1. Introduction
There is increasing interest in the exploitation of various mucosal routes such as the vaginal mucosa for the delivery of drugs, which are poorly absorbed via the gastrointestinal route or where local rather than systemic action is required. The vaginal environment and its physiological characteristics possess structures and specific features that help in sexual arousal and also play a role in reproduction [
1]. The vagina is characterized by a dense network of blood vessels and a large surface area, which provides an ideal route for administering drugs to achieve therapeutic action, both locally and systemically [
2].
To achieve systemic bioavailability, different transport (transcellular and intracellular) pathways, as well as receptor-mediated mechanisms, are involved. The key advantages of delivering drugs via the vaginal route compared to the gastrointestinal tract and skin include by-passing the first-pass metabolic effect of the liver, easy administration, and higher rates of permeation for drugs with low molecular weight [
3]. However, the vaginal mucosa has not been extensively targeted due to wide individual variations that affect important parameters such as pH, as well as limited vaginal secretions that vary depending on factors such as age and the menstrual cycle. Other challenges that need to be taken into consideration when designing vaginal formulations include cultural sensitivities, hygiene, gender specificity, irritation, and the effects of intercourse. The rate and extent of absorption of drugs delivered via the vaginal route are significantly affected by changes in the thickness of the vaginal epithelium, which are caused by changes in hormone levels during the menstrual cycle. In addition, variations in endopeptidase and aminopeptidase activities cause difficulties in sustaining absorption of the administered drug [
4].
The vaginal mucosa has also been shown to be a suitable site for delivering drugs that act locally within the vagina, including steroids, prostaglandins, and spermicidal, antibacterial, antiprotozoal, antifungal, and antiviral agents [
5]. This notwithstanding, there are only a limited number of vaginal dosage forms currently available on the market. Vaginal formulations, such as tablets, irrigations, foams, and creams, present various disadvantages, including leaking, which causes messiness. Further, these formulations have a short residence time owing to the ability of the vaginal tract to cleanse itself, thus requiring repeated daily administration of the drug to achieve the required pharmacological action [
6]. Pessaries are ovoid-shaped solid dosage forms specifically designed for vaginal administration and prepared by melting, molding, and compression. Though they are characterized by ease of production and a low cost, the inconvenience of vaginal pessaries, such as messiness upon application, poor retention in the vagina, and poor stability, are key limitations. Administered vaginal pessaries dissolve in the vaginal fluids or melt at body temperature, resulting in the rapid release of drugs; therefore, more sustained-release formulations have been proposed to circumvent this problem [
7].
Various approaches for developing innovative vaginal formulations that could satisfy the needs of both patients and clinicians have been undertaken [
8]. Considerable attention has been directed towards developing formulations that can control the release of a loaded drug, which can afford high amounts of a drug at therapeutic levels, and that allow for prolonged drug release, and therefore result in the need for only a single dose. Novel systems include liposomes [
9], vaginal rings [
10], cubic gels [
11], and systems designed using polystyrene and silicone-based elastomers [
8]. One group of auxiliary agents that is important for successful vaginal delivery is the mucoadhesive polymer group, which now underpins the design and development of novel systems. In recent years, attention has been directed towards various polymers that are used in the form of hydrogels to impart bioadhesive properties so that the formulations remain in the vaginal cavity long enough to elicit the intended therapeutic action. Subsequently, bioadhesive vaginal formulations have been designed that allow drug release at a controlled rate for treating both local and systemic diseases [
12].
Common bioadhesive materials include natural (e.g., chitosan, carrageenan, sodium alginate, and cellulose derivatives) as well as synthetic (e.g., Carbopol) polymers. The vaginal mucus forms a cross-linked interaction with bioadhesive polymers, which causes the formulation to swell and to release the active substance in a controlled manner to achieve the desired bioavailability [
13]. In addition, some bioadhesive polymers are more effective than others depending on their properties, such as their interactions with cervical mucus or the vaginal pH. Therefore, in the formulation of vaginal drug-delivery systems, the characteristics of the polymer, the vaginal environment, and the dosage form characteristics must be taken into consideration [
14]. Various marketed products have used bioadhesive delivery systems for treatments, using polymers with ideal properties such as a low cost of production, avoidance of the use of solvents, ease of self-administration without using an applicator, the maintenance of structural integrity when hydrated, the absence of local irritation, quick mucoadhesion, the ability to remain in the vaginal cavity for long periods even with no physiological secretions, a reduced dosing frequency, and improved chemical and physical stability [
13]. Among the natural biopolymers, chitosan is one of the most used in this regard due to its functional properties such as biocompatibility and ease of biodegradation, as well as a strong bioadhesive behavior. The latter property is attributed to its cationic nature, which allows strong ionic bond formation with mucosal surfaces [
15], and this can be further enhanced via derivatization using side chains such as thiol groups. For vaginal drug delivery, chitosan’s known biological (antimicrobial and anti-inflammatory) actions make it a suitable candidate to use as a drug-delivery platform for treating local vaginal infections [
16]. Fitaihi et al [
17] reported on the formulation of chitosan-based vaginal tablets loaded with fluconazole. For synthetic polymers, nonoxynol-9, which is a spermicidal agent, has been formulated as a gel using Carbopol 934P as the bioadhesive polymer and succeeded in releasing high amounts of the drug within 2 min after insertion and then maintaining a controlled release over 6 h [
18].
Microorganisms such as bacteria occur naturally in various parts of the body, including mucosal cell linings such as the mouth and vagina. Commonly occurring bacterial strains in the vaginal cavity include
Gardnerella,
Lactobacilli,
Bacteroides,
Mobiluncus,
Mycoplasma, and
Streptococci. The uncontrolled growth of these bacterial strains results in itchiness, smelly discharge, and painful intercourse, and this form of vaginal infection is known as bacterial vaginosis [
19]. Vaginal infections are commonly treated using antifungal and antibacterial agents formulated in the form of tablets, gels, foams, powders, and creams. However, most of these formulations have limited residence time in the vaginal tract and therefore require an increased drug dose and increased application frequency. Based on these challenges, vaginal delivery systems must be developed and optimized to improve the retention of the dosage form and loaded drug in the genital tract over a longer period.
This study therefore involved the development and optimization of freeze-dried polymer-based wafers as platforms for delivering metronidazole via the vaginal mucosa with the aim of prolonging the residence time to achieve local therapeutic effects for the treatment of bacterial vaginosis.
2. Materials and Methods
2.1. Materials
Carbopol 974P NF and carrageenan (Gelcarin GP 379) NF were donated by IMCD Ltd. (Sutton, UK); polyethylene glycol 200, sodium alginate W201502, mucin from porcine stomach, urea, gelatin, and calcium hydroxide were purchased from Sigma-Aldrich (Gillingham, UK). Sodium chloride, potassium hydroxide, lactic acid, glucose, hydroxypropylmethyl cellulose, metronidazole (99%), bovine serum albumin, ethanol, acetic acid, acetonitrile, and glycerol were obtained from Fisher Scientific (Loughborough, UK).
2.2. Formulation Development of Wafers
2.2.1. Blank (BLK) Wafers
Composite formulations were prepared using Carbopol (CARB) as the base polymer in all cases. CARB was mixed with sodium alginate (SA) or carrageenan (CARR) at different ratios and concentrations, while polyethylene glycol 200 (PEG) and hydroxypropylmethyl cellulose (HPMC) were also added to some of the composite formulations as modifiers to improve their physical properties as part of the formulation development process (
Table 1,
Table 2 and
Table 3). Gels were prepared by first dissolving CARB in deionized water, and the other polymers were then added slowly to avoid lump formation. Once homogeneous gels were obtained, 3 g of each formulation was poured into Eppendorf tubes, which were placed on a rack and loaded into a freeze-drying machine. The wafers were prepared by freeze-drying the gels using a Virtis Advantage XL 70 freeze-dryer (Biopharma Process System, Winchester, UK). The freeze-dryer was programmed to undergo freezing, primary drying, and secondary drying processes in automatic mode.
2.2.2. Drug-Loaded (DL) Wafers
The formulations for drug loading were selected based on the results of a physico-chemical characterization (especially hardness). The selected optimized formulations for drug loading were: (i) 6% CARB:CARR 1:3 + 1% HPMC (W14), (ii) 6% CARB:CARR 1:1 + 1% HPMC (W16), (iii) 8% CARB:SA 1:2 + 8% PEG (W20), (iv) 8% CARB:SA 1:4 (W10), and (v) 8% CARB:SA 1:9 (W13). The gels were prepared by first dissolving metronidazole (MTz) in deionized water, and then the polymers were added slowly to avoid lump formation. The composition of the MTz-loaded wafers has been summarized in
Table 4. Wafers were obtained by freeze-drying the gels in Eppendorf tubes using the automated lyophilization cycle described for the BLK wafers above.
