
Understanding Long COVID; Mitochondrial Health and Adaptation—Old Pathways, New Problems

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. From Alkaline Vents to Mitochondria and SARS-CoV-2
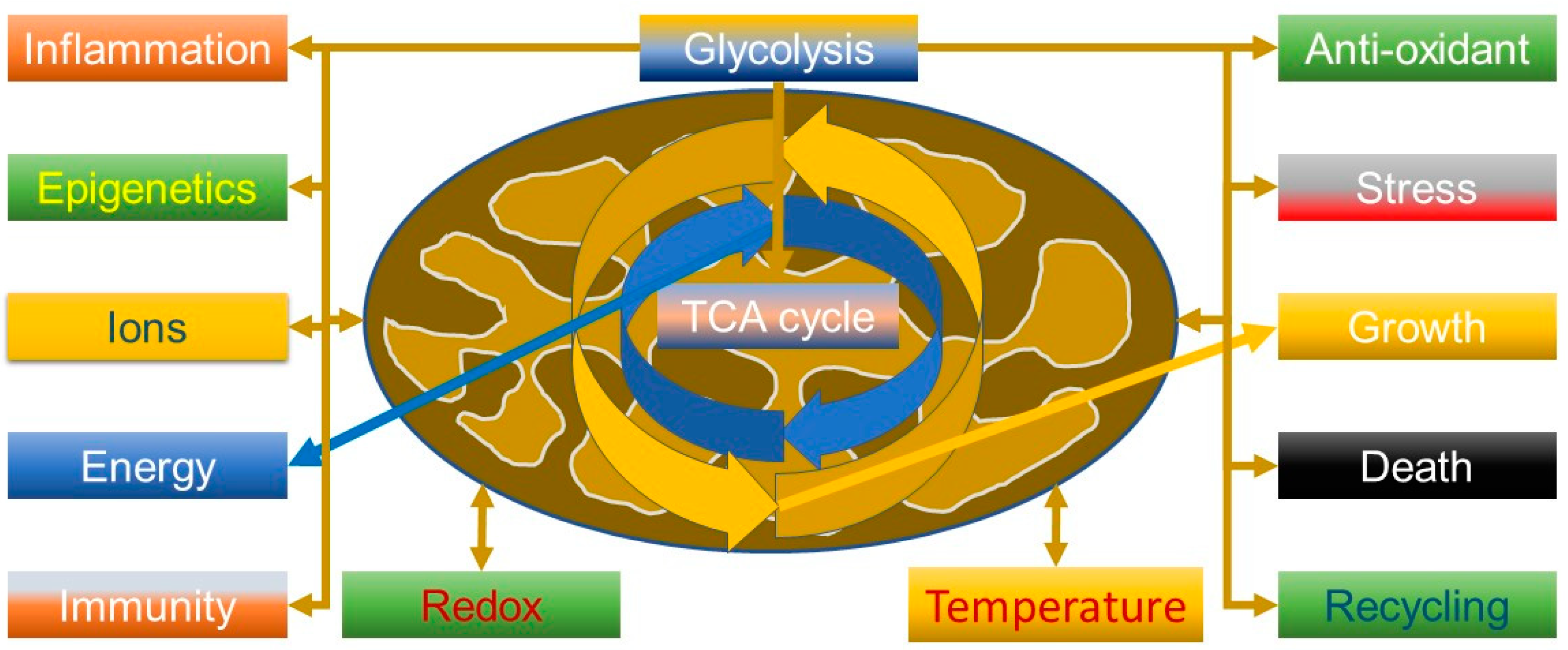
2.1. Beyond the Powerhouse—Mitochondria Do a Lot More Than Produce Energy
2.2. SARS-CoV-2 Can Modulate Mitochondria and Glycolysis
2.3. Taking the Viral Viewpoint; Is Hypoxia Good or Bad for the Virus?
2.4. Viruses, Mitochondria and Lipids; a Nutritional Arms Race
3. Long COVID and Mitochondria—A Tipping Point
3.1. The Sub-Optimal Mitochondrial Function Theory and Acute SARS-CoV-2 Severity
3.2. Temporal-Compartmental Effects of the Virus vs. the Host on Metabolic Reprogramming
3.3. The Importance of Mitochondrial DNA Copy Number—A Tipping Point?
4. Mitochondrial Health and Platelets in Long COVID
4.1. Platelets and Health
4.2. SARS-CoV-2, Platelets and Mitochondria
4.3. The Optimal Platelet Mitochondrion—Exercise as a Medicine
5. Discussion and Implications
5.1. Can We Say What Long COVID Is?
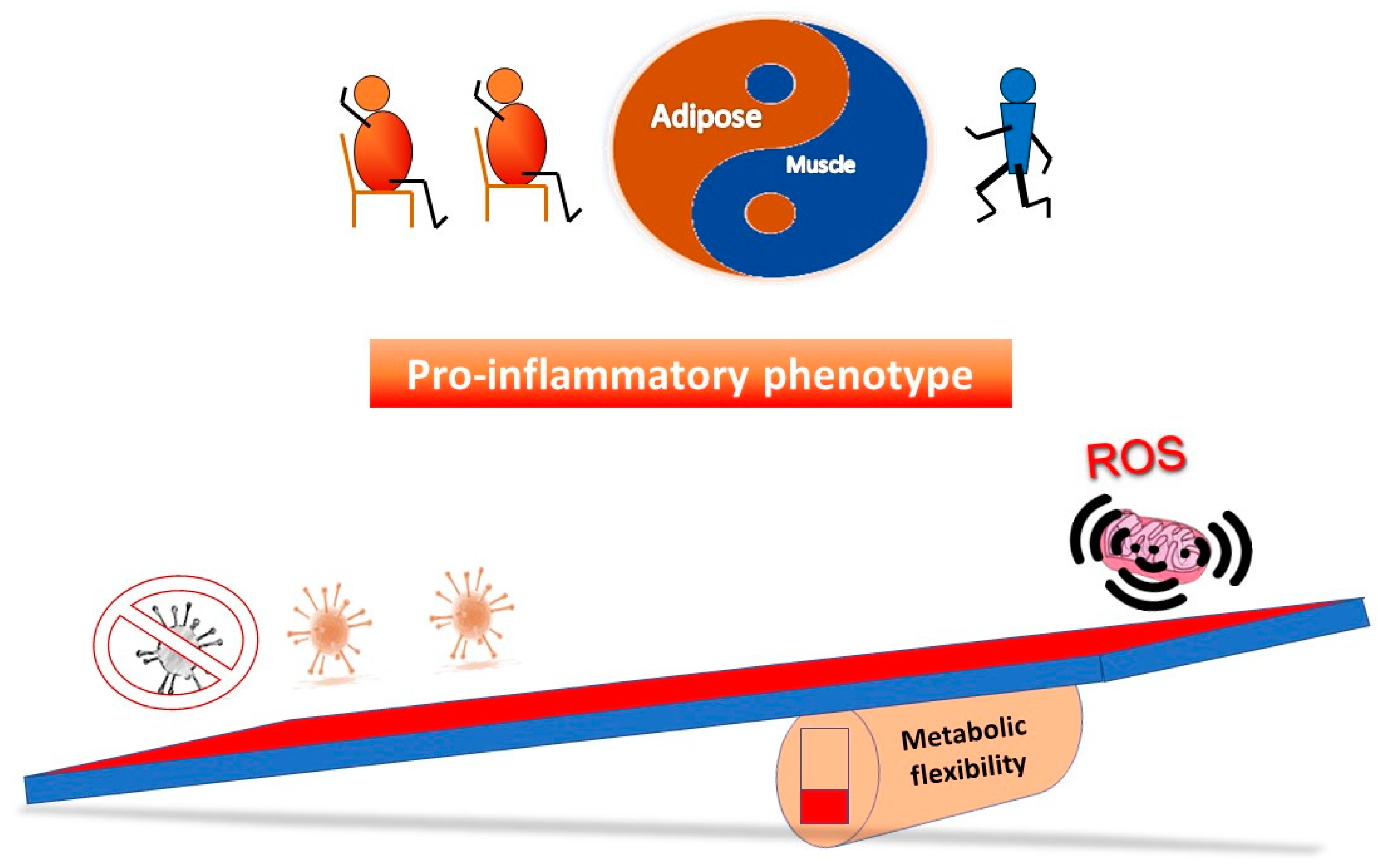
5.2. Restoring Metabolic Flexibility—A Hormetic Approach?
5.2.1. A Healthy Plant-Based Diet?
5.2.2. Antioxidants and Oxidants—The Metformin Experience
5.2.3. Learnings from Physical Activity
5.2.4. Calorie Restriction and the Ketogenic Diet—Sirtuins to the Rescue?
5.2.5. Emerging Nonchemical Modalities
5.2.6. Mitochondrial Transplant
5.2.7. Can We Measure Mitochondrial Health?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wise, J. COVID-19: Two million people in the UK are estimated to be experiencing long COVID, says ONS. BMJ 2022, 377, o1391. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Haupert, S.R.; Zimmermann, L.; Shi, X.; Fritsche, L.G.; Mukherjee, B. Global Prevalence of Post COVID-19 Condition or Long COVID: A Meta-Analysis and Systematic Review. J. Infect. Dis. 2022, 226, 1593–1607. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. How common is long COVID? Why studies give different answers. Nature 2022, 606, 852–853. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Bowe, B.; Xie, Y. Long COVID after breakthrough SARS-CoV-2 infection. Nat. Med. 2022, 28, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Morrone, M.C.; Patrono, C.; Santoro, M.G.; Schiaffino, S.; Remuzzi, G.; Bussolati, G. COVID-19 Commission of the Accademia Nazionale dei Lincei. Long COVID: Where we stand and challenges ahead. Cell Death Differ. 2022, 29, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Durstenfeld, M.S.; Sun, K.; Tahir, P.; Peluso, M.J.; Deeks, S.G.; Aras, M.A.; Grandis, D.J.; Long, C.S.; Beatty, A.; Hsue, P.Y. Use of Cardiopulmonary Exercise Testing to Evaluate Long COVID-19 Symptoms in Adults: A Systematic Review and Meta-analysis. JAMA Netw. Open 2022, 5, e2236057. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef]
- Mahony, L.O.; Buwalda, T.; Blair, M.; Forde, B.; Lunjani, N.; Ambikan, A.; Neogi, U.; Barrett, P.; Geary, E.; O’Connor, N.; et al. Impact of Long COVID on health and quality of life. HRB Open Res 2022, 5, 31. [Google Scholar] [CrossRef]
- Wulf Hanson, S.; Abbafati, C.; Aerts, J.G.; Al-Aly, Z.; Ashbaugh, C.; Ballouz, T.; Blyuss, O.; Bobkova, P.; Bonsel, G.; Borzakova, S.; et al. A global systematic analysis of the occurrence, severity, and recovery pattern of long COVID in 2020 and 2021. medRxiv 2022. preprint. [Google Scholar] [CrossRef]
- Trapani, G.; Verlato, G.; Bertino, E.; Maiocco, G.; Vesentini, R.; Spadavecchia, A.; Dessi, A.; Fanos, V. Long COVID-19 in children: An Italian cohort study. Ital. J. Pediatr. 2022, 48, 83. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Clues to long COVID. Science 2022, 376, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Xie, Y.; Al-Aly, Z. Long-term neurologic outcomes of COVID-19. Nat. Med. 2022, 28, 2406–2415. [Google Scholar] [CrossRef]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.; Vendramini, P.H.; Valenca, A.G.F.; Brandao-Teles, C.; Zuccoli, G.D.S.; et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2200960119. [Google Scholar] [CrossRef] [PubMed]
- Asakura, H.; Ogawa, H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int. J. Hematol. 2021, 113, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.M.; Kruger, A.; Proal, A.; Kell, D.B.; Pretorius, E. The Occurrence of Hyperactivated Platelets and Fibrinaloid Microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Pharmaceuticals 2022, 15, 931. [Google Scholar] [CrossRef] [PubMed]
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Front. Neurol. 2022, 13, 877772. [Google Scholar] [CrossRef]
- Ambikan, A.T.; Yang, H.; Krishnan, S.; Svensson Akusjärvi, S.; Gupta, S.; Lourda, M.; Sperk, M.; Arif, M.; Zhang, C.; Nordqvist, H.; et al. Multi-omics personalized network analyses highlight progressive disruption of central metabolism associated with COVID-19 severity. Cell Syst. 2022, 13, 665–681.e664. [Google Scholar] [CrossRef]
- Tomas, C.; Brown, A.; Strassheim, V.; Elson, J.L.; Newton, J.; Manning, P. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 2017, 12, e0186802. [Google Scholar] [CrossRef] [Green Version]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef] [Green Version]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef] [PubMed]
- Nunn, A.V.W.; Guy, G.W.; Brysch, W.; Botchway, S.W.; Frasch, W.; Calabrese, E.J.; Bell, J.D. SARS-CoV-2 and mitochondrial health: Implications of lifestyle and ageing. Immun. Ageing 2020, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef]
- Burtscher, J.; Cappellano, G.; Omori, A.; Koshiba, T.; Millet, G.P. Mitochondria: In the Cross Fire of SARS-CoV-2 and Immunity. iScience 2020, 23, 101631. [Google Scholar] [CrossRef]
- Ganji, R.; Reddy, P.H. Impact of COVID-19 on Mitochondrial-Based Immunity in Aging and Age-Related Diseases. Front. Aging Neurosci. 2020, 12, 614650. [Google Scholar] [CrossRef] [PubMed]
- Captur, G.; Moon, J.C.; Topriceanu, C.-C.; Joy, G.; Swadling, L. Plasma proteomic signature predicts who will get persistent symptoms following SARS-CoV-2 infection. EBioMedicine 2022, 85, 104293. [Google Scholar] [CrossRef]
- Del Nonno, F.; Nardacci, R.; Colombo, D.; Visco-Comandini, U.; Cicalini, S.; Antinori, A.; Marchioni, L.; D’Offizi, G.; Piacentini, M.; Falasca, L. Hepatic Failure in COVID-19: Is Iron Overload the Dangerous Trigger? Cells 2021, 10, 1103. [Google Scholar] [CrossRef] [PubMed]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [Green Version]
