.R. Abnormalities of the brain in nonketotic hyperglycinemia: MR manifestations. AJNR Am. J. Neuroradiol. 1989, 10, 315–321. [Google Scholar]
- Santos, C.; Pai, Y.J.; Mahmood, M.R.; Leung, K.-Y.; Savery, D.; Waddington, S.N.; Copp, A.J.; Greene, N.D. Impaired folate 1-carbon metabolism causes formate-preventable hydrocephalus in glycine decarboxylase–deficient mice. J. Clin. Investig. 2020, 130, 1446–1452. [Google Scholar] [CrossRef]
- Farris, J.; Alam, S.; Rajashekara, A.M.; Haldar, K. Genomic analyses of glycine decarboxylase neurogenic mutations yield a large-scale prediction model for prenatal disease. PLoS Genet. 2021, 17, e1009307. [Google Scholar] [CrossRef] [PubMed]
- Molinari, E.; Sayer, J.A. Disease Modeling To Understand the Pathomechanisms of Human Genetic Kidney Disorders. Clin. J. Am. Soc. Nephrol. 2020, 15, 855–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlach, G.F.; Wingert, R.A. Kidney organogenesis in the zebrafish: Insights into vertebrate nephrogenesis and regeneration. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 559–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poureetezadi, S.J.; Wingert, R.A. Little fish, big catch: Zebrafish as a model for kidney disease. Kidney Int. 2016, 89, 1204–1210. [Google Scholar] [CrossRef] [Green Version]
- Desgrange, A.; Cereghini, S. Nephron Patterning: Lessons from Xenopus, Zebrafish, and Mouse Studies. Cells 2015, 4, 483–499. [Google Scholar] [CrossRef] [Green Version]
- Drummond, I.A.; Majumdar, A.; Hentschel, H.; Elger, M.; Solnica-Krezel, L.; Schier, A.F.; Neuhauss, S.C.; Stemple, D.L.; Zwartkruis, F.; Rangini, Z.; et al. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development 1998, 125, 4655–4667. [Google Scholar] [CrossRef] [PubMed]
- Wingert, R.A.; Selleck, R.; Yu, J.; Song, H.-D.; Chen, Z.; Song, A.; Zhou, Y.; Thisse, B.; Thisse, C.; McMahon, A.P.; et al. The cdx Genes and Retinoic Acid Control the Positioning and Segmentation of the Zebrafish Pronephros. PLoS Genet. 2007, 3, 1922–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingert, R.A.; Davidson, A.J. The zebrafish pronephros: A model to study nephron segmentation. Kidney Int. 2008, 73, 1120–1127. [Google Scholar] [CrossRef] [Green Version]
- Wingert, R.A.; Davidson, A.J. Zebrafish nephrogenesis involves dynamic spatiotemporal expression changes in renal progenitors and essential signals from retinoic acid and irx3b. Dev. Dyn. 2011, 240, 2011–2027. [Google Scholar] [CrossRef] [Green Version]
- Fatma, S.; Nayak, U.; Swain, R.K. Methods to generate and evaluate zebrafish models of human kidney diseases. Int. J. Dev. Biol. 2021, 65, 475–485. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.N.; Li, Y.; Marra, A.N.; Verdun, V.; Wingert, R.A. Flat Mount Preparation for Observation and Analysis of Zebrafish Embryo Specimens Stained by Whole Mount In situ Hybridization. J. Vis. Exp. 2014, 89, e51604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, A.N.; Ulrich, M.; White, A.; Springer, M.; Wingert, R.A. Visualizing Multiciliated Cells in the Zebrafish Through a Combined Protocol of Whole Mount Fluorescent In Situ Hybridization and Immunofluorescence. J. Vis. Exp. 2017, 129, e56261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, A.N.; Chambers, B.E.; Chambers, J.M.; Drummond, B.E.; Adeeb, B.D.; Wesselman, H.M.; Morales, E.E.; Handa, N.; Pettini, T.; Ronshaugen, M.; et al. Visualizing gene expression during zebrafish pronephros development and regeneration. Methods Cell. Biol. 2019, 154, 183–215. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, K.K.; Springer, K.N.; Wingert, R.A. Analysis of nephron composition and function in the adult zebrafish kidney. J. Vis. Exp. 2014, 90, e51644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroeger, P.T.; Drummond, B.E.; Miceli, R.; McKernan, M.; Gerlach, G.F.; Marra, A.N.; Fox, A.; McCampbell, K.K.; Leshchiner, I.; Rodriguez-Mari, A.; et al. The zebrafish kidney mutant zeppelin reveals that brca2/fancd1 is essential for pronephros development. Dev. Biol. 2017, 428, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Lardelli, M. A Rapid Apoptosis Assay Measuring Relative Acridine Orange Fluorescence in Zebrafish Embryos. Zebrafish 2007, 4, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Neuhauss, S.C.F.; Solnica-Krezel, L.; Schier, A.F.; Zwartkruis, F.; Stemple, D.L.; Malicki, J.; Driever, W. Mutations affecting craniofacial development in zebrafish. Development 1996, 123, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Sive, H. Initial formation of zebrafish brain ventricles occurs independently of circulation and requires the nagie oko and snakehead/atp1a1a.1 gene products. Development 2005, 132, 2057–2067. [Google Scholar] [CrossRef] [Green Version]
- Poureetezadi, S.J.; Donahue, E.K.; Wingert, R.A. A manual small molecule screen approaching high-throughput using zebrafish embryos. J. Vis. Exp. 2014, 93, e52063. [Google Scholar] [CrossRef] [Green Version]
- Poureetezadi, S.J.; Cheng, C.N.; Chambers, J.M.; Drummond, B.E.; Wingert, R.A. Prostaglandin signaling regulates nephron segment patterning of renal progenitors during zebrafish kidney development. eLife 2016, 5, e17551. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.M.; Poureetezadi, S.J.; Addiego, A.; Lahne, M.; A Wingert, R. ppargc1a controls nephron segmentation during zebrafish embryonic kidney ontogeny. eLife 2018, 7, e40266. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.N.; Adeeb, B.D.; Chambers, B.E.; Drummond, B.E.; Ulrich, M.; Addiego, A.; Springer, M.; Poureetezadi, S.J.; Chambers, J.M.; Ronshaugen, M.; et al. Prostaglandin signaling regulates renal multiciliated cell specification and maturation. Proc. Natl. Acad. Sci. USA 2019, 116, 8409–8418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji-Tamura, K.; Sato, M.; Fujita, M.; Tamura, M. Glycine exerts dose-dependent biphasic effects on vascular development of zebrafish embryos. Biochem. Biophys. Res. Commun. 2020, 527, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Riché, R.; Liao, M.; Pena, I.A.; Leung, K.-Y.; Lepage, N.; Greene, N.D.; Sarafoglou, K.; Schimmenti, L.A.; Drapeau, P.; Samarut, É. Glycine decarboxylase deficiency–induced motor dysfunction in zebrafish is rescued by counterbalancing glycine synaptic level. J. Clin. Investig. 2018, 3, e124642. [Google Scholar] [CrossRef] [Green Version]