2.3. Physico-Chemical Characterization
2.3.1. Mechanical Properties
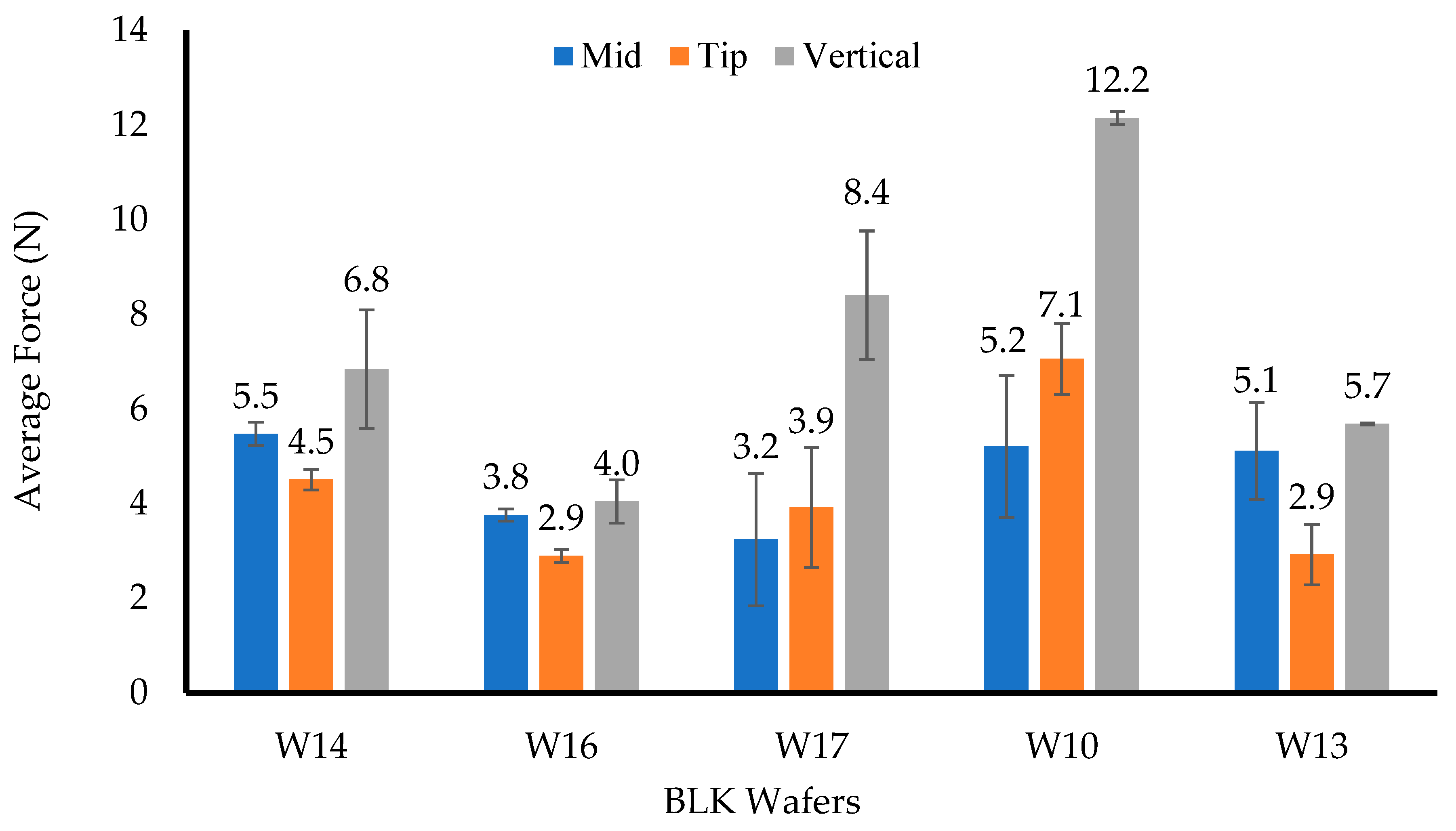
The resistance to compressive deformation (hardness) was evaluated under ambient conditions using a TA HD Texture Analyser (Stable Microsystem Ltd., Surrey, UK) with a load cell of 5 kg. The results were processed using the Texture Exponent 32 software program (Stable Microsystem Ltd., Surrey, UK). The hardness was measured by compressing the formulations (n = 3) in the middle, at the tip, and vertically with the help of a 6 mm-diameter (P/6) probe using a trigger force of 0.001 N, a penetration depth of 2 mm, a test speed of 1 mm/s, and a return distance of 10 mm.
2.3.2. Adhesive Properties
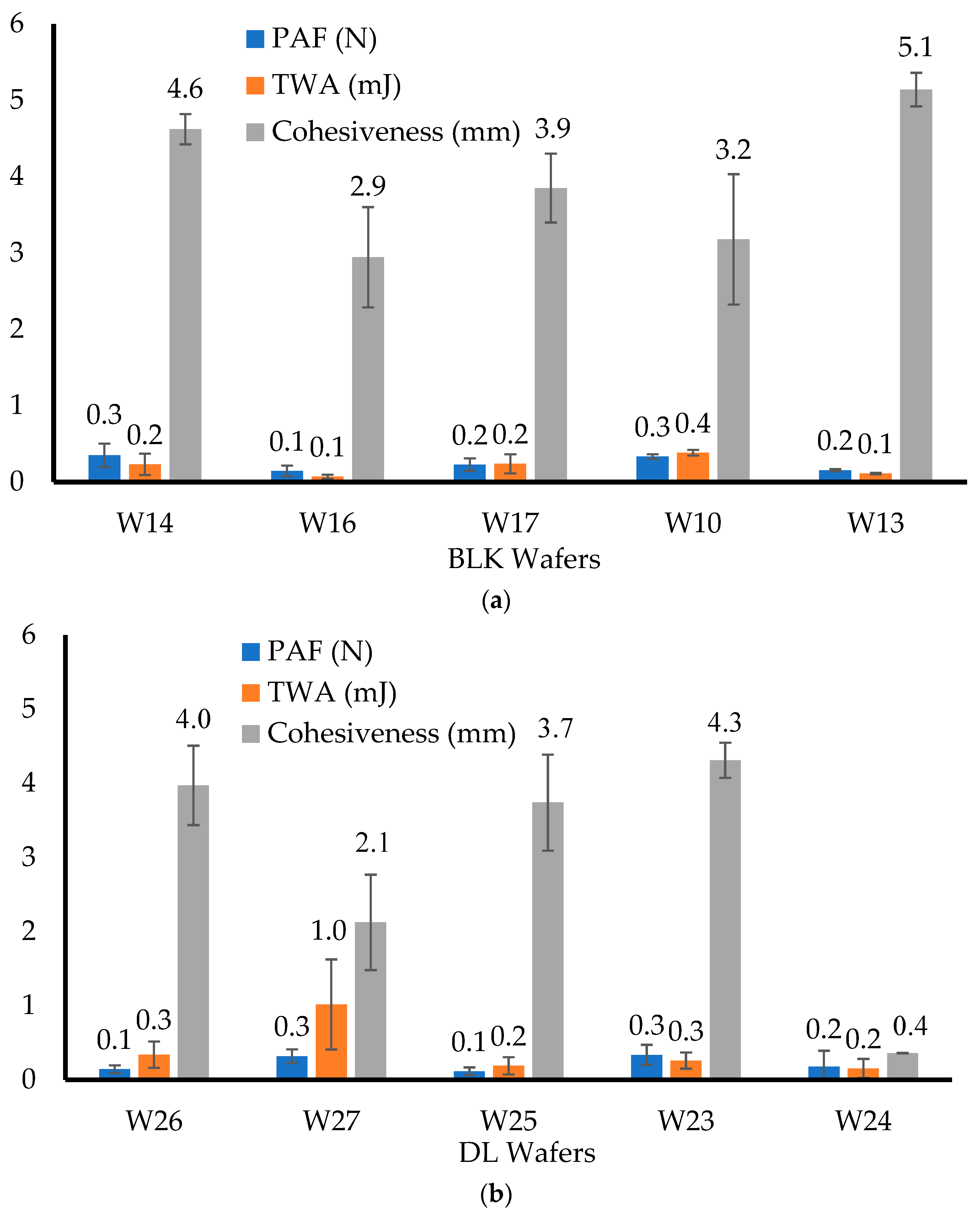
The mucoadhesive properties (peak adhesive force (PAF); total work of adhesion (TWA) and cohesiveness) of the lyophilized wafers were investigated using the same Texture Analyser mentioned above, but in tension mode. The wafers (n = 3) were attached to an adhesion rig (75 mm in diameter) using double-sided adhesive tape. The mucoadhesive surface was prepared by pouring 20 g of hot gelatin gel (6.67% w/w) into a Petri dish (90 mm diameter) and allowed to set by cooling. The surface of the set gelatin was then equilibrated with simulated vaginal fluid (SVF) (0.5 mL) to represent the vaginal mucosa. The probe was lowered until the attached wafer contacted the gelatin surface and was kept in place for 60 s using a force of 1 N to ensure proper contact. The probe was detached using pre-test and test speeds of 0.5 mm/s and post-test speed of 1 mm/s, a 0.05 N trigger force, and a 10 mm return distance. The force required to break the adhesive bond between the wafer and gelatin surface was determined by the maximum force (PAF). The TWA was obtained by calculating the area under the force–distance curve while the cohesiveness was the total distance (in mm) travelled by the probe before the wafer was completely detached from the gelatin surface. Each formulation was tested three times (n = 3).
2.3.3. Dislodge Test
The dislodge test was used to estimate how long the wafers would stay in the vagina before detachment from the vaginal wall. An amount of 10 mL of SVF solution was maintained at 37 °C. The wafer was gently removed from the Eppendorf tube using a pair of tweezers. A syringe was used to transfer mucin solution (1 mL) into the Eppendorf tube, followed by 3–4 droplets of SVF solution. The Eppendorf tube was closed and shaken to allow the SVF and mucin solution to coat and wet the inside of the tube, after which the wafer was placed back into the moistened Eppendorf tube. The Eppendorf tube was inverted vertically with its cap opened and placed on a flat surface, and the narrow tip was cut off to allow for fluid to be added. At specific time intervals (1 h), 2 to 3 drops of SVF solution were introduced into the Eppendorf tube through the cut tip and regularly observed to determine when the hydrated wafer dislodged from the tube. Each formulation was tested in triplicate (n = 3).