- Grillo, A.S.; Kelly, C.; Ha, V.T.; Bodart, C.M.; Huff, S.; Couch, R.K.; Herrel, N.T.; Kim, H.D.; Zimmermann, A.O.; Shattuck, J.; et al. Iron Status Influences Mitochondrial Disease Progression in Complex I-Deficient Mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Stenberg, S.; Li, J.; Gjuvsland, A.B.; Persson, K.; Demitz-Helin, E.; Gonzalez Pena, C.; Yue, J.X.; Gilchrist, C.; Arengard, T.; Ghiaci, P.; et al. Genetically controlled mtDNA deletions prevent ROS damage by arresting oxidative phosphorylation. Elife 2022, 11, e76095. [Google Scholar] [CrossRef]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantin-Teodosiu, D.; Constantin, D.; Pelsers, M.M.; Verdijk, L.B.; van Loon, L.; Greenhaff, P.L. Mitochondrial DNA copy number associates with insulin sensitivity and aerobic capacity, and differs between sedentary, overweight middle-aged males with and without type 2 diabetes. Int. J. Obes. 2020, 44, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.; Rauseo, A.M.; Liu, Z.; Sahu, S.K.; et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 2021, 6, e143299. [Google Scholar] [CrossRef] [PubMed]
- Targhetta, V.P.; Amaral, M.A.; Camara, N.O.S. Through DNA sensors and hidden mitochondrial effects of SARS-CoV-2. J. Venom. Anim. Toxins Incl. Trop. Dis. 2021, 27, e20200183. [Google Scholar] [CrossRef]
- Mehra, C.; Pernas, L. Move it to lose it: Mitocytosis expels damaged mitochondria. Dev. Cell 2021, 56, 2014–2015. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Dasgupta, A.; Chen, K.-H.; Wu, D.; Baid, K.; Mamatis, J.E.; Gonzalez, V.; Read, A.; Bentley, R.E.T.; Martin, A.Y.; et al. SARS-CoV-2 mitochondriopathy in COVID-19 pneumonia exacerbates hypoxemia. Redox Biol. 2022, 58, 102508. [Google Scholar] [CrossRef]
- Lane, N. Transformer: The Deep Chemistry of Life and Death; W. W. Norton: New York, NY, USA, 2022. [Google Scholar]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; Martinez-Outschoorn, U.E.; Howell, A.; et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: Similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging 2010, 2, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Hiltunen, M.; Kauppinen, A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: Novel insights into mitochondrial regulation of aging process. Cell Signal. 2014, 26, 1598–1603. [Google Scholar] [CrossRef]
- Kee, J.; Thudium, S.; Renner, D.M.; Glastad, K.; Palozola, K.; Zhang, Z.; Li, Y.; Lan, Y.; Cesare, J.; Poleshko, A.; et al. SARS-CoV-2 disrupts host epigenetic regulation via histone mimicry. Nature 2022, 610, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L. Cellular metabolism in the defense against microbes. J. Cell Sci. 2021, 134, jcs252023. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ahn, D.G.; Syed, G.H.; Siddiqui, A. The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion 2018, 41, 21–27. [Google Scholar] [CrossRef]
- Aguilar-Lopez, B.A.; Moreno-Altamirano, M.M.B.; Dockrell, H.M.; Duchen, M.R.; Sanchez-Garcia, F.J. Mitochondria: An Integrative Hub Coordinating Circadian Rhythms, Metabolism, the Microbiome, and Immunity. Front. Cell Dev. Biol. 2020, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Scholey, J.; Aburto, J.M.; Kashnitsky, I.; Kniffka, M.S.; Zhang, L.; Jaadla, H.; Dowd, J.B.; Kashyap, R. Life expectancy changes since COVID-19. Nat. Hum. Behav. 2022. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Metaxaki, M.; Wills, M.R.; Sithole, N. Reduced Incidence of Long Coronavirus Disease Referrals to the Cambridge University Teaching Hospital Long Coronavirus Disease Clinic. Clin. Infect. Dis. 2022. ahead of print. [Google Scholar] [CrossRef]
- Ostendorf, B.N.; Patel, M.A.; Bilanovic, J.; Hoffmann, H.H.; Carrasco, S.E.; Rice, C.M.; Tavazoie, S.F. Common human genetic variants of APOE impact murine COVID-19 mortality. Nature 2022, 611, 346–351. [Google Scholar] [CrossRef]
- Yin, J.; Reiman, E.M.; Beach, T.G.; Serrano, G.E.; Sabbagh, M.N.; Nielsen, M.; Caselli, R.J.; Shi, J. Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology 2020, 94, e2404–e2411. [Google Scholar] [CrossRef]
- Arena, R.; Pronk, N.P.; Laddu, D.; Whitsel, L.P.; Sallis, J.F.; Lavie, C.J.; Network, H.-P. Mapping One Million COVID-19 Deaths and Unhealthy Lifestyle Behaviors in the United States: Recognizing the Syndemic Pattern and Taking Action. Am. J. Med. 2022, 135, 1288–1295. [Google Scholar] [CrossRef]
- Smith, E.; Morowitz, H. The Origin and Nature of Life on Earth; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Trefil, J.; Morowitz, H.J.; Smith, E. The origins of life: The case is made for the descent of electrons. Am. Sci. 2009, 97, 206–213. [Google Scholar] [CrossRef]
- Blackstone, N.W. The impact of mitochondrial endosymbiosis on the evolution of calcium signaling. Cell Calcium 2015, 57, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta 2016, 1857, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Guillon, A.; Brea-Diakite, D.; Cezard, A.; Wacquiez, A.; Baranek, T.; Bourgeais, J.; Picou, F.; Vasseur, V.; Meyer, L.; Chevalier, C.; et al. Host succinate inhibits influenza virus infection through succinylation and nuclear retention of the viral nucleoprotein. EMBO J. 2022, 41, e108306. [Google Scholar] [CrossRef] [PubMed]
- Roca, F.J.; Whitworth, L.J.; Prag, H.A.; Murphy, M.P.; Ramakrishnan, L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376, eabh2841. [Google Scholar] [CrossRef]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Kamunde, C.; Sharaf, M.; MacDonald, N. H2O2 metabolism in liver and heart mitochondria: Low emitting-high scavenging and high emitting-low scavenging systems. Free. Radic. Biol. Med. 2018, 124, 135–148. [Google Scholar] [CrossRef]
- Drechsel, D.A.; Patel, M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 2010, 285, 27850–27858. [Google Scholar] [CrossRef] [Green Version]
- Munro, D.; Treberg, J.R. A radical shift in perspective: Mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 2017, 220, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Munro, D.; Baldy, C.; Pamenter, M.E.; Treberg, J.R. The exceptional longevity of the naked mole-rat may be explained by mitochondrial antioxidant defenses. Aging Cell 2019, 18, e12916. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877. [Google Scholar] [CrossRef]