- Applegarth, D.A.; Toone, J.R. Glycine encephalopathy (nonketotic hyperglycinaemia): Review and update. J. Inherit. Metab. Dis. 2004, 27, 417–422. [Google Scholar] [CrossRef]
- Narisawa, A.; Komatsuzaki, S.; Kikuchi, A.; Niihori, T.; Aoki, Y.; Fujiwara, K.; Tanemura, M.; Hata, A.; Suzuki, Y.; Relton, C.L.; et al. Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum. Mol. Genet. 2012, 21, 1496–1503. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.N.; Wingert, R.A. Nephron proximal tubule patterning and corpuscles of Stannius formation are regulated by the sim1a transcription factor and retinoic acid in zebrafish. Dev. Biol. 2015, 399, 100–116. [Google Scholar] [CrossRef] [Green Version]
- Marra, A.N.; Wingert, R.A. Epithelial cell fate in the nephron tubule is mediated by the ETS transcription factors etv5a and etv4 during zebrafish kidney development. Dev. Biol. 2016, 411, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Hanke, N.; Staggs, L.; Schroder, P.; Litteral, J.; Fleig, S.; Kaufeld, J.; Pauli, C.; Haller, H.; Schiffer, M. “Zebrafishing” for novel genes relevant to the glomerular filtration barrier. BioMed. Res. Int. 2013, 2013, 658270. [Google Scholar] [CrossRef] [Green Version]
- Visel, A.; Thaller, C.; Eichele, G. GenePaint.org: An atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004, 32, 552–556. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P.; Aronow, B.J.; Davidson, D.R.; Davies, J.; Gaido, K.W.; Grimmond, S.; Lessard, J.L.; Little, M.; Potter, S.S.; Wilder, E.L.; et al. GUDMAP: The Genitourinary Developmental Molecular Anatomy Project. J. Am. Soc. Nephrol. 2008, 19, 667–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, S.D.; Armit, C.; Armstrong, J.; Brennan, J.; Cheng, Y.; Haggarty, B.; Houghton, D.; Lloyd-MacGilp, S.; Pi, X.; Roochun, Y.; et al. The GUDMAP database—An online resource for genitourinary research. Development 2011, 138, 2845–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayrak, H.; Yıldız, Y.; Olgaç, A.; Kasapkara, Ç.S.; Küçükcongar, A.; Zenciroğlu, A.; Yüksel, D.; Ceylaner, S.; Kılıç, M. Genotypic and phenotypic features in Turkish patients with classic nonketotic hyperglycinemia. Metab. Brain Dis. 2021, 36, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Alfallaj, R.; Alfadhel, M. Glycine Transporter 1 Encephalopathy From Biochemical Pathway to Clinical Disease: Review. Child Neurol. Open 2019, 6, 2329048X19831486. [Google Scholar] [CrossRef] [Green Version]
- Méndez, K.; Vega-López, G.A.; Aybar, M.; Velasco, I. Neurogenesis From Neural Crest Cells: Molecular Mechanisms in the Formation of Cranial Nerves and Ganglia. Front. Cell Dev. Biol. 2020, 8, 635. [Google Scholar] [CrossRef]
- Li, Y.; Cheng, C.N.; Verdun, V.A.; Wingert, R.A. Zebrafish nephrogenesis is regulated by interactions between retinoic acid, mecom, and Notch signaling. Dev. Biol. 2014, 386, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Naylor, R.W.; Przepiorski, A.; Ren, Q. HNF1B is essential for nephron segmentation during nephrogenesis. J. Am. Soc. Nephrol. 2013, 24, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, G.F.; Wingert, R.A. Zebrafish pronephros tubulogenesis and epithelial identity maintenance are reliant on the polarity proteins Prkc iota and zeta. Dev. Biol. 2014, 396, 183–200. [Google Scholar] [CrossRef] [Green Version]