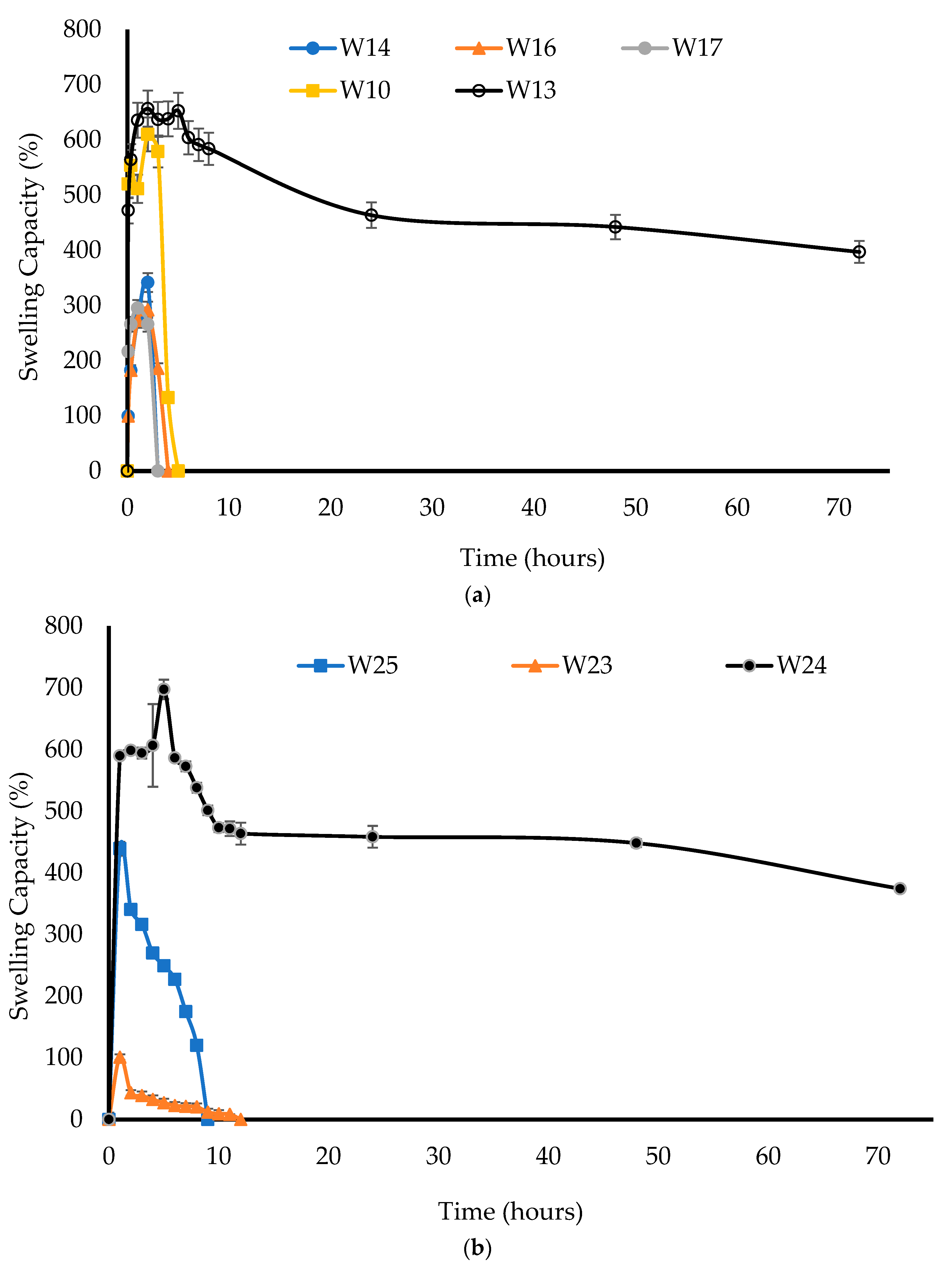
2.3.4. Swelling (Hydration) Capacity
The swelling capacity was measured to investigate the hydration and swelling ability of each formulation. SVF was heated to 37 °C to mimic the temperature of vaginal fluid and approximately 20 mL was transferred into a pre-weighed Petri dish. The wafers were weighed (dry weight) and then placed into the Petri dish containing SVF. The wafers and Petri dishes were then reweighed after removing the free SVF and swabbing the Petri dishes with paper towel to remove any excess SVF at 0.5, 1, 2, 3, 6, 24, 48, and 72 h intervals until total disintegration. After each weighing of the Petri dishes containing the swollen wafer, a fresh solution of SVF was added. The percentage water uptake (swelling capacity) (
n = 3) was calculated using Equation (1) [
20]:
where Ws is the weight of the hydrated wafer and W is the initial weight of the wafer.
2.3.5. Scanning Electron Microscopy (SEM)
A Hitachi UHR FE-SEM SU8030 microscope (Hitachi High-Technologies, Krefeld, Germany) was used to evaluate the surface morphology of the formulations. The wafers were cut into small slices and placed onto aluminum pin-type stubs (G301) (Agar Scientific Limited, Stansted, UK) using double-sided adhesive carbon tape (G3347N) (Agar Scientific Limited, Stansted, UK). Each sample was coated with gold using an Edwards 188 Sputter Coater S1508 (Leica Microsystems (UK) Ltd., Milton Keynes, UK) and analyzed at an accelerating voltage of 5.0 kV [
21]. The emission was set to 10 µA with normal probe current, and 5 condensers were used with the working distance (Z height) set to 8 mm.
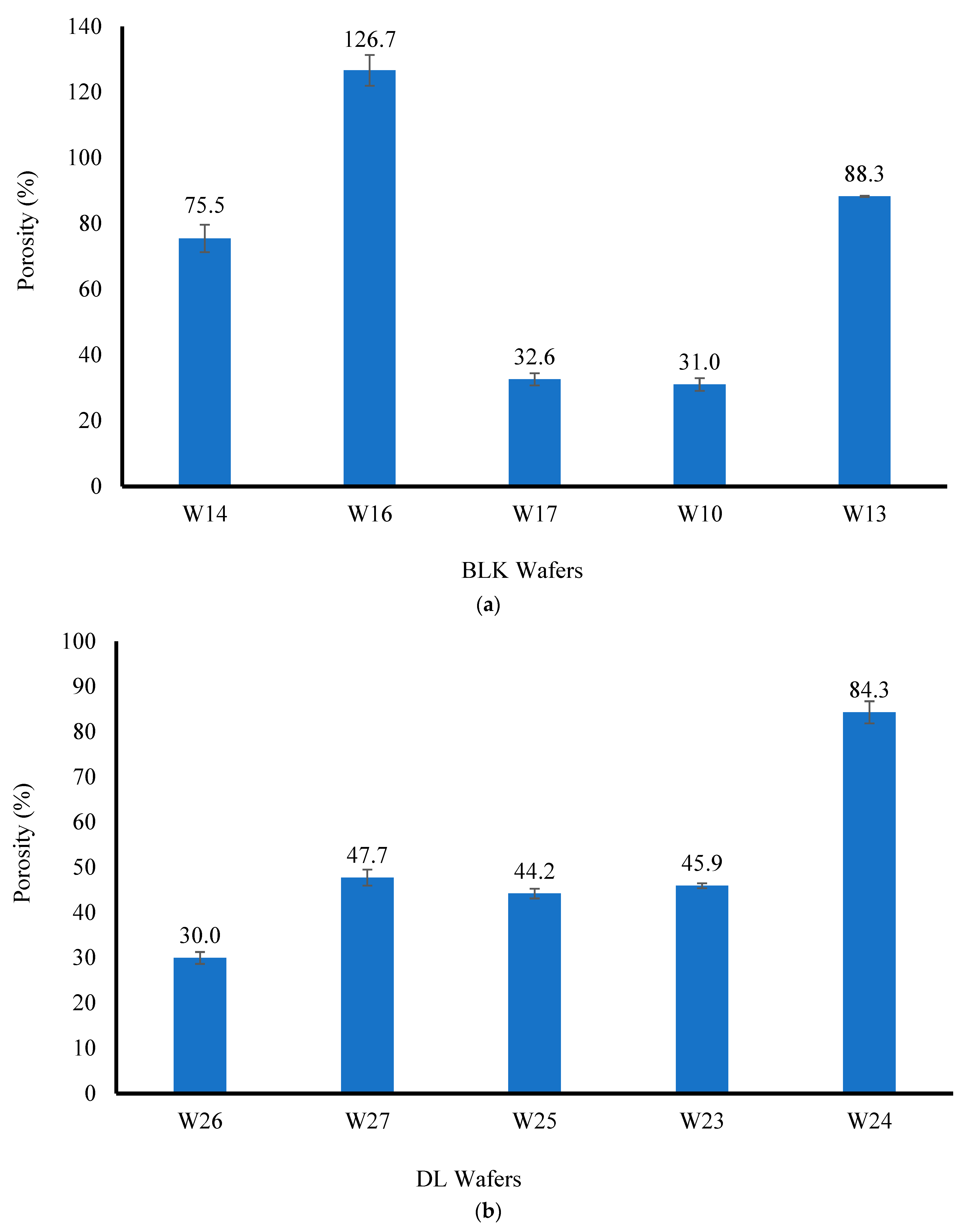
2.3.6. Porosity
The solvent displacement method was employed to determine the porosity of the wafer micro-structure. The wafers were accurately weighed and the weighed sample was subsequently immersed in absolute ethanol (20 mL) in a 50 mL Falcon tube. The set up was left for 3 h to ensure complete saturation and then degassed to remove all air bubbles from the wafer formulations. The wet samples were removed from the solvent, wiped gently with tissue paper to remove excess solvent, and immediately re-weighed to avoid the loss of ethanol via evaporation. The porosity (%) of each wafer formulation (
n = 3) was calculated using Equation (2) [
22]:
where:
Vg = wafer’s geometrical volume;
Wf = final weight of wafer;
Wi = initial weight of wafer;
ρe = ethanol density (0.789 g/cm3).