- Brook, C.E.; Dobson, A.P. Bats as ‘special’ reservoirs for emerging zoonotic pathogens. Trends Microbiol. 2015, 23, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, F.J.; Perez-Hernandez, C.A.; Rodriguez-Murillo, M.; Moreno-Altamirano, M.M.B. The Role of Tricarboxylic Acid Cycle Metabolites in Viral Infections. Front. Cell. Infect. Microbiol. 2021, 11, 725043. [Google Scholar] [CrossRef] [PubMed]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef]
- Shi, D.; Yan, R.; Lv, L.; Jiang, H.; Lu, Y.; Sheng, J.; Xie, J.; Wu, W.; Xia, J.; Xu, K.; et al. The serum metabolome of COVID-19 patients is distinctive and predictive. Metabolism 2021, 118, 154739. [Google Scholar] [CrossRef] [PubMed]
- Barberis, E.; Timo, S.; Amede, E.; Vanella, V.V.; Puricelli, C.; Cappellano, G.; Raineri, D.; Cittone, M.G.; Rizzi, E.; Pedrinelli, A.R.; et al. Large-Scale Plasma Analysis Revealed New Mechanisms and Molecules Associated with the Host Response to SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 8623. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, C.; Bizkarguenaga, M.; Gil-Redondo, R.; Diercks, T.; Arana, E.; Garcia de Vicuna, A.; Seco, M.; Bosch, A.; Palazon, A.; San Juan, I.; et al. SARS-CoV-2 Infection Dysregulates the Metabolomic and Lipidomic Profiles of Serum. iScience 2020, 23, 101645. [Google Scholar] [CrossRef]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; et al. COVID-19 infection results in alterations of the kynurenine pathway and fatty acid metabolism that correlate with IL-6 levels and renal status. medRxiv 2020. [Google Scholar] [CrossRef]
- Paez-Franco, J.C.; Torres-Ruiz, J.; Sosa-Hernandez, V.A.; Cervantes-Diaz, R.; Romero-Ramirez, S.; Perez-Fragoso, A.; Meza-Sanchez, D.E.; German-Acacio, J.M.; Maravillas-Montero, J.L.; Mejia-Dominguez, N.R.; et al. Metabolomics analysis reveals a modified amino acid metabolism that correlates with altered oxygen homeostasis in COVID-19 patients. Sci. Rep. 2021, 11, 6350. [Google Scholar] [CrossRef]
- Hogberg, C.; Gidlof, O.; Tan, C.; Svensson, S.; Nilsson-Ohman, J.; Erlinge, D.; Olde, B. Succinate independently stimulates full platelet activation via cAMP and phosphoinositide 3-kinase-beta signaling. J. Thromb. Haemost. 2011, 9, 361–372. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Y.; Meng, X.; Wang, Z.; Younis, M.; Liu, Y.; Wang, P.; Huang, X. SARS-CoV-2 membrane protein causes the mitochondrial apoptosis and pulmonary edema via targeting BOK. Cell Death Differ. 2022, 29, 1395–1408. [Google Scholar] [CrossRef]
- Diaz-Resendiz, K.J.G.; Benitez-Trinidad, A.B.; Covantes-Rosales, C.E.; Toledo-Ibarra, G.A.; Ortiz-Lazareno, P.C.; Giron-Perez, D.A.; Bueno-Duran, A.Y.; Perez-Diaz, D.A.; Barcelos-Garcia, R.G.; Giron-Perez, M.I. Loss of mitochondrial membrane potential (DeltaPsim) in leucocytes as post-COVID-19 sequelae. J. Leukoc. Biol. 2022, 112, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Grossini, E.; Concina, D.; Rinaldi, C.; Russotto, S.; Garhwal, D.; Zeppegno, P.; Gramaglia, C.; Kul, S.; Panella, M. Association Between Plasma Redox State/Mitochondria Function and a Flu-Like Syndrome/COVID-19 in the Elderly Admitted to a Long-Term Care Unit. Front. Physiol. 2021, 12, 707587. [Google Scholar] [CrossRef] [PubMed]
- Gabanella, F.; Barbato, C.; Corbi, N.; Fiore, M.; Petrella, C.; de Vincentiis, M.; Greco, A.; Ferraguti, G.; Corsi, A.; Ralli, M.; et al. Exploring Mitochondrial Localization of SARS-CoV-2 RNA by Padlock Assay: A Pilot Study in Human Placenta. Int. J. Mol. Sci. 2022, 23, 2100. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A. COVID-19 and Mitochondrial Non-Coding RNAs: New Insights From Published Data. Front. Physiol. 2021, 12, 805005. [Google Scholar] [CrossRef]
- Miller, B.; Silverstein, A.; Flores, M.; Cao, K.; Kumagai, H.; Mehta, H.H.; Yen, K.; Kim, S.J.; Cohen, P. Host mitochondrial transcriptome response to SARS-CoV-2 in multiple cell models and clinical samples. Sci. Rep. 2021, 11, 3. [Google Scholar] [CrossRef]
- Freundt, E.C.; Yu, L.; Goldsmith, C.S.; Welsh, S.; Cheng, A.; Yount, B.; Liu, W.; Frieman, M.B.; Buchholz, U.J.; Screaton, G.R.; et al. The open reading frame 3a protein of severe acute respiratory syndrome-associated coronavirus promotes membrane rearrangement and cell death. J. Virol. 2010, 84, 1097–1109. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Liu, W.; Li, X.; Zhao, P.; Shereen, M.A.; Zhu, C.; Huang, S.; Liu, S.; Yu, X.; Yue, M.; et al. HIF-1alpha promotes SARS-CoV-2 infection and aggravates inflammatory responses to COVID-19. Signal Transduct. Target. Ther. 2021, 6, 308. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Wang, G.; Huang, X.; Lee, H.; Lee, J.G.; Yang, P.; van de Leemput, J.; Huang, W.; Kane, M.A.; Yang, P.; et al. SARS-CoV-2 Nsp6 damages Drosophila heart and mouse cardiomyocytes through MGA/MAX complex-mediated increased glycolysis. Commun. Biol. 2022, 5, 1039. [Google Scholar] [CrossRef]
- Eaton, A.F.; Merkulova, M.; Brown, D. The H(+)-ATPase (V-ATPase): From proton pump to signaling complex in health and disease. Am. J. Physiol. Cell Physiol. 2021, 320, C392–C414. [Google Scholar] [CrossRef]
- Qi, Z.; Zhai, X.; Ding, S. How to explain exercise-induced phenotype from molecular data: Rethink and reconstruction based on AMPK and mTOR signaling. Springerplus 2013, 2, 693. [Google Scholar] [CrossRef]
- Khan, M.M.; Lee, S.; Couoh-Cardel, S.; Oot, R.A.; Kim, H.; Wilkens, S.; Roh, S.H. Oxidative stress protein Oxr1 promotes V-ATPase holoenzyme disassembly in catalytic activity-independent manner. EMBO J. 2022, 41, e109360. [Google Scholar] [CrossRef]
- Banerjee, S.; Kane, P.M. Regulation of V-ATPase Activity and Organelle pH by Phosphatidylinositol Phosphate Lipids. Front. Cell Dev. Biol. 2020, 8, 510. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, T.; Ding, Y.; Yu, T.; Cui, Y.; Nie, H. Expression profiles of respiratory V-ATPase and calprotectin in SARS-CoV-2 infection. Cell Death Discov. 2022, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Icho, S.; Rujas, E.; Muthuraman, K.; Tam, J.; Liang, H.; Landreth, S.; Liao, M.; Falzarano, D.; Julien, J.P.; Melnyk, R.A. Dual Inhibition of Vacuolar-ATPase and TMPRSS2 Is Required for Complete Blockade of SARS-CoV-2 Entry into Cells. Antimicrob. Agents Chemother. 2022, 66, e0043922. [Google Scholar] [CrossRef] [PubMed]