- McKee, R.; Gerlach, G.F.; Jou, J.; Cheng, C.N.; Wingert, R.A. Temporal and spatial expression of tight junction genes during zebrafish pronephros development. Gene Expr. Patterns 2014, 16, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Jiang, Y.-J. Jagged2a-Notch Signaling Mediates Cell Fate Choice in the Zebrafish Pronephric Duct. PLoS Genet. 2007, 3, e18. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pathak, N.; Kramer-Zucker, A.; Drummond, I.A. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development 2007, 134, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, A.N.; Li, Y.; Wingert, R.A. Antennas of organ morphogenesis: The roles of cilia in vertebrate kidney development. Genesis 2016, 54, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Boutin, C.; Kodjabachian, L. Biology of multiciliated cells. Curr. Opin. Genet. Dev. 2019, 56, 1–7. [Google Scholar] [CrossRef]
- Marra, A.N.; Cheng, C.N.; Adeeb, B.; Addiego, A.; Wesselman, H.M.; Chambers, B.E.; Chambers, J.M.; Wingert, R.A. Iroquois transcription factor irx2a is required for multiciliated and transporter cell fate decisions during zebrafish pronephros development. Sci. Rep. 2019, 9, 6454. [Google Scholar] [CrossRef] [Green Version]
- Hentschel, D.M.; Park, K.M.; Cilenti, L.; Zervos, A.S.; Drummond, I.; Bonventre, J.V. Acute renal failure in zebrafish: A novel system to study a complex disease. Am. J. Physiol. Physiol. 2005, 288, F923–F929. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.S.; Holzemer, N.F.; Wingert, R.A. Laser Ablation of the Zebrafish Pronephros to Study Renal Epithelial Regeneration. J. Vis. Exp. 2011, 54, e2845. [Google Scholar] [CrossRef] [Green Version]
- Bolten, J.S.; Pratsinis, A.; Alter, C.L.; Fricker, G.; Huwyler, J. Zebrafish (Danio rerio) larva as an in vivo vertebrate model to study renal function. Am. J. Physiol. Physiol. 2022, 322, F280–F294. [Google Scholar] [CrossRef]
- Reggiani, L.; Raciti, D.; Airik, R.; Kispert, A.; Brändli, A.W. The prepattern transcription factor Irx3 directs nephron segment identity. Genes Dev. 2007, 21, 2358–2370. [Google Scholar] [CrossRef] [Green Version]
- Chambers, B.E.; Gerlach, G.F.; Clark, E.G.; Chen, K.H.; Levesque, A.E.; Leshchiner, I.; Goessling, W.; Wingert, R.A. Tfap2a is a novel gatekeeper of nephron differentiation during kidney development. Development 2019, 146, dev172387. [Google Scholar] [CrossRef] [Green Version]
- Morishita, K.; Parganas, E.; Parham, D.M.; Matsugi, T.; Ihle, J.N. The Evi-1 zinc finger myeloid transforming gene is normally expressed in the kidney and in developing oocytes. Oncogene 1990, 5, 1419–1423. [Google Scholar] [PubMed]