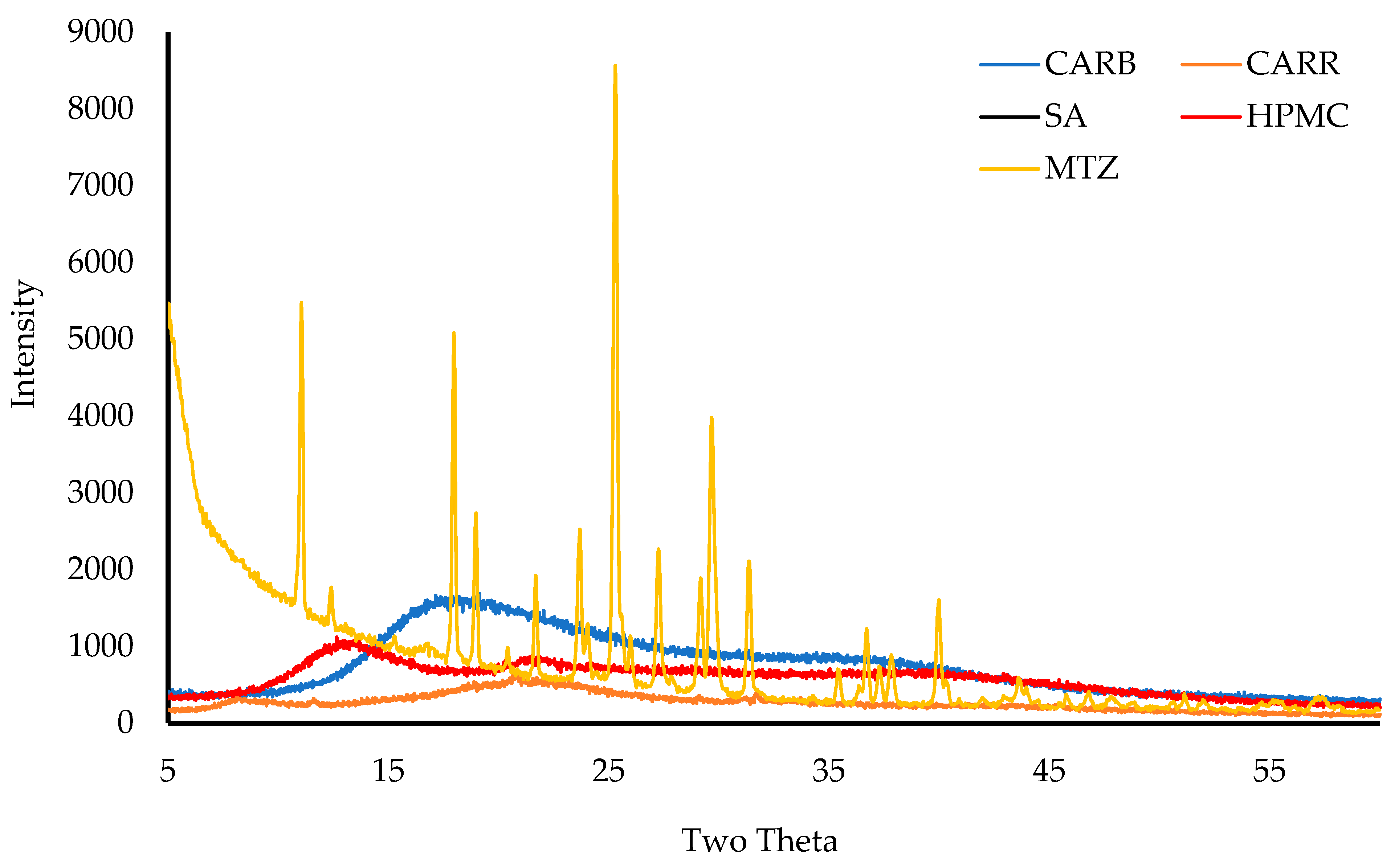
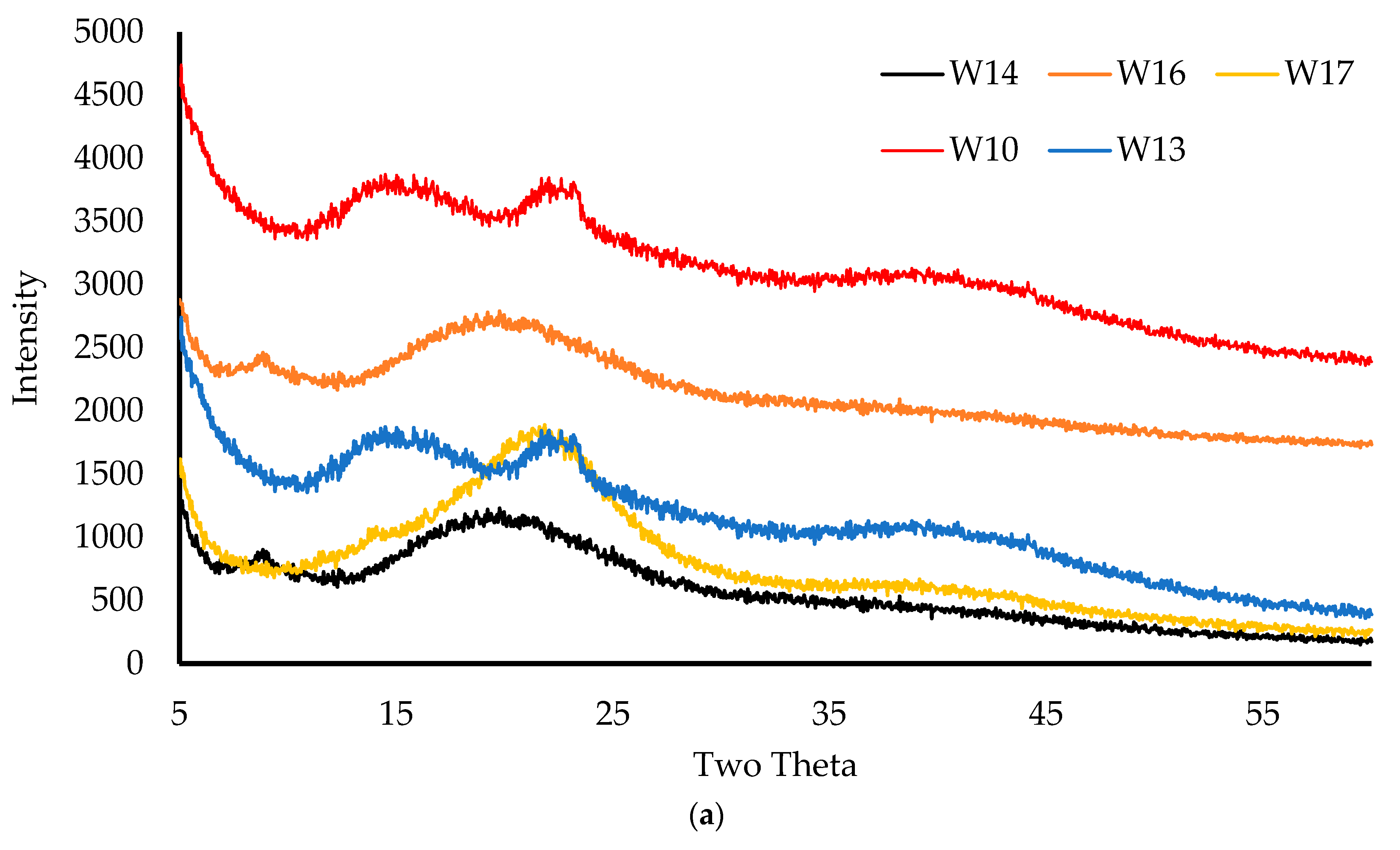
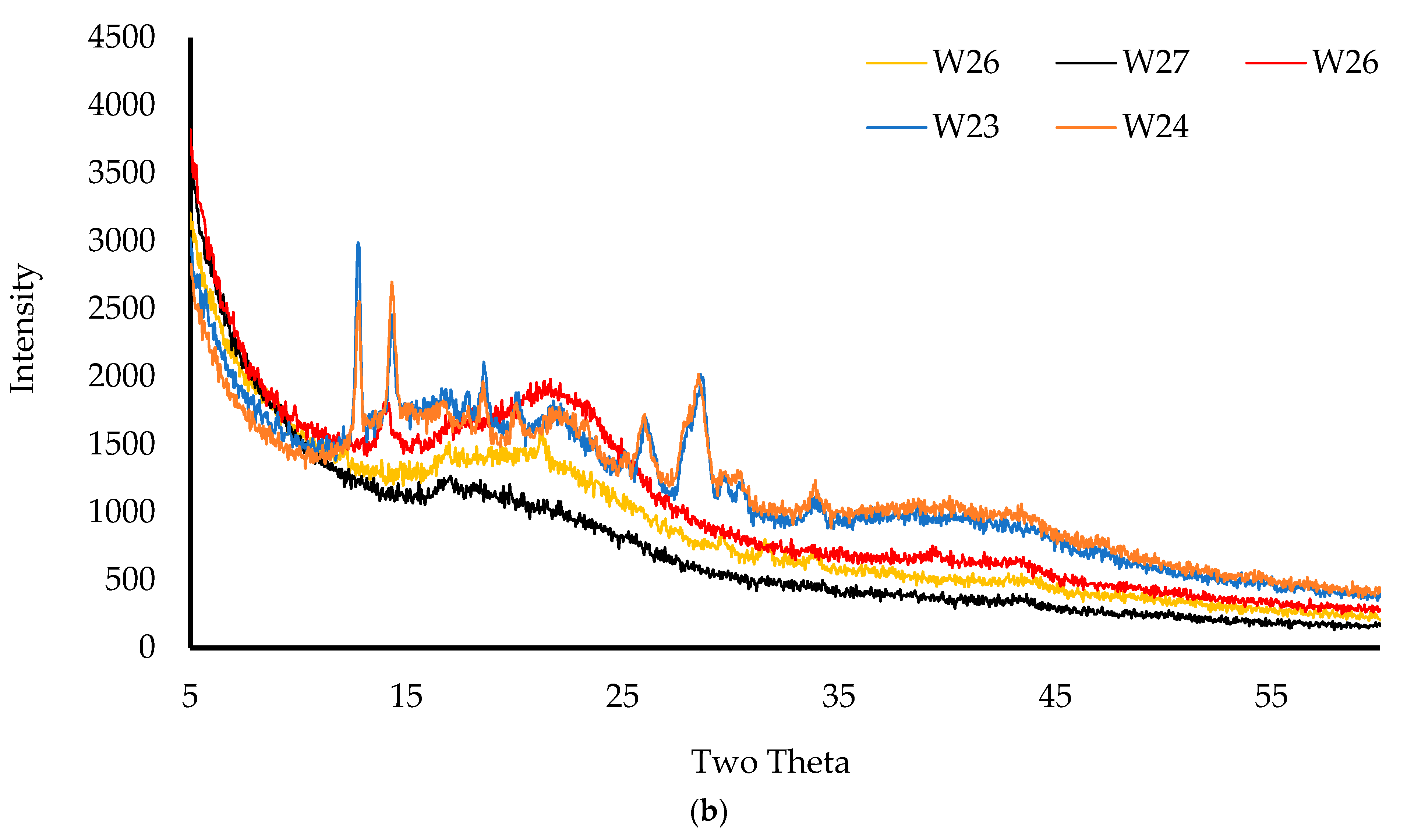
2.3.7. X-ray Diffraction (XRD)
The physical form (crystalline/amorphous) of the pure starting materials and wafers was analyzed on a D8 Advance X-ray Diffractometer (Billerica, MA, USA). Before analyzing, the wafers were compressed using two clean glasses, placed in the holder, and mounted on the sample cell. For starting materials, the powders were held together using mylar before placement on the sample cell. The samples were analyzed in transmission mode with a diffraction angle range of 5° to 50° 2θ, a 0.04° step size, and a scan speed of 0.4 s/step [
21].