- Miles, A.L.; Burr, S.P.; Grice, G.L.; Nathan, J.A. The vacuolar-ATPase complex and assembly factors, TMEM199 and CCDC115, control HIF1alpha prolyl hydroxylation by regulating cellular iron levels. Elife 2017, 6, e22693. [Google Scholar] [CrossRef]
- Hayek, S.R.; Rane, H.S.; Parra, K.J. Reciprocal Regulation of V-ATPase and Glycolytic Pathway Elements in Health and Disease. Front. Physiol. 2019, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Cuninghame, S.; Jackson, R.; Zehbe, I. Hypoxia-inducible factor 1 and its role in viral carcinogenesis. Virology 2014, 456–457, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Wing, P.A.C.; Keeley, T.P.; Zhuang, X.; Lee, J.Y.; Prange-Barczynska, M.; Tsukuda, S.; Morgan, S.B.; Harding, A.C.; Argles, I.L.A.; Kurlekar, S.; et al. Hypoxic and pharmacological activation of HIF inhibits SARS-CoV-2 infection of lung epithelial cells. Cell Rep. 2021, 35, 109020. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.d.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef]
- Farley, S.E.; Kyle, J.E.; Leier, H.C.; Bramer, L.M.; Weinstein, J.B.; Bates, T.A.; Lee, J.Y.; Metz, T.O.; Schultz, C.; Tafesse, F.G. A global lipid map reveals host dependency factors conserved across SARS-CoV-2 variants. Nat. Commun. 2022, 13, 3487. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, S.M.; Solmonson, A.; Rusin, S.F.; Maschek, J.A.; Bensard, C.L.; Fogarty, S.; Jeong, M.Y.; Lettlova, S.; Berg, J.A.; Morgan, J.T.; et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. Elife 2020, 9, e58041. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, J.K.; Autio, K.J.; Schonauer, M.S.; Kursu, V.A.; Dieckmann, C.L.; Kastaniotis, A.J. Mitochondrial fatty acid synthesis and respiration. Biochim. Biophys. Acta 2010, 1797, 1195–1202. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.R.; Koivisto, H.; Jokivarsi, K.; Miinalainen, I.J.; Autio, K.J.; Manninen, A.; Poutiainen, P.; Tanila, H.; Hiltunen, J.K.; Kastaniotis, A.J. Impaired Mitochondrial Fatty Acid Synthesis Leads to Neurodegeneration in Mice. J. Neurosci. 2018, 38, 9781–9800. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Ding, L.; Liang, J.; Yang, X.; Liu, Y.; Wang, Y.; Ding, M.; Huang, X. NAD kinase sustains lipogenesis and mitochondrial metabolismthrough fatty acid synthesis. Cell Rep. 2021, 37, 110157. [Google Scholar] [CrossRef]
- Pan, J.H.; Tang, J.; Kim, Y.J.; Lee, J.H.; Shin, E.C.; Zhao, J.; Kim, K.H.; Hwang, K.A.; Huang, Y.; Kim, J.K. IDH2 Deficiency Is Critical in Myogenesis and Fatty Acid Metabolism in Mice Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 5596. [Google Scholar] [CrossRef]
- Nunn, A.V.; Bell, J.; Barter, P. The integration of lipid-sensing and anti-inflammatory effects: How the PPARs play a role in metabolic balance. Nucl. Recept. 2007, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, X.; Yao, H.; Chen, X.; Shang, L.; Li, P.; Cui, X.; Zeng, J. Peroxisome-generated succinate induces lipid accumulation and oxidative stress in the kidneys of diabetic mice. J. Biol. Chem. 2022, 298, 101660. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, X.; Wang, Z.; Quan, J.; Zhao, X.; Tang, H.; Wu, H.; Di, Q.; Wu, Z.; Chen, W. Succinate Is a Natural Suppressor of Antiviral Immune Response by Targeting MAVS. Front. Immunol. 2022, 13, 816378. [Google Scholar] [CrossRef]
- Lane, N.; Martin, W.F. The origin of membrane bioenergetics. Cell 2012, 151, 1406–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.K.; Sarode, S.C. Do compromised mitochondria aggravate severity and fatality by SARS-CoV-2? Curr. Med. Res. Opin. 2022, 38, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Alhoufie, S.T.S.; Hifny, A.; Schwartz, L.; Alqahtani, A.S.; Ahmed, S.B.M.; Alqahtani, A.M.; Alqahtani, S.S.; Muddathir, A.K.; Ali, H.; et al. Of mitochondrion and COVID-19. J. Enzym. Inhib. Med. Chem. 2021, 36, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Nunn, A.V.W.; Guy, G.W.; Botchway, S.W.; Bell, J.D. SARS-CoV-2 and EBV; the cost of a second mitochondrial “whammy”? Immun. Ageing 2021, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.E.; Okyay, R.A.; Licht, W.E.; Hurley, D.J. Investigation of Long COVID Prevalence and Its Relationship to Epstein-Barr Virus Reactivation. Pathogens 2021, 10, 763. [Google Scholar] [CrossRef]
- Pizzamiglio, C.; Machado, P.M.; Thomas, R.H.; Gorman, G.S.; McFarland, R.; Hanna, M.G.; Pitceathly, R.D.S.; Mito, C.-S.G. COVID-19-Related Outcomes in Primary Mitochondrial Diseases: An International Study. Neurology 2022, 98, 576–582. [Google Scholar] [CrossRef]
- Sharma, A.; Smith, H.J.; Yao, P.; Mair, W.B. Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 2019, 20, e48395. [Google Scholar] [CrossRef]
- Guarnieri, J.W.; Dybas, J.M.; Fazelinia, H.; Kim, M.S.; Frere, J.; Zhang, Y.; Albrecht, Y.S.; Murdock, D.G.; Angelin, A.; Singh, L.N.; et al. Targeted Down Regulation Of Core Mitochondrial Genes During SARS-CoV-2 Infection. bioRxiv 2022. [Google Scholar] [CrossRef]
- Demetrius, L.A.; Magistretti, P.J.; Pellerin, L. Alzheimer’s disease: The amyloid hypothesis and the Inverse Warburg effect. Front. Physiol. 2014, 5, 522. [Google Scholar] [CrossRef] [Green Version]
- Claus, C.; Liebert, U.G. A renewed focus on the interplay between viruses and mitochondrial metabolism. Arch. Virol. 2014, 159, 1267–1277. [Google Scholar] [CrossRef]
- Speelman, T.; Dale, L.; Louw, A.; Verhoog, N.J.D. The Association of Acute Phase Proteins in Stress and Inflammation-Induced T2D. Cells 2022, 11, 2163. [Google Scholar] [CrossRef]
- Richter, K.; Amati, A.L.; Padberg, W.; Grau, V. Negative regulation of ATP-induced inflammasome activation and cytokine secretion by acute-phase proteins: A mini review. Front. Pharmacol. 2022, 13, 981276. [Google Scholar] [CrossRef] [PubMed]
- Gavkare, A.M.; Nanaware, N.; Rayate, A.S.; Mumbre, S.; Nagoba, B.S. COVID-19 associated diabetes mellitus: A review. World J. Diabetes 2022, 13, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Bjornstad, E.C.; Cutter, G.; Guru, P.; Menon, S.; Aldana, I.; House, S.; Tofil, N.M.; St Hill, C.A.; Tarabichi, Y.; Banner-Goodspeed, V.M.; et al. SARS-CoV-2 infection increases risk of acute kidney injury in a bimodal age distribution. BMC Nephrol. 2022, 23, 63. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, M.; Honavar, S.G.; Sharma, N.; Sachdev, M.S. COVID-19 and Eye: A Review of Ophthalmic Manifestations of COVID-19. Indian J. Ophthalmol. 2021, 69, 488–509. [Google Scholar] [CrossRef]
