
Deep Learning in Phosphoproteomics: Methods and Application in Cancer Drug Discovery

Abstract
:1. Introduction
2. Methods for Phosphorylation Site Prediction
2.1. Algorithm-Based Computational Approaches
2.2. Machine Learning (ML)-Based Computational Approaches
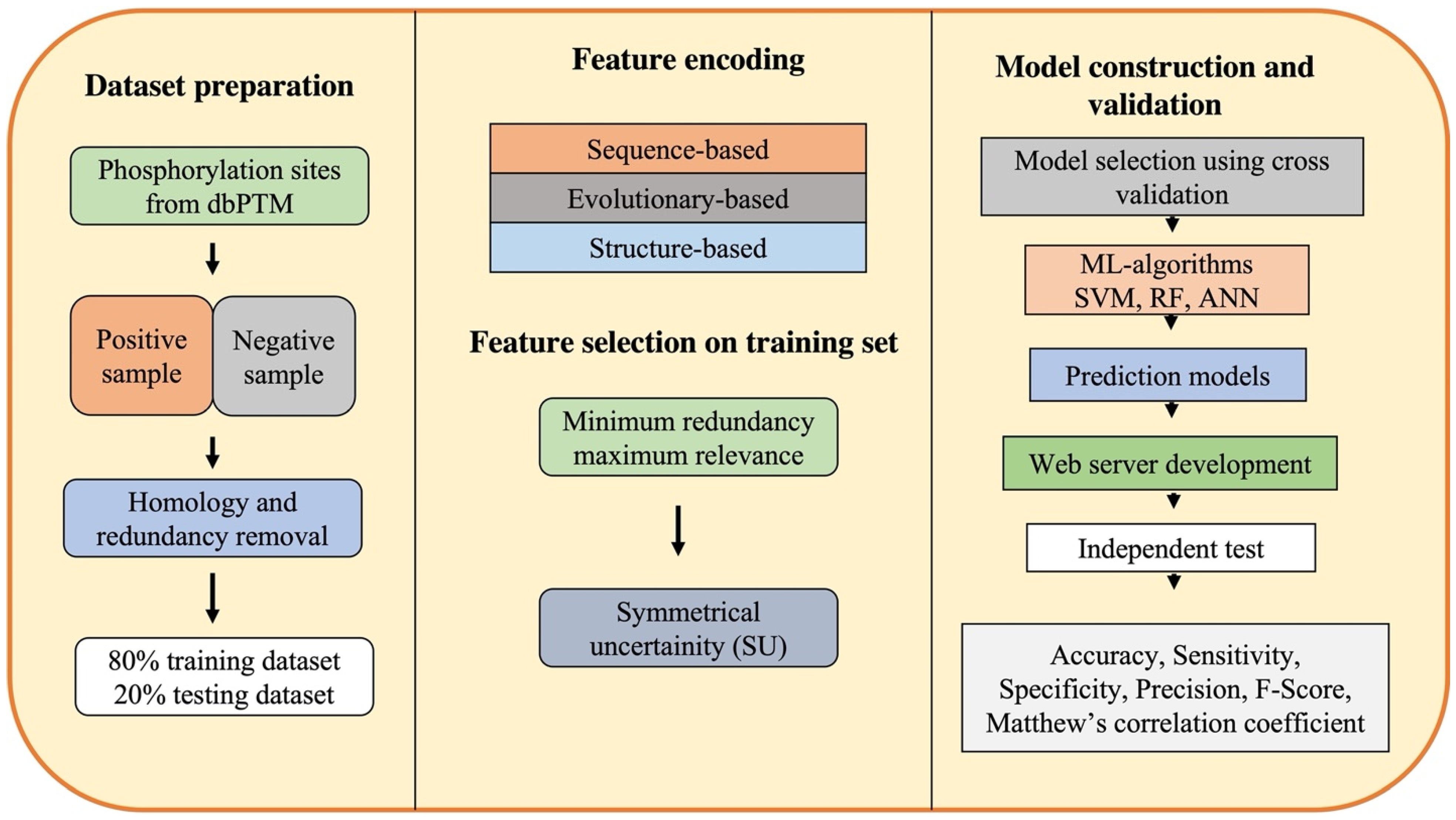
3. Framework of ML-Based Approaches for Phosphorylation Site Prediction
3.1. Dataset Preparation
3.2. Feature Encoding and Selection
3.3. Model Construction and Validation