2.3.8. Attenuated Total Reflectance Fourier-Transform Infrared (ATR-FTIR) Spectroscopy
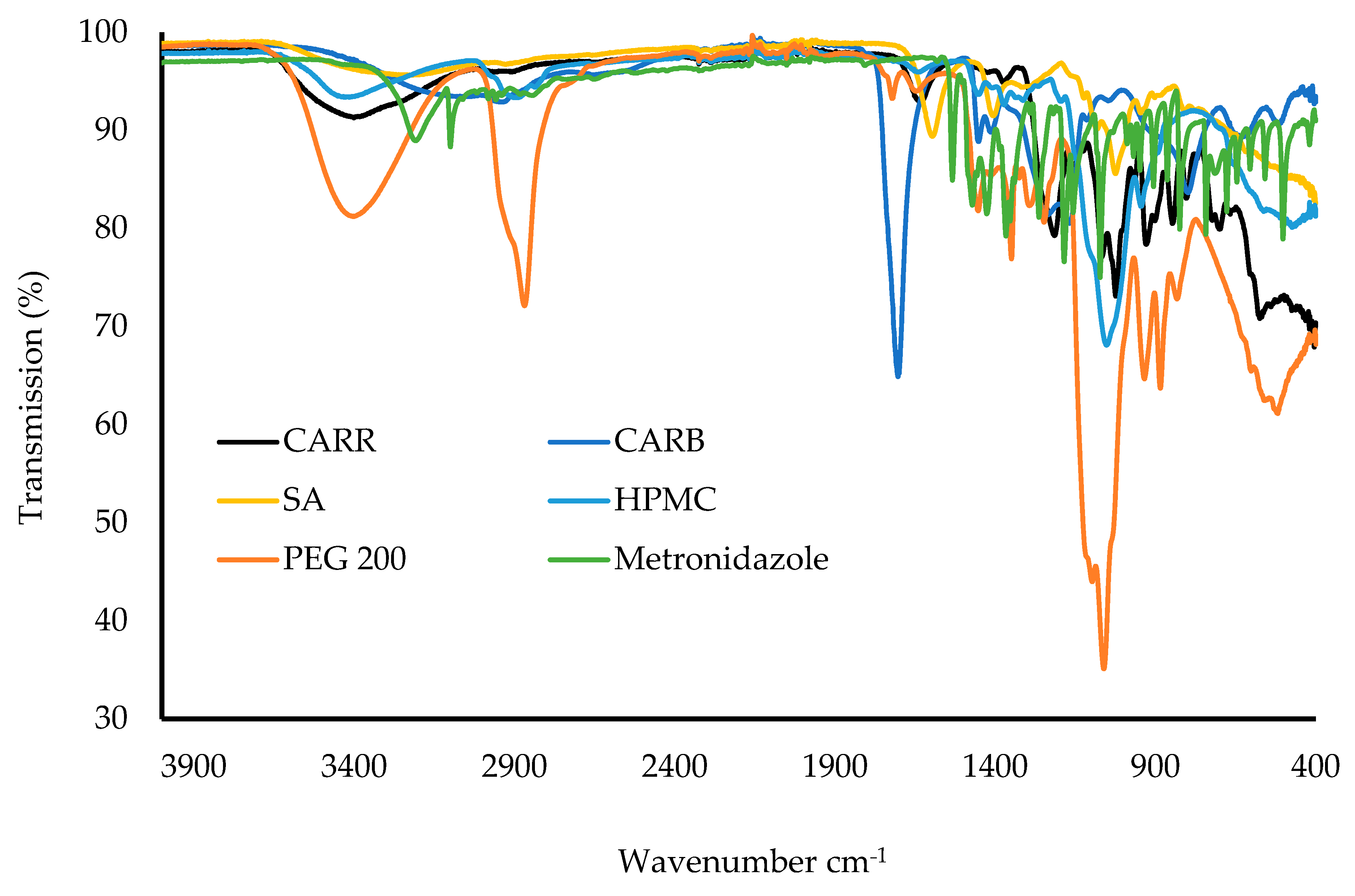
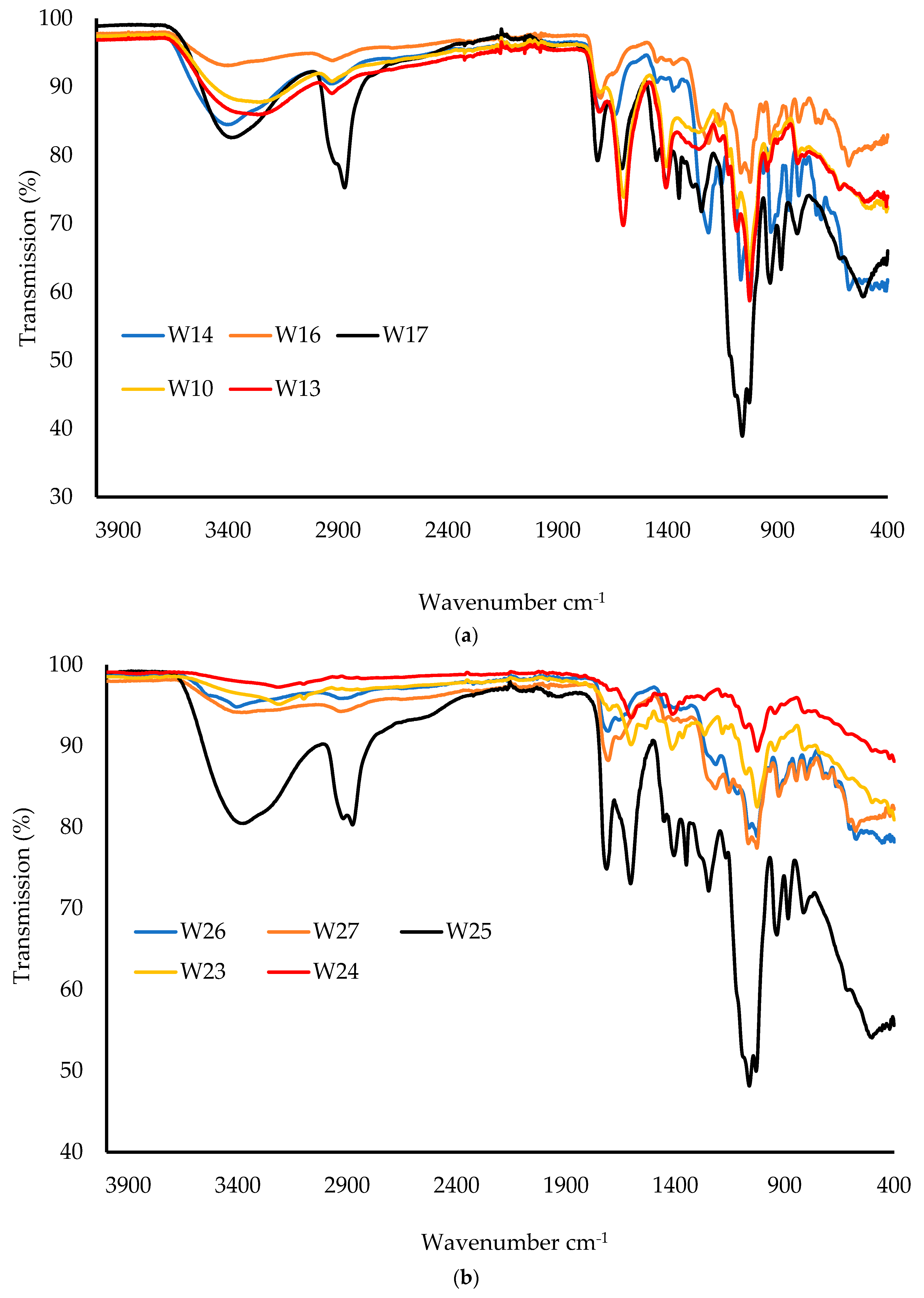
Samples were analyzed for their FTIR spectral profiles using a Perkin Elmer Spectrum instrument (Waltham, MA, USA) equipped with a diamond universal ATR unit. The wafers were cut into small pieces whilst the starting materials were used in powder form, and the samples were placed on the ATR diamond crystal. A pressure clamp was used to apply force on each sample to ensure adequate contact between the diamond crystal and the samples. The FTIR spectra were recorded for each sample (wafer and pure starting material) using a resolution of 4 cm−1 within the range of 400–4000 cm−1. Background spectra were subtracted to obtain a consistent absorbance for each sample.