- Soares, M.N.; Eggelbusch, M.; Naddaf, E.; Gerrits, K.H.L.; van der Schaaf, M.; van den Borst, B.; Wiersinga, W.J.; van Vugt, M.; Weijs, P.J.M.; Murray, A.J.; et al. Skeletal muscle alterations in patients with acute COVID-19 and post-acute sequelae of COVID-19. J. Cachexia Sarcopenia Muscle 2022, 13, 11–22. [Google Scholar] [CrossRef]
- Monlun, M.; Hyernard, C.; Blanco, P.; Lartigue, L.; Faustin, B. Mitochondria as Molecular Platforms Integrating Multiple Innate Immune Signalings. J. Mol. Biol. 2017, 429, 1–13. [Google Scholar] [CrossRef]
- Ebrahim Nakhli, R.; Shanker, A.; Sarosiek, I.; Boschman, J.; Espino, K.; Sigaroodi, S.; Al Bayati, I.; Elhanafi, S.; Sadeghi, A.; Sarosiek, J.; et al. Gastrointestinal symptoms and the severity of COVID-19: Disorders of gut-brain interaction are an outcome. Neurogastroenterol. Motil. 2022, 34, e14368. [Google Scholar] [CrossRef]
- Cortes, G.M.; Marcialis, M.A.; Bardanzellu, F.; Corrias, A.; Fanos, V.; Mussap, M. Inflammatory Bowel Disease and COVID-19: How Microbiomics and Metabolomics Depict Two Sides of the Same Coin. Front. Microbiol. 2022, 13, 856165. [Google Scholar] [CrossRef]
- Clark, A.; Mach, N. The Crosstalk between the Gut Microbiota and Mitochondria during Exercise. Front. Physiol. 2017, 8, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008, 4, e1000018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filograna, R.; Koolmeister, C.; Upadhyay, M.; Pajak, A.; Clemente, P.; Wibom, R.; Simard, M.L.; Wredenberg, A.; Freyer, C.; Stewart, J.B.; et al. Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci. Adv. 2019, 5, eaav9824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zole, E.; Ranka, R. Mitochondria, its DNA and telomeres in ageing and human population. Biogerontology 2018, 19, 189–208. [Google Scholar] [CrossRef]
- Kaufman, B.A.; Picard, M.; Sondheimer, N. Mitochondrial DNA, nuclear context, and the risk for carcinogenesis. Environ. Mol. Mutagen. 2018, 60, 455–462. [Google Scholar] [CrossRef]
- Lane, N. Power, Sex, Suicide; Mitochondria and the Meaning of Life; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Picard, M. Blood mitochondrial DNA copy number: What are we counting? Mitochondrion 2021, 60, 1–11. [Google Scholar] [CrossRef]
- Longchamps, R.J.; Yang, S.Y.; Castellani, C.A.; Shi, W.; Lane, J.; Grove, M.L.; Bartz, T.M.; Sarnowski, C.; Liu, C.; Burrows, K.; et al. Genome-wide analysis of mitochondrial DNA copy number reveals loci implicated in nucleotide metabolism, platelet activation, and megakaryocyte proliferation. Hum. Genet. 2022, 141, 127–146. [Google Scholar] [CrossRef]
- Corsi, S.; Iodice, S.; Vigna, L.; Cayir, A.; Mathers, J.C.; Bollati, V.; Byun, H.M. Platelet mitochondrial DNA methylation predicts future cardiovascular outcome in adults with overweight and obesity. Clin. Epigenetics 2020, 12, 29. [Google Scholar] [CrossRef] [Green Version]
- Gorog, D.A.; Storey, R.F.; Gurbel, P.A.; Tantry, U.S.; Berger, J.S.; Chan, M.Y.; Duerschmied, D.; Smyth, S.S.; Parker, W.A.E.; Ajjan, R.A.; et al. Current and novel biomarkers of thrombotic risk in COVID-19: A Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nat. Rev. Cardiol. 2022, 19, 475–495. [Google Scholar] [CrossRef]
- Obydennyy, S.I.; Sveshnikova, A.N.; Ataullakhanov, F.I.; Panteleev, M.A. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 2016, 14, 1867–1881. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.P.; Tiwari, A.; Singh, N.; Gautam, D.; Sonkar, V.K.; Agarwal, V.; Dash, D. Aerobic glycolysis fuels platelet activation: Small-molecule modulators of platelet metabolism as anti-thrombotic agents. Haematologica 2019, 104, 806–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obydennyi, S.I.; Artemenko, E.O.; Sveshnikova, A.N.; Ignatova, A.A.; Varlamova, T.V.; Gambaryan, S.; Lomakina, G.Y.; Ugarova, N.N.; Kireev, I.I.; Ataullakhanov, F.I.; et al. Mechanisms of increased mitochondria-dependent necrosis in Wiskott-Aldrich syndrome platelets. Haematologica 2020, 105, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Kholmukhamedov, A.; Janecke, R.; Choo, H.J.; Jobe, S.M. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J. Thromb. Haemost. 2018, 16, 2315–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koseoglu, S.; Dilks, J.R.; Peters, C.G.; Fitch-Tewfik, J.L.; Fadel, N.A.; Jasuja, R.; Italiano, J.E., Jr.; Haynes, C.L.; Flaumenhaft, R. Dynamin-related protein-1 controls fusion pore dynamics during platelet granule exocytosis. Arter. Thromb. Vasc. Biol. 2013, 33, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Hu, W.; Song, X.; Zhao, Y. Immune Modulation of Platelet-Derived Mitochondria on Memory CD4(+) T Cells in Humans. Int. J. Mol. Sci. 2020, 21, 6295. [Google Scholar] [CrossRef]
- Levoux, J.; Prola, A.; Lafuste, P.; Gervais, M.; Chevallier, N.; Koumaiha, Z.; Kefi, K.; Braud, L.; Schmitt, A.; Yacia, A.; et al. Platelets facilitate the wound-healing capability of mesenchymal stem cells by mitochondrial transfer and metabolic reprogramming. Cell Metab. 2021, 33, 688–690. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhou, Y.; Hilton, T.; Li, F.; Han, C.; Liu, L.; Yuan, H.; Li, Y.; Xu, X.; Wu, X.; et al. Extracellular mitochondria released from traumatized brains induced platelet procoagulant activity. Haematologica 2020, 105, 209–217. [Google Scholar] [CrossRef]
- Baaten, C.; Moenen, F.; Henskens, Y.M.C.; Swieringa, F.; Wetzels, R.J.H.; van Oerle, R.; Heijnen, H.F.G.; Ten Cate, H.; Holloway, G.P.; Beckers, E.A.M.; et al. Impaired mitochondrial activity explains platelet dysfunction in thrombocytopenic cancer patients undergoing chemotherapy. Haematologica 2018, 103, 1557–1567. [Google Scholar] [CrossRef] [Green Version]
- Heber, S.; Volf, I. Effects of Physical (In)activity on Platelet Function. Biomed. Res. Int. 2015, 2015, 165078. [Google Scholar] [CrossRef]
- Hoppel, F.; Garcia-Souza, L.F.; Kantner-Rumplmair, W.; Burtscher, M.; Gnaiger, E.; Pesta, D.; Calabria, E. Human Platelet Mitochondrial Function Reflects Systemic Mitochondrial Alterations: A Protocol for Application in Field Studies. Cells 2021, 10, 2088. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.P.S.; Chan, W.S.; Chan, C.B. Mitochondria Homeostasis and Oxidant/Antioxidant Balance in Skeletal Muscle-Do Myokines Play a Role? Antioxidants 2021, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Lemes, I.R.; Smaira, F.I.; Ribeiro, W.J.D.; Favero, N.K.; Matos, L.; Pinto, A.L.S.; Dolan, E.; Gualano, B.; Coalition, S.-C. Acute and post-acute COVID-19 presentations in athletes: A systematic review and meta-analysis. Br. J. Sports Med. 2022, 56, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.A.; Boilard, E.; Rondina, M.T. Is there a role for the ACE2 receptor in SARS-CoV-2 interactions with platelets? J. Thromb. Haemost. 2021, 19, 46–50. [Google Scholar] [CrossRef]