- Van Campenhout, C.; Nichane, M.; Antoniou, A.; Pendeville, H.; Bronchain, O.J.; Marine, J.-C.; Mazabraud, A.; Voz, M.L.; Bellefroid, E.J. Evi1 is specifically expressed in the distal tubule and duct of the Xenopus pronephros and plays a role in its formation. Dev. Biol. 2006, 294, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.-S.; Choi, S.-C.; Park, E.C.; Han, J.-K. Role of Tbx2 in defining the territory of the pronephric nephron. Development 2011, 138, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, B.E.; Li, Y.; Marra, A.N.; Cheng, C.N.; Wingert, R.A. The tbx2a/b transcription factors direct pronephros segmentation and corpuscle of Stannius formation in zebrafish. Dev. Biol. 2017, 421, 52–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyati, U.J.; Cooper, M.S.; Davidson, A.J.; Nechiporuk, A.; Kimelman, D. Sustained Bmp signaling is essential for cloaca development in zebrafish. Development 2006, 133, 2275–2284. [Google Scholar] [CrossRef] [Green Version]
- Prasad, M.; Iqbal, M.; Mordekar, S.R. Nonketotic hyperglycinemia case series. J. Pediatr. Neurosci. 2015, 10, 355–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, M.A.; Miller, K.; Young, S.P.; Tong, S.; Ghaloul-Gonzalez, L.; Neira-Fresneda, J.; Schlichting, L.; Peck, C.; Gabel, L.; Friederich, M.W.; et al. Cerebrospinal fluid amino acids glycine, serine, and threonine in nonketotic hyperglycinemia. J. Inherit. Metab. Dis. 2022, 45, 734–747. [Google Scholar] [CrossRef]
- Bowery, N.G.; Smart, T.G. GABA and glycine as neurotransmitters: A brief history. Br. J. Pharmacol. 2006, 147, S109–S119. [Google Scholar] [CrossRef] [Green Version]
- Ito, S. GABA and glycine in the developing brain. J. Physiol. Sci. 2016, 66, 375–379. [Google Scholar] [CrossRef]
- Autuori, M.C.; Pai, Y.J.; Stuckey, D.J.; Savery, D.; Marconi, A.M.; Massa, V.; Lythgoe, M.F.; Copp, A.J.; David, A.L.; Greene, N.D. Use of high-frequency ultrasound to study the prenatal development of cranial neural tube defects and hydrocephalus in Gldc-deficient mice. Prenat. Diagn. 2017, 37, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Leung, K.-Y.; Pai, Y.J.; Chen, Q.; Santos, C.; Calvani, E.; Sudiwala, S.; Savery, D.; Ralser, M.; Gross, S.S.; Copp, A.J.; et al. Partitioning of One-Carbon Units in Folate and Methionine Metabolism Is Essential for Neural Tube Closure. Cell Rep. 2017, 21, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Moens, C.B.; Prince, V.E. Constructing the hindbrain: Insights from the zebrafish. Dev. Dyn. 2002, 224, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kanekar, S.; Byler, D. Characteristic MRI findings in neonatal nonketotic hyperglycinemia due to sequence changes in GLDC gene encoding the enzyme glycine decarboxylase. Metab. Brain Dis. 2013, 28, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, A.; Kure, S.; Mikawa, S.; Ueki, T.; Kojima, K.; Fujiwara, K.; Iinuma, K.; Matsubara, Y.; Sato, K. Glycine cleavage system in neurogenic regions. Eur. J. Neurosci. 2004, 19, 2365–2370. [Google Scholar] [CrossRef]
- Jog, R.; Chen, G.; Wang, J.; Leff, T. Hormonal regulation of glycine decarboxylase and its relationship to oxidative stress. Physiol. Rep. 2021, 9, e14991. [Google Scholar] [CrossRef]
- Wang, W.; Wu, Z.; Dai, Z.; Yang, Y.; Wang, J.; Wu, G. Glycine metabolism in animals and humans: Implications for nutrition and health. Amino Acids 2013, 45, 463–477. [Google Scholar] [CrossRef]
- Thisse, B.; Thisse, C. Fast Release Clones: A High Throughput Expression Analysis. ZFIN Direct Data Submission. 2004. Available online: http://zfin.org (accessed on 20 September 2020).