2.4. In Vitro Drug Dissolution and Release Profiles
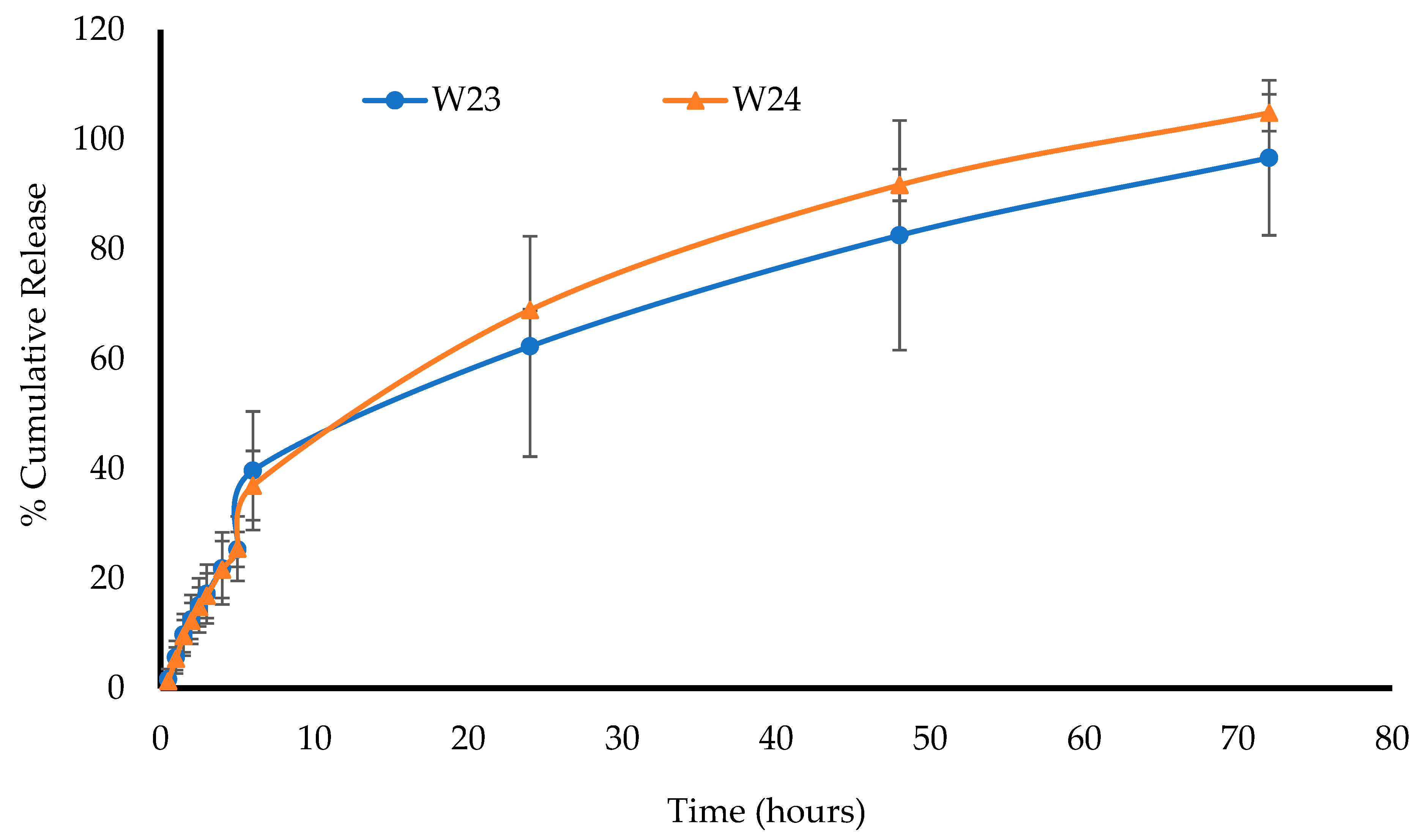
The in vitro drug dissolution of the MTz-loaded wafers was performed using an Agilent BIO-DIS reciprocating cylinder cell apparatus (Agilent Technologies, Cheshire, UK). The formulation was covered with a dialysis membrane with MWCO 3.5 KDa and a diameter of 29 mm and placed in chambers containing wire meshes with a pore size of 0.4 mm. The cylindrical compartment was filled with 200 mL of SVF at a pH of 4.2 ± 0.1 with the mesh on the receptor surface. The system was placed in a water bath at 37 °C and the automated chambers were dipped into the SVF at a rate of 5 dips/min. Aliquots (2 mL) of the SVF were withdrawn at predetermined time intervals using a 3 mL syringe (and replaced with the same volume of fluid at 37 °C), filtered through a 0.45 μm cellulose acetate membrane, transferred into glass vials, and analyzed by high-performance liquid chromatography (HPLC). The percentage of cumulative drug released from the wafers (n = 3) at each time point was calculated and plotted against time.
2.4.1. HPLC Analysis
The saturation solubility of the drug within the DL wafers was investigated to determine the minimum volume of SVF needed for drug dissolution, to ensure sink conditions. This was carried out by dissolving a whole DL wafer in 50 mL of SVF for 48 h and then injecting onto the HPLC column. By using the standard calibration equation, the minimum volume of SVF needed for the drug dissolution test was calculated. The MTz released during the drug dissolution was analyzed using an Agilent 1200 HPLC system equipped with an auto sampler and data analyses were performed using a Chemstation® software program (Agilent Technologies, Cheshire, UK). The stationary phase used was a HICROM S10-0DS1-3634 column (250 × 4.6 mm) with a 10 μm particle size (Waters, Leicestershire, UK). The mobile phase consisted of a mixture of acetonitrile and 0.01% trifluoroacetic acid (TFA) in the ratio 80:20 v/v. The flow rate of the mobile phase was maintained at 1.5 mL/min and the wavelength for MTz detection was set at 315 nm. A volume of 5 μL was injected during each run with a column temperature of 60 °C.
Eight calibration solutions (0.08 to 1.0 mg/mL) were prepared, analyzed in triplicate, and used to plot the linear calibration curve. The intraday accuracy and precision of the assay were evaluated by analyzing three replicates of the standard MTz calibration solutions at each concentration.
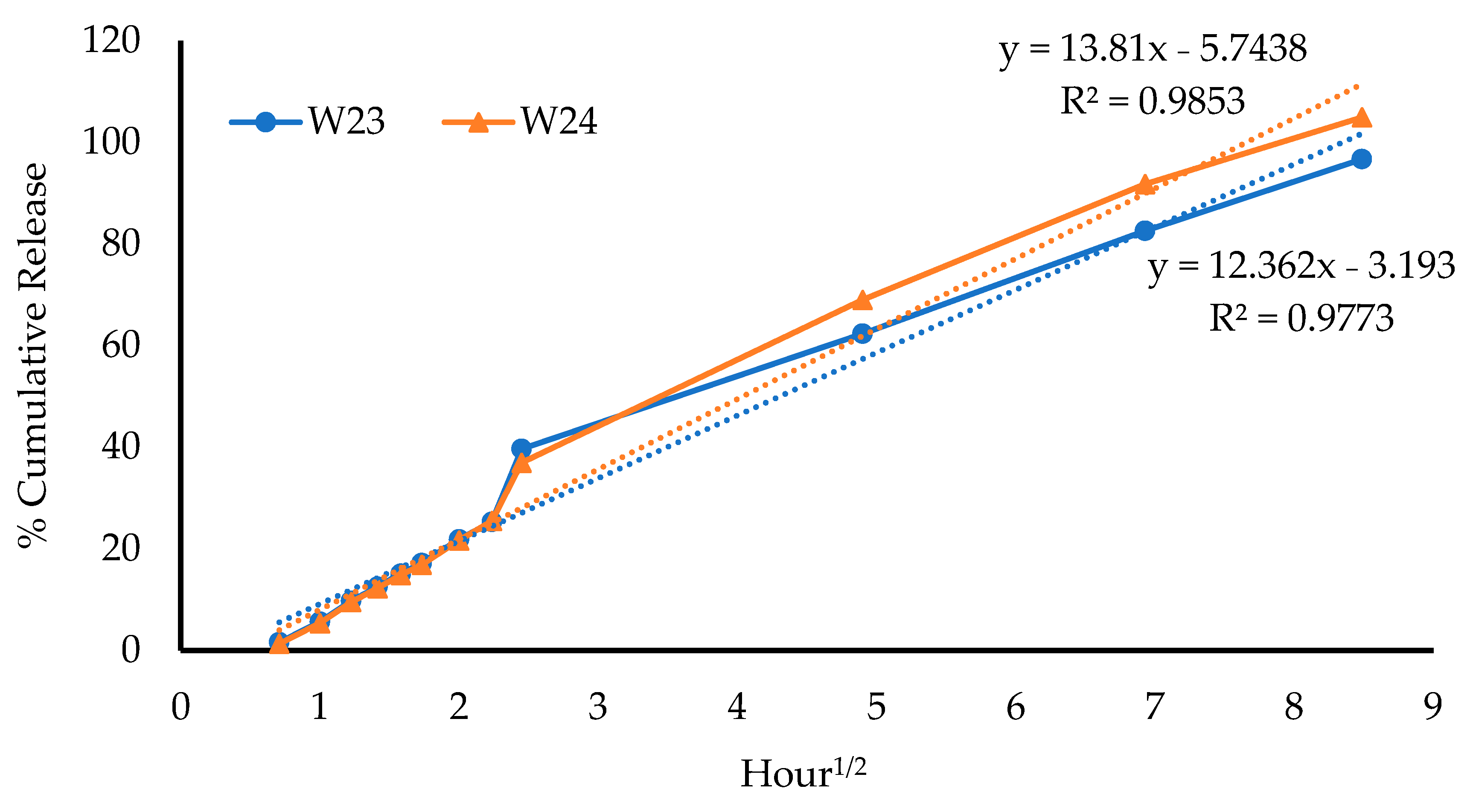
2.4.2. Evaluation of Drug-Release Mechanisms
Release kinetic equations are mathematical models employed to investigate the mechanisms of drug release from different formulations. The various equations are compared to determine the model that fits the release data best, based on the model that gives the highest correlation coefficient (R
2) [
23]. In this study, four kinetic models were used to investigate the mechanism(s) of the MTz release from the wafers, as shown in
Table 5.