- Nazy, I.; Jevtic, S.D.; Moore, J.C.; Huynh, A.; Smith, J.W.; Kelton, J.G.; Arnold, D.M. Platelet-activating immune complexes identified in critically ill COVID-19 patients suspected of heparin-induced thrombocytopenia. J. Thromb. Haemost. 2021, 19, 1342–1347. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Tanriverdi, K.; Somasundaran, M.; Liu, P.; Soofi, S.; Bhandari, R.; Godwin, M.; Parsi, K.M.; et al. SARS-CoV-2 Initiates Programmed Cell Death in Platelets. Circ. Res. 2021, 129, 631–646. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Ouseph, M.M.; Huang, Y.; Banerjee, M.; Joshi, S.; MacDonald, L.; Zhong, Y.; Liu, H.; Li, X.; Xiang, B.; Zhang, G.; et al. Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood 2015, 126, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Thushara, R.M.; Hemshekhar, M.; Basappa; Kemparaju, K.; Rangappa, K.S.; Girish, K.S. Biologicals, platelet apoptosis and human diseases: An outlook. Crit. Rev. Oncol. Hematol. 2015, 93, 149–158. [Google Scholar] [CrossRef]
- Williamson, C.D.; DeBiasi, R.L.; Colberg-Poley, A.M. Viral product trafficking to mitochondria, mechanisms and roles in pathogenesis. Infect. Disord. Drug Targets 2012, 12, 18–37. [Google Scholar] [CrossRef]
- Krishnan, S.; Nordqvist, H.; Ambikan, A.T.; Gupta, S.; Sperk, M.; Svensson-Akusjarvi, S.; Mikaeloff, F.; Benfeitas, R.; Saccon, E.; Ponnan, S.M.; et al. Metabolic perturbation associated with COVID-19 disease severity and SARS-CoV-2 replication. Mol. Cell Proteom. 2021, 20, 100159. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Jeon, M.T.; Kim, K.S.; Lee, S.; Kim, S.; Kim, D.G. Spike Proteins of SARS-CoV-2 Induce Pathological Changes in Molecular Delivery and Metabolic Function in the Brain Endothelial Cells. Viruses 2021, 13, 2021. [Google Scholar] [CrossRef] [PubMed]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.; Rodriguez-Perez, A.I.; Costa-Besada, M.A.; Rivas-Santisteban, R.; Garrido-Gil, P.; Lopez-Lopez, A.; Navarro, G.; Lanciego, J.L.; Franco, R.; Labandeira-Garcia, J.L. An ACE2/Mas-related receptor MrgE axis in dopaminergic neuron mitochondria. Redox Biol. 2021, 46, 102078. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Bihl, J. Exosome-Mediated Transfer of ACE2 (Angiotensin-Converting Enzyme 2) from Endothelial Progenitor Cells Promotes Survival and Function of Endothelial Cell. Oxid Med. Cell. Longev. 2020, 2020, 4213541. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Zimba, O.; Gasparyan, A.Y. Thrombosis in Coronavirus disease 2019 (COVID-19) through the prism of Virchow’s triad. Clin. Rheumatol. 2020, 39, 2529–2543. [Google Scholar] [CrossRef]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Ding, T.; Zuo, Z.; Xu, Z.; Deng, J.; Wei, Z. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front. Immunol. 2020, 11, 1030. [Google Scholar] [CrossRef]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrocola, R.; Aragno, M.; Alloatti, G.; Collino, M.; Penna, C.; Pagliaro, P. Metaflammation: Tissue-Specific Alterations of the NLRP3 Inflammasome Platform in Metabolic Syndrome. Curr. Med. Chem. 2018, 25, 1294–1310. [Google Scholar] [CrossRef] [PubMed]
- Lien, T.S.; Sun, D.S.; Wu, C.Y.; Chang, H.H. Exposure to Dengue Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Endothelial Dysfunction and Hemorrhage in Mice. Front. Immunol. 2021, 12, 617251. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Y.; Li, G.; Feng, Q.; Hou, M.; Peng, J. Reduced intracellular antioxidant capacity in platelets contributes to primary immune thrombocytopenia via ROS-NLRP3-caspase-1 pathway. Thromb. Res. 2021, 199, 1–9. [Google Scholar] [CrossRef]
- Denson, J.L.; Gillet, A.S.; Zu, Y.; Brown, M.; Pham, T.; Yoshida, Y.; Mauvais-Jarvis, F.; Douglas, I.S.; Moore, M.; Tea, K.; et al. Metabolic Syndrome and Acute Respiratory Distress Syndrome in Hospitalized Patients With COVID-19. JAMA Netw. Open. 2021, 4, e2140568. [Google Scholar] [CrossRef]
- Arazi, H.; Falahati, A.; Suzuki, K. Moderate Intensity Aerobic Exercise Potential Favorable Effect Against COVID-19: The Role of Renin-Angiotensin System and Immunomodulatory Effects. Front. Physiol. 2021, 12, 747200. [Google Scholar] [CrossRef]
- Burtscher, J.; Burtscher, M.; Millet, G.P. The central role of mitochondrial fitness on antiviral defenses: An advocacy for physical activity during the COVID-19 pandemic. Redox Biol. 2021, 43, 101976. [Google Scholar] [CrossRef]
- Cerqueira, E.; Marinho, D.A.; Neiva, H.P.; Lourenco, O. Inflammatory Effects of High and Moderate Intensity Exercise-A Systematic Review. Front. Physiol. 2019, 10, 1550. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Willenborg, S.; Sanin, D.E.; Jais, A.; Ding, X.; Ulas, T.; Nuchel, J.; Popovic, M.; MacVicar, T.; Langer, T.; Schultze, J.L.; et al. Mitochondrial metabolism coordinates stage-specific repair processes in macrophages during wound healing. Cell. Metab. 2021, 33, 2398–2414.e2399. [Google Scholar] [CrossRef]
- O’Hearn, M.; Lauren, B.N.; Wong, J.B.; Kim, D.D.; Mozaffarian, D. Trends and Disparities in Cardiometabolic Health Among U.S. Adults, 1999–2018. J. Am. Coll. Cardiol. 2022, 80, 138–151. [Google Scholar] [CrossRef]
- Kolodziej, F.; O’Halloran, K.D. Re-Evaluating the Oxidative Phenotype: Can Endurance Exercise Save the Western World? Antioxidants 2021, 10, 609. [Google Scholar] [CrossRef] [PubMed]
- Nunn, A.V.; Bell, J.D.; Guy, G.W. Lifestyle-induced metabolic inflexibility and accelerated ageing syndrome: Insulin resistance, friend or foe? Nutr.Metab. 2009, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattan, S.I. Mechanisms of hormesis through mild heat stress on human cells. Ann. N. Y. Acad. Sci. 2004, 1019, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Patrick, R.P.; Johnson, T.L. Sauna use as a lifestyle practice to extend healthspan. Exp. Gerontol. 2021, 154, 111509. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.E.; Pasiakos, S.M.; Blesso, C.N.; Fussell, M.A.; Rodriguez, N.R. Divergent Roles of Inflammation in Skeletal Muscle Recovery From Injury. Front. Physiol. 2020, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef]