- Weinberg, J.M.; Davis, J.A.; Abarzua, M.; Kiani, T.; Kunkel, R. Protection by glycine of proximal tubules from injury due to inhibitors of mitochondrial ATP production. Am. J. Physiol. Physiol. 1990, 258, C1127–C1140. [Google Scholar] [CrossRef]
- Takayanagi, M.; Kure, S.; Sakata, Y.; Kurihara, Y.; Ohya, Y.; Kajita, M.; Tada, K.; Matsubara, Y.; Narisawa, K. Human glycine decarboxylase gene (GLDC) and its highly conserved processed pseudogene (ψGLDC): Their structure and expression, and the identification of a large deletion in a family with nonketotic hyperglycinemia. Hum. Genet. 2000, 106, 298–305. [Google Scholar]
- Steiman, G.S.; Yudkoff, M.; Berman, P.H.; Blazer-Yost, B.; Segal, S. Late-onset nonketotic hyperglycinemia and spinocerebellar degeneration. J. Pediatr. 1979, 94, 907–911. [Google Scholar] [CrossRef]
- Mount, D.B. Thick Ascending Limb of the Loop of Henle. Clin. J. Am. Soc. Nephrol. 2014, 9, 1974–1986. [Google Scholar] [CrossRef] [Green Version]
- Subramanya, A.R.; Ellison, D.H. Distal convoluted tubule. Clin. J. Am. Soc. Nephrol. 2014, 9, 2147–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, B.E.; Clark, E.G.; Gatz, A.E.; Wingert, R.A. Kctd15 regulates nephron segment development by repressing Tfap2a activity. Development 2020, 147, dev191973. [Google Scholar] [CrossRef] [PubMed]
- Morales, E.E.; Handa, N.; Drummond, B.E.; Chambers, J.M.; Marra, A.; Addiego, A.; Wingert, R.A. Homeogene emx1 is required for nephron distal segment development in zebrafish. Sci. Rep. 2018, 8, 18038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieke, J.M.; Zhang, R.; Braun, D.; Yilmaz, Ö.; Japp, A.S.; Lopes, F.M.; Pleschka, M.; Hilger, A.C.; Schneider, S.; Newman, W.G.; et al. SLC20A1 Is Involved in Urinary Tract and Urorectal Development. Front. Cell Dev. Biol. 2020, 8, 567. [Google Scholar] [CrossRef] [PubMed]
- Korman, S.H.; Gutman, A. Pitfalls in the diagnosis of glycine encephalopathy (non-ketotic hyperglycinemia). Dev. Med. Child Neurol. 2002, 44, 712–720. [Google Scholar] [CrossRef]
- Theisen, U.; Hey, S.; Hennig, C.D.; Schnabel, R.; Köster, R.W. Glycine is able to induce both a motility speed in- and decrease during zebrafish neuronal migration. Commun. Integr. Biol. 2018, 11, 1–7. [Google Scholar] [CrossRef]
- Moura, A.P.; Parmeggiani, B.; Gasparotto, J.; Grings, M.; Cardoso, G.M.F.; Seminotti, B.; Moreira, J.C.F.; Gelain, D.P.; Wajner, M.; Leipnitz, G. Glycine Administration Alters MAPK Signaling Pathways and Causes Neuronal Damage in Rat Brain: Putative Mechanisms Involved in the Neurological Dysfunction in Nonketotic Hyperglycinemia. Mol. Neurobiol. 2018, 55, 741–750. [Google Scholar] [CrossRef]
- Moura, A.P.; Grings, M.; Parmeggiani, B.D.S.; Marcowich, G.F.; Tonin, A.M.; Viegas, C.M.; Zanatta, A.; Ribeiro, C.A.J.; Wajner, M.; Leipnitz, G. Glycine Intracerebroventricular Administration Disrupts Mitochondrial Energy Homeostasis in Cerebral Cortex and Striatum of Young Rats. Neurotox. Res. 2013, 24, 502–511. [Google Scholar] [CrossRef]
- Dumitrescu, E.; Karunaratne, D.P.; Prochaska, M.K.; Liu, X.; Wallace, K.N.; Andreescu, S. Developmental toxicity of glycine-coated silica nanoparticles in embryonic zebrafish. Environ. Pollut. 2017, 229, 439–447. [Google Scholar] [CrossRef]
- Tsuji-Tamura, K.; Sato, M.; Fujita, M.; Tamura, M. The role of PI3K/Akt/mTOR signaling in dose-dependent biphasic effects of glycine on vascular development. Biochem. Biophys. Res. Commun. 2020, 529, 596–602. [Google Scholar] [CrossRef]