2.5. Antibacterial Study
2.5.1. Minimum Inhibitory and Bactericidal Concentrations
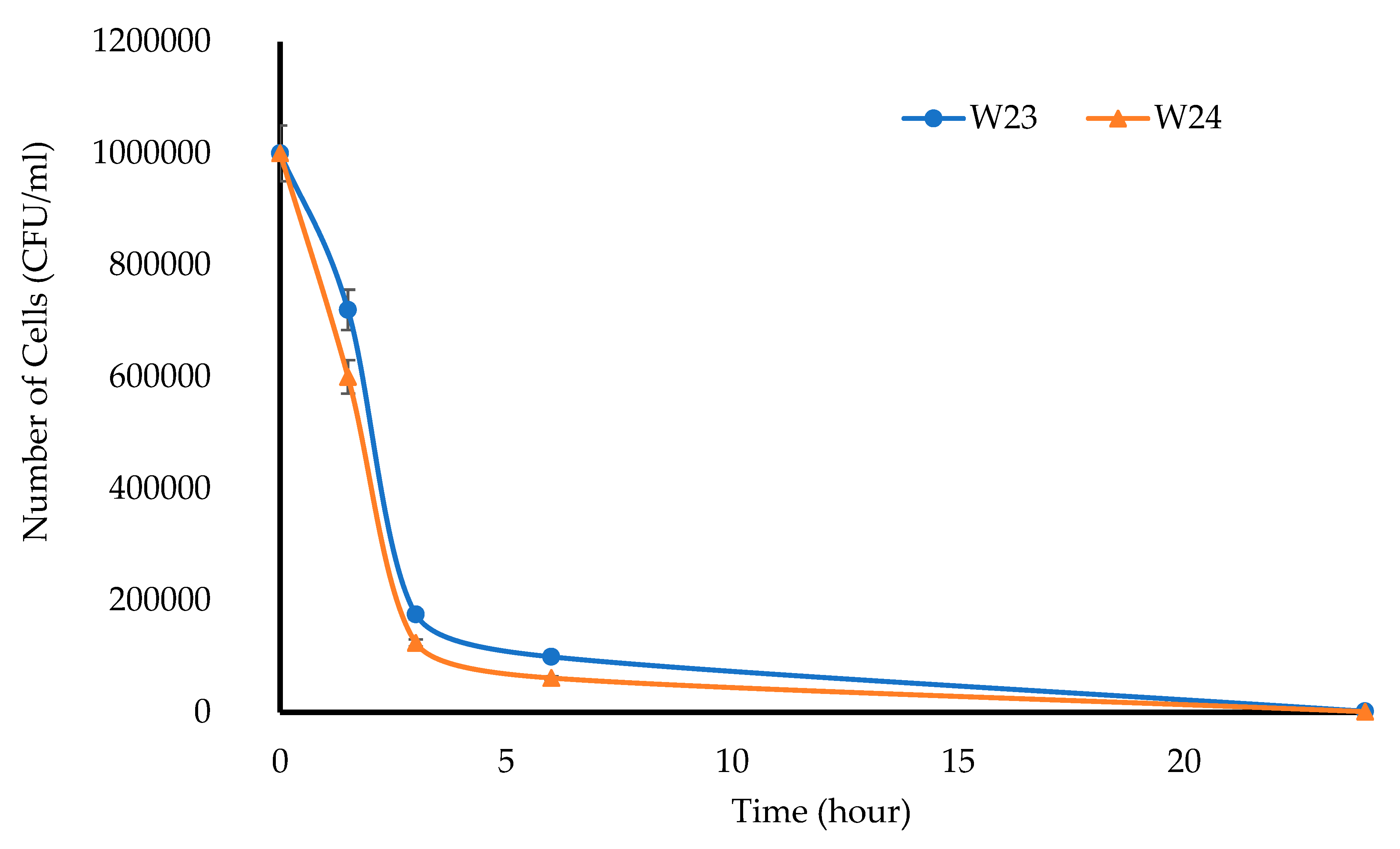
In vitro antibacterial testing of the MTz-loaded wafers was performed using turbidimetric and Kirby Bauer disk diffusion assays. Bacterial cultures were obtained in a brain heart infusion (BHI) broth medium at 37 °C using
E. coli as the model bacterial organism. The minimum inhibitory concentrations (MICs) and minimum bactericidal concentrations (MBCs) for MTz were measured in Muller Hinton (MH) broth using the broth dilution method. Serial dilutions of MTz (4–10 mg/mL) were prepared in sterile MH broth and inoculated to achieve a final bacteria concentration of 10
6 CFU/mL (
n = 3). After 24 h, samples from each treatment and relevant dilutions were plated on MH agar plates, and the colony-forming units (CFUs) were counted after incubation for another 24 h at 37 °C and used to determine the MBC. The number of viable cells was plotted against the antibiotic concentration and the number of cells per ml was calculated using Equation (3):
2.5.2. Turbidimetric Assay
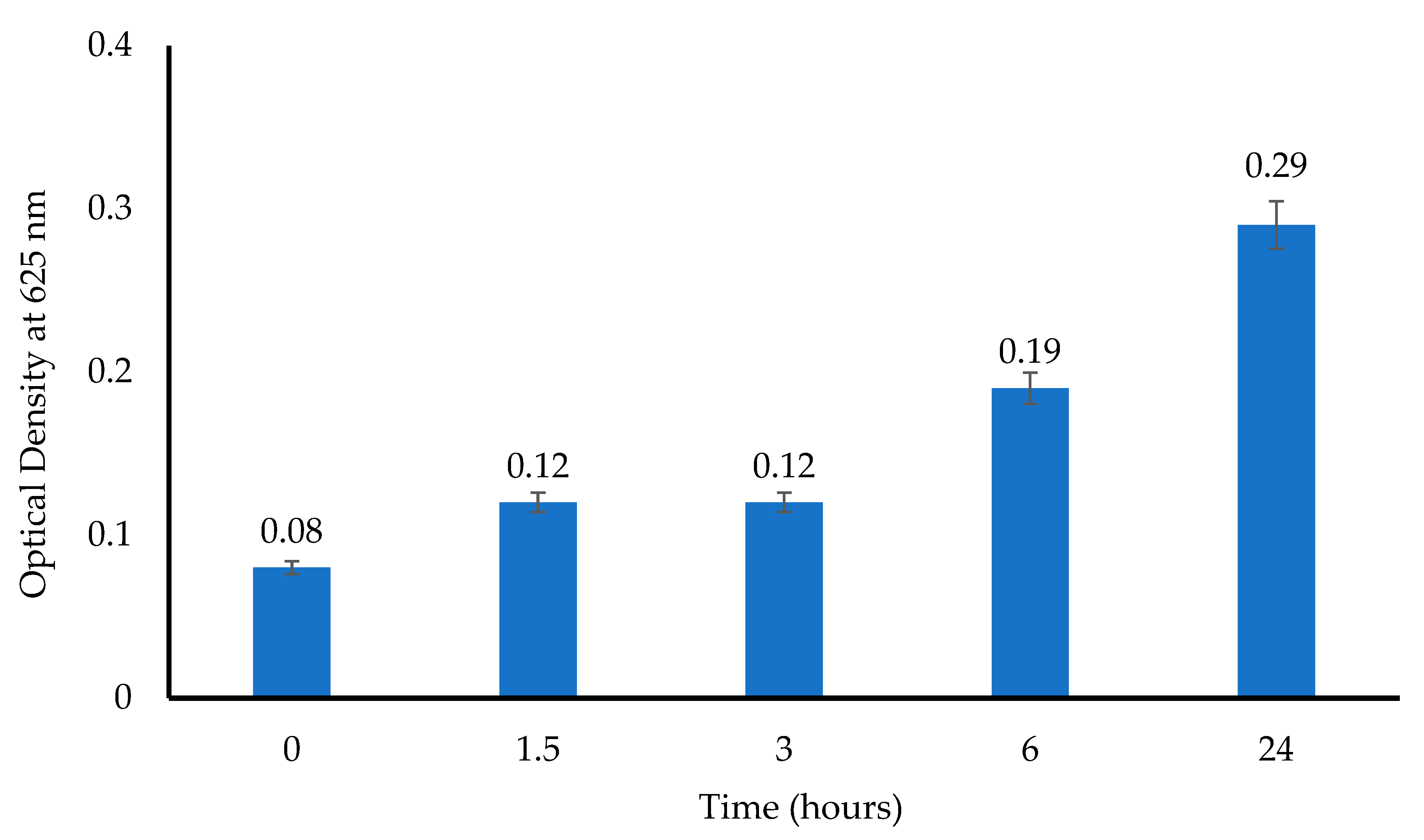
For the turbidimetric assay, 10 mL of a prepared
E. coli suspension (10
6 CFU/mL) was transferred into sterile test tubes that contained MTz-loaded wafers. Tubes that were filled with only the
E. coli suspension and a pure MTz solution were used as the negative and positive controls, respectively. The tubes were incubated (37 °C, 180 rpm), aliquots were sampled after 1.5, 3, 6, 12, and 24 h, and the absorbance was measured (625 nm). Further, 0.1 mL aliquots were plated directly or after serial dilution to count the number of viable bacterial colonies, with only the plates having 30 to 300 colonies accepted for determining the number of CFU/mL [
24]. The experiments were conducted in triplicate (
n = 3).