- Naik, S.; Fuchs, E. Inflammatory memory and tissue adaptation in sickness and in health. Nature 2022, 607, 249–255. [Google Scholar] [CrossRef]
- Santoro, A.; Martucci, M.; Conte, M.; Capri, M.; Franceschi, C.; Salvioli, S. Inflammaging, hormesis and the rationale for anti-aging strategies. Ageing Res. Rev. 2020, 64, 101142. [Google Scholar] [CrossRef]
- Paul, B.D.; Lemle, M.D.; Komaroff, A.L.; Snyder, S.H. Redox imbalance links COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2021, 118, e2024358118. [Google Scholar] [CrossRef]
- Kabacik, S.; Lowe, D.; Fransen, L.; Leonard, M.; Ang, S.-L.; Whiteman, C.; Corsi, S.; Cohen, H.; Felton, S.; Bali, R.; et al. The relationship between epigenetic age and the hallmarks of aging in human cells. Nat. Aging 2022, 2, 484–493. [Google Scholar] [CrossRef]
- Anderson, J.J.; Susser, E.; Arbeev, K.G.; Yashin, A.I.; Levy, D.; Verhulst, S.; Aviv, A. Telomere-length dependent T-cell clonal expansion: A model linking ageing to COVID-19 T-cell lymphopenia and mortality. EBioMedicine 2022, 78, 103978. [Google Scholar] [CrossRef] [PubMed]
- Sepe, S.; Rossiello, F.; Cancila, V.; Iannelli, F.; Matti, V.; Cicio, G.; Cabrini, M.; Marinelli, E.; Alabi, B.R.; di Lillo, A.; et al. DNA damage response at telomeres boosts the transcription of SARS-CoV-2 receptor ACE2 during aging. EMBO Rep. 2022, 23, e53658. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Tang, B.S.; Guo, J.F.; Li, J.C. Telomere Length and COVID-19 Outcomes: A Two-Sample Bidirectional Mendelian Randomization Study. Front. Genet. 2022, 13, 805903. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Martucci, M.; Chiariello, A.; Franceschi, C.; Salvioli, S. Mitochondria, immunosenescence and inflammaging: A role for mitokines? Semin. Immunopathol. 2020, 42, 607–617. [Google Scholar] [CrossRef]
- Simpson, R.J.; Kunz, H.; Agha, N.; Graff, R. Exercise and the Regulation of Immune Functions. Prog. Mol. Biol. Transl. Sci. 2015, 135, 355–380. [Google Scholar] [CrossRef]
- Wu, J.; Weisshaar, N.; Hotz-Wagenblatt, A.; Madi, A.; Ma, S.; Mieg, A.; Hering, M.; Mohr, K.; Schlimbach, T.; Borgers, H.; et al. Skeletal muscle antagonizes antiviral CD8(+) T cell exhaustion. Sci. Adv. 2020, 6, eaba3458. [Google Scholar] [CrossRef]
- Nunn, A.V.; Guy, G.W.; Bell, J.D. The intelligence paradox; will ET get the metabolic syndrome? Lessons from and for Earth. Nutr. Metab. 2014, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Jarrott, B.; Head, R.; Pringle, K.G.; Lumbers, E.R.; Martin, J.H. “LONG COVID”-A hypothesis for understanding the biological basis and pharmacological treatment strategy. Pharmacol. Res. Perspect 2022, 10, e00911. [Google Scholar] [CrossRef]
- Ceban, F.; Leber, A.; Jawad, M.Y.; Yu, M.; Lui, L.M.W.; Subramaniapillai, M.; Di Vincenzo, J.D.; Gill, H.; Rodrigues, N.B.; Cao, B.; et al. Registered clinical trials investigating treatment of long COVID: A scoping review and recommendations for research. Infect. Dis. 2022, 54, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Cash, A.; Kaufman, D.L. Oxaloacetate Treatment For Mental And Physical Fatigue In Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and Long-COVID fatigue patients: A non-randomized controlled clinical trial. J. Transl. Med. 2022, 20, 295. [Google Scholar] [CrossRef] [PubMed]
- Raciti, L.; De Luca, R.; Raciti, G.; Arcadi, F.A.; Calabro, R.S. The Use of Palmitoylethanolamide in the Treatment of Long COVID: A Real-Life Retrospective Cohort Study. Med. Sci. 2022, 10, 37. [Google Scholar] [CrossRef]
- Kahleova, H.; Barnard, N.D. Can a plant-based diet help mitigate COVID-19? Eur. J. Clin. Nutr. 2022, 76, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Nunn, A.V.W.; Guy, G.W.; Botchway, S.W.; Bell, J.D. From sunscreens to medicines: Can a dissipation hypothesis explain the beneficial aspects of many plant compounds? Phytother. Res. 2020, 34, 1868–1888. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.W.; Cirillo, P.M.; Ryan, J.C.; Krigbaum, N.Y.; Badathala, A.; Cohn, B.A. Association of the patterns of use of medications with mortality of COVID-19 infection: A hospital-based observational study. BMJ Open. 2021, 11, e050051. [Google Scholar] [CrossRef]
- Bramante, C.; Ingraham, N.; Murray, T.; Marmor, S.; Hoversten, S.; Gronski, J.; McNeil, C.; Feng, R.; Guzman, G.; Abdelwahab, N.; et al. Observational Study of Metformin and Risk of Mortality in Patients Hospitalized with COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Luo, P.; Qiu, L.; Liu, Y.; Liu, X.L.; Zheng, J.L.; Xue, H.Y.; Liu, W.H.; Liu, D.; Li, J. Metformin Treatment Was Associated with Decreased Mortality in COVID-19 Patients with Diabetes in a Retrospective Analysis. Am. J. Trop. Med. Hyg. 2020, 103, 69–72. [Google Scholar] [CrossRef]
- Xian, H.; Liu, Y.; Rundberg Nilsson, A.; Gatchalian, R.; Crother, T.R.; Tourtellotte, W.G.; Zhang, Y.; Aleman-Muench, G.R.; Lewis, G.; Chen, W.; et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 2021, 54, 1463–1477.e1411. [Google Scholar] [CrossRef] [PubMed]
- Randriamboavonjy, V.; Mann, W.A.; Elgheznawy, A.; Popp, R.; Rogowski, P.; Dornauf, I.; Drose, S.; Fleming, I. Metformin reduces hyper-reactivity of platelets from patients with polycystic ovary syndrome by improving mitochondrial integrity. Thromb. Haemost. 2015, 114, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Casuso, R.A.; Huertas, J.R. Mitochondrial Functionality in Inflammatory Pathology-Modulatory Role of Physical Activity. Life 2021, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Axelrod, C.L.; Reynolds, C.F., 3rd; Greenway, F.L.; Kirwan, J.P. Exercise as a Moderator of Persistent Neuroendocrine Symptoms of COVID-19. Exerc. Sport Sci. Rev. 2022, 50, 65–72. [Google Scholar] [CrossRef]
- Wang, M.; Baker, J.S.; Quan, W.; Shen, S.; Fekete, G.; Gu, Y. A Preventive Role of Exercise Across the Coronavirus 2 (SARS-CoV-2) Pandemic. Front. Physiol. 2020, 11, 572718. [Google Scholar] [CrossRef]
- Torjesen, I. NICE advises against using graded exercise therapy for patients recovering from COVID-19. BMJ 2020, 370, m2912. [Google Scholar] [CrossRef]
- Salman, D.; Vishnubala, D.; Le Feuvre, P.; Beaney, T.; Korgaonkar, J.; Majeed, A.; McGregor, A.H. Returning to physical activity after COVID-19. BMJ 2021, 372, m4721. [Google Scholar] [CrossRef]