- Cataltepe, S.; van Marter, L.J.; Kozakewich, H.; Wessel, D.L.; Lee, P.J.; Levy, H.L. Pulmonary hypertension associated with nonketotic hyperglycinemia. J. Inherit. Metab. Dis. 2000, 23, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Del Toro, M.; Arranz, J.A.; Macaya, A.; Riudor, E.; Raspall, M.; Moreno-Galdó, A.; Vazquez, E.; Ortega, A.; Matsubara, Y.; Kure, S.; et al. Progressive vacuolating glycine leukoencephalopathy with pulmonary hypertension. Ann. Neurol. 2006, 60, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.E.; Arranz, J.A.; Colomé, N.; Bech-Serra, J.J.; Canals, F.; Del Toro, M.; Riudor, E. Proteomic analysis in cerebrospinal fluid of patients with atypical nonketotic hyperglycinemia and pulmonary hypertension—A pilot study. Proteom. Clin. Appl. 2009, 3, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Suso, J.J.M.; Marín, M.J.D.C.; Martínez-Romillo, P.D.; de Lera, C.L.; García-Moya, L.F.; González, J.I.R. Nonketotic Hyperglycinemia Presenting as Pulmonary Hypertensive Vascular Disease and Fatal Pulmonary Edema in Response to Pulmonary Vasodilator Therapy. J. Pediatr. 2012, 161, 557–559. [Google Scholar] [CrossRef]
- Perens, E.A.; Garavito-Aguilar, Z.V.; Guio-Vega, G.P.; Peña, K.T.; Schindler, Y.L.; Yelon, D. Hand2 inhibits kidney specification while promoting vein formation within the posterior mesoderm. eLife 2016, 5, e19941. [Google Scholar] [CrossRef] [PubMed]
- Perens, E.A.; Diaz, J.T.; Quesnel, A.; Askary, A.; Crump, J.G.; Yelon, D. osr1 couples intermediate mesoderm cell fate with temporal dynamics of vessel progenitor cell differentiation. Development 2021, 148, dev198408. [Google Scholar] [CrossRef]
- Drummond, B.E.; Chambers, B.E.; Wesselman, H.M.; Gibson, S.; Arceri, L.; Ulrich, M.N.; Gerlach, G.F.; Kroeger, P.T.; Leshchiner, I.; Goessling, W.; et al. osr1 Maintains Renal Progenitors and Regulates Podocyte Development by Promoting wnt2ba via the Antagonism of hand2. Biomedicines 2022, 10, 2868. [Google Scholar] [CrossRef]
- Yin, M.; Zhong, Z.; Connor, H.D.; Bunzendahl, H.; Finn, W.F.; Rusyn, I.; Li, X.; Raleigh, J.A.; Mason, R.P.; Thurman, R.G. Protective effect of glycine on renal injury induced by ischemia-reperfusion in vivo. Am. J. Physiol. Physiol. 2002, 282, F417–F423. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, K.; Nielsen, C.B.; Flyvbjerg, A. Effects Of Glycine On Glomerular Filtration Rate And Segmental Tubular Handling Of Sodium In Conscious Rats. Clin. Exp. Pharmacol. Physiol. 2002, 29, 449–454. [Google Scholar] [CrossRef]
- Morales, E.E.; Wingert, R.A. Zebrafish as a Model of Kidney Disease. Results Probl. Cell Differ. 2017, 60, 55–75. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weaver, N.E.; Healy, A.; Wingert, R.A. gldc Is Essential for Renal Progenitor Patterning during Kidney Development. Biomedicines 2022, 10, 3220. https://doi.org/10.3390/biomedicines10123220
Weaver NE, Healy A, Wingert RA. gldc Is Essential for Renal Progenitor Patterning during Kidney Development. Biomedicines. 2022; 10(12):3220. https://doi.org/10.3390/biomedicines10123220
Chicago/Turabian StyleWeaver, Nicole E., Allison Healy, and Rebecca A. Wingert. 2022. "gldc Is Essential for Renal Progenitor Patterning during Kidney Development" Biomedicines 10, no. 12: 3220. https://doi.org/10.3390/biomedicines10123220