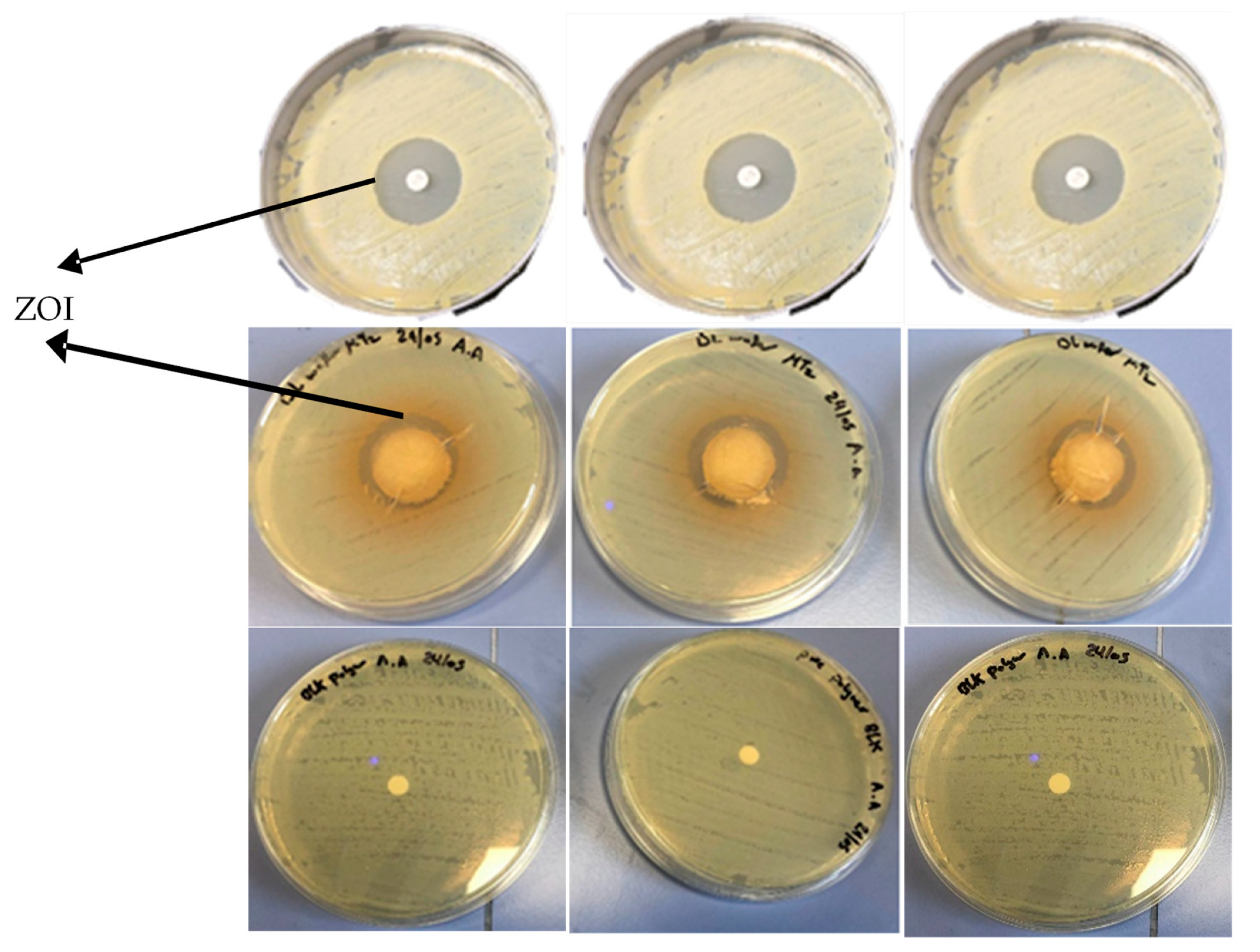
2.5.3. Kirby Bauer Disk Diffusion Assay
MH agar plates were prepared as previously reported [
25]. Briefly, MH agar was autoclaved for 45 min at 121 °C and allowed to cool to ambient temperature, and then 25 mL was poured into a Petri dish to obtain a 5 mm layer of solid agar slant. A sterile swab was placed into the tube containing the
E. coli suspension (10
6 CFU/mL) and streaked over the surface of the entire MH agar plate three times with clockwise rotation. The wafers were placed at the center of the colonized agar plates and the plates were incubated at 37 °C for 24 h, after which the zone of inhibition (ZOI) in mm was measured with a Vernier caliper. A 6 mm filter disk loaded with 20 μg of pure MTz and a BLK wafer were used as the positive and negative controls, respectively.
2.6. Statistical Analysis
Statistical analyses were performed to compare the results of the quantitative data, including hardness, mucoadhesion, hydration capacity, and porosity, using one-way ANOVA. The results are expressed as the mean ± standard deviation and the level of significance was set at p < 0.05.
4. Conclusions
This project aimed to develop, characterize, and optimize polymer-based freeze-dried wafers for the delivery of antimicrobial agents (using MTz as a model antibacterial drug) via the vaginal mucosa over a prolonged period to treat local infections. Composite wafers were initially formulated using GRAS mucoadhesive polymers, which were chosen depending on their ability to attach to the vaginal mucosa and achieve controlled release of the drug. Composite wafers comprising two natural polymers, CARR and SA, did not yield the expected optimal functional physical properties, especially hardness. Therefore, a synthetic polymer, CARB, was added to the formulations to improve the physical properties. The combination of CARB with CARR and CARB with SA at optimal concentrations showed an improved cone (rocket) shaped structure for easy vaginal insertion, improved porosity, and improved mechanical properties, which were then selected for loading the drug. The XRPD results indicated the amorphous nature of the BLK wafers; however, MTz maintained its crystalline nature in the DL wafers. The SEM micrographs showed a highly porous microstructure for the BLK wafers, while the MTz-loaded wafers showed a more compact structure with dispersed drug crystals on the pore walls. Based on the hardness profiles, W23 and W24 were chosen for the in vitro drug dissolution study. The drug release was controlled over 72 h, suggesting the potential to achieve the prolonged release of the drug, which would reduce the need for regular insertion, thereby reducing the incidence of non-compliance. The MTz-loaded optimized wafers also showed better inhibition of the model infection-causing bacteria (E. coli) compared to the BLK wafers. Overall, the MTz CARB:SA wafers show potential for vaginal mucosal delivery to treat local bacterial infections within the vaginal cavity. Though interesting, the results from the functional characterization are limited by being in vitro. Further studies, including testing mucoadhesion and drug permeation using an ex vivo (e.g., porcine) vaginal mucosal model, as well as an in vivo bioadhesion and antibacterial infection model, will be required in the future.























{kind=link}
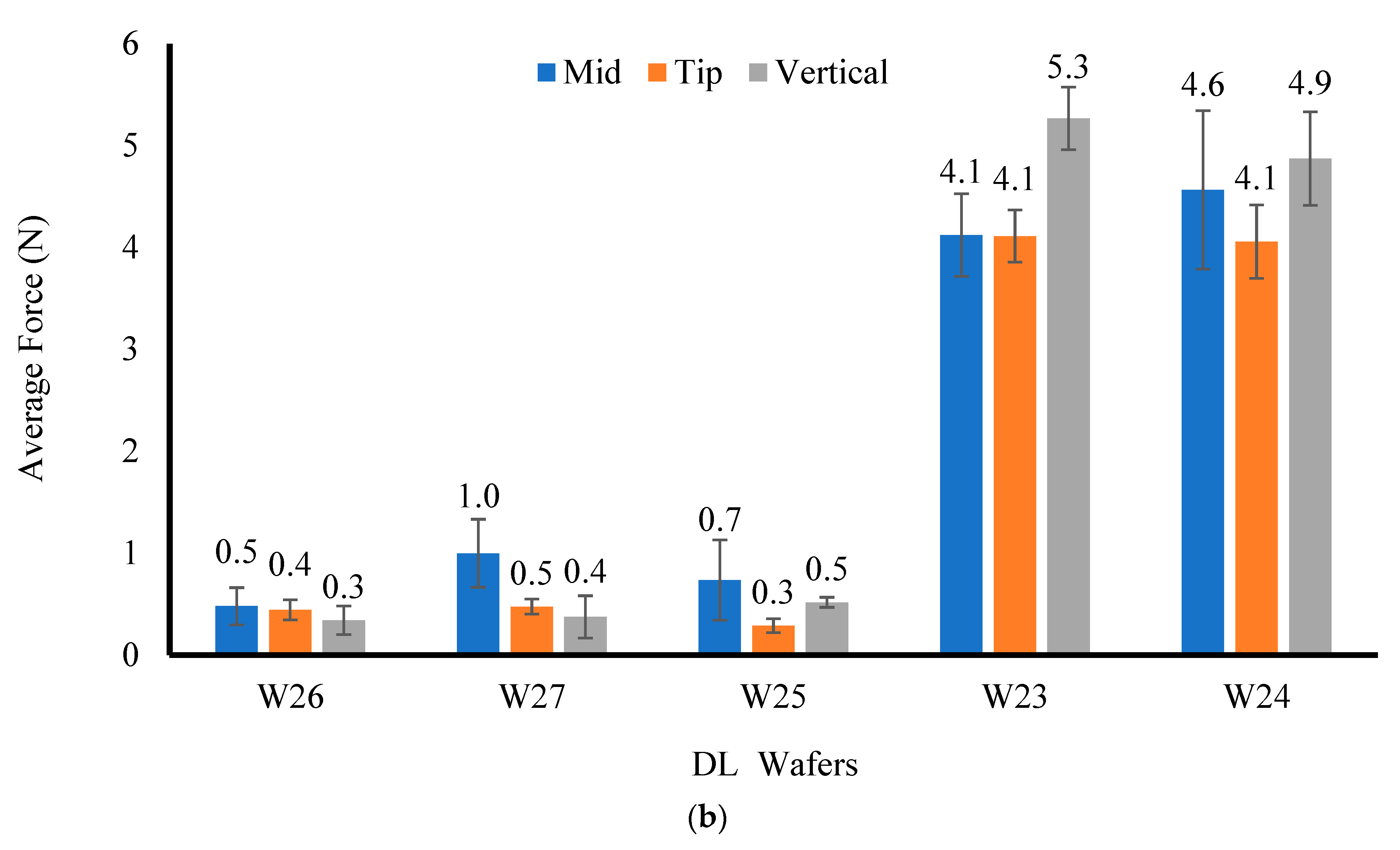{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}