- Ji, L.L.; Dickman, J.R.; Kang, C.; Koenig, R. Exercise-induced hormesis may help healthy aging. Dose Response 2010, 8, 73–79. [Google Scholar] [CrossRef]
- Gubert, C.; Hannan, A.J. Exercise mimetics: Harnessing the therapeutic effects of physical activity. Nat. Rev. Drug Discov. 2021, 20, 862–879. [Google Scholar] [CrossRef]
- Guerrieri, D.; Moon, H.Y.; van Praag, H. Exercise in a Pill: The Latest on Exercise-Mimetics. Brain Plast. 2017, 2, 153–169. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Sokolowski, A.A.; Graier, W.F. Dosis Facit Sanitatem-Concentration-Dependent Effects of Resveratrol on Mitochondria. Nutrients 2017, 9, 1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoli, A.; Gorini, S.; Caprio, M. The dark side of the spoon—Glucose, ketones and COVID-19: A possible role for ketogenic diet? J. Transl. Med. 2020, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Hannan, M.A.; Rahman, M.A.; Rahman, M.S.; Sohag, A.A.M.; Dash, R.; Hossain, K.S.; Farjana, M.; Uddin, M.J. Intermittent fasting, a possible priming tool for host defense against SARS-CoV-2 infection: Crosstalk among calorie restriction, autophagy and immune response. Immunol. Lett. 2020, 226, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Chung, H.Y. The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients 2019, 11, 2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabande-Rodriguez, E.; Gomez de Las Heras, M.M.; Mittelbrunn, M. Control of Inflammation by Calorie Restriction Mimetics: On the Crossroad of Autophagy and Mitochondria. Cells 2019, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Mehmel, M.; Jovanovic, N.; Spitz, U. Nicotinamide Riboside-The Current State of Research and Therapeutic Uses. Nutrients 2020, 12, 1616. [Google Scholar] [CrossRef]
- Sack, M.N.; Finkel, T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb. Perspect. Biol. 2012, 4, a013102. [Google Scholar] [CrossRef] [Green Version]
- Koyuncu, E.; Budayeva, H.G.; Miteva, Y.V.; Ricci, D.P.; Silhavy, T.J.; Shenk, T.; Cristea, I.M. Sirtuins are evolutionarily conserved viral restriction factors. mBio 2014, 5, e02249-14. [Google Scholar] [CrossRef] [Green Version]
- Heer, C.D.; Sanderson, D.J.; Voth, L.S.; Alhammad, Y.M.O.; Schmidt, M.S.; Trammell, S.A.J.; Perlman, S.; Cohen, M.S.; Fehr, A.R.; Brenner, C. Coronavirus infection and PARP expression dysregulate the NAD metabolome: An actionable component of innate immunity. J. Biol. Chem. 2020, 295, 17986–17996. [Google Scholar] [CrossRef]
- Shenoy, S. Coronavirus (COVID-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1077–1085. [Google Scholar] [CrossRef]
- Hamblin, M.R. Mechanisms and applications of the anti-inflammatory effects of photobiomodulation. AIMS Biophys. 2017, 4, 337–361. [Google Scholar] [CrossRef] [PubMed]
- Amaroli, A.; Pasquale, C.; Zekiy, A.; Utyuzh, A.; Benedicenti, S.; Signore, A.; Ravera, S. Photobiomodulation and Oxidative Stress: 980 nm Diode Laser Light Regulates Mitochondrial Activity and Reactive Oxygen Species Production. Oxidative Med. Cell. Longev. 2021, 2021, 6626286. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.A.; Molena, K.F.; Martins, C.; Corona, S.A.M.; Borsatto, M.C. Photobiomodulation (PBMT) and antimicrobial photodynamic therapy (aPDT) in oral manifestations of patients infected by SARS-CoV-2: Systematic review and meta-analysis. Bull. Natl. Res. Cent. 2022, 46, 140. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S.; Huang, S.C.; Searby, C.C.; Cassaidy, B.; Miller, M.J.; Grzesik, W.J.; Piorczynski, T.B.; Pak, T.K.; Walsh, S.A.; Acevedo, M.; et al. Exposure to Static Magnetic and Electric Fields Treats Type 2 Diabetes. Cell Metab. 2020, 32, 1076. [Google Scholar] [CrossRef]
- Fels, D.; Cifra, M.; Scholkmann, F.E. Fields of the Cell; Fels, D., Cifra, M., Scholkmann, F., Eds.; Research Signpost: Thiruvananthapuram, India, 2015. [Google Scholar]
- Park, A.; Oh, M.; Lee, S.J.; Oh, K.J.; Lee, E.W.; Lee, S.C.; Bae, K.H.; Han, B.S.; Kim, W.K. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 4793. [Google Scholar] [CrossRef]
- Matsuyama, T.; Yoshinaga, S.K.; Shibue, K.; Mak, T.W. Comorbidity-associated glutamine deficiency is a predisposition to severe COVID-19. Cell Death Differ. 2021, 28, 3199–3213. [Google Scholar] [CrossRef]
- Rogeri, P.S.; Gasparini, S.O.; Martins, G.L.; Costa, L.K.F.; Araujo, C.C.; Lugaresi, R.; Kopfler, M.; Lancha, A.H., Jr. Crosstalk between Skeletal Muscle and Immune System: Which Roles Do IL-6 and Glutamine Play? Front. Physiol. 2020, 11, 582258. [Google Scholar] [CrossRef]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Carvalho, E.; Diaz, E.C.; Cotter, M.; Bennuri, S.C.; Azhar, G.; Frye, R.E.; Adams, S.H.; Borsheim, E. A comparative study of mitochondrial respiration in circulating blood cells and skeletal muscle fibers in women. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E503–E512. [Google Scholar] [CrossRef]
- DeConne, T.M.; Munoz, E.R.; Sanjana, F.; Hobson, J.C.; Martens, C.R. Cardiometabolic risk factors are associated with immune cell mitochondrial respiration in humans. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H481–H487. [Google Scholar] [CrossRef]
- Hoppel, F.; Calabria, E.; Pesta, D.H.; Kantner-Rumplmair, W.; Gnaiger, E.; Burtscher, M. Effects of Ultramarathon Running on Mitochondrial Function of Platelets and Oxidative Stress Parameters: A Pilot Study. Front. Physiol. 2021, 12, 632664. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, P.M.; Kalinina, S.; Rueck, A.; von Arnim, C.A.F.; von Einem, B. NADH Autofluorescence-A Marker on its Way to Boost Bioenergetic Research. Cytom. A 2019, 95, 34–46. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunn, A.V.W.; Guy, G.W.; Brysch, W.; Bell, J.D. Understanding Long COVID; Mitochondrial Health and Adaptation—Old Pathways, New Problems. Biomedicines 2022, 10, 3113. https://doi.org/10.3390/biomedicines10123113
Nunn AVW, Guy GW, Brysch W, Bell JD. Understanding Long COVID; Mitochondrial Health and Adaptation—Old Pathways, New Problems. Biomedicines. 2022; 10(12):3113. https://doi.org/10.3390/biomedicines10123113
Chicago/Turabian StyleNunn, Alistair V. W., Geoffrey W. Guy, Wolfgang Brysch, and Jimmy D. Bell. 2022. "Understanding Long COVID; Mitochondrial Health and Adaptation—Old Pathways, New Problems" Biomedicines 10, no. 12: 3113. https://doi.org/10.3390/biomedicines10123113