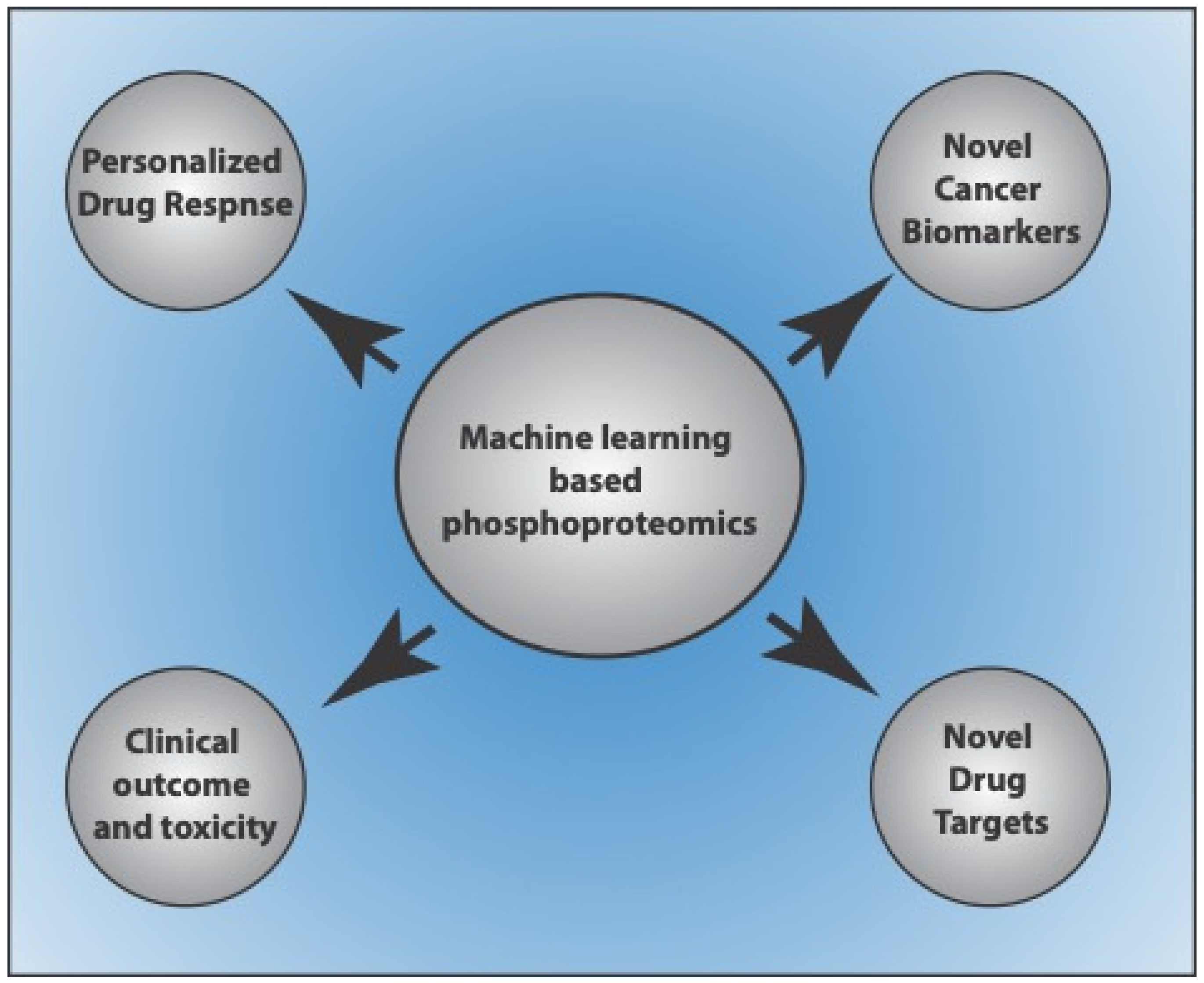
4. Use of Machine Learning-Based Approaches for Phosphoproteome Prediction in Cancers
4.1. Machine Learning-Based Approaches for Phosphoproteome-Based Biomarker Prediction
4.2. Machine Learning-Based Approaches for Phosphoproteome-Based Patient-Specific Drug Targets and Responses
5. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ubersax, J.A.; Ferrell, J.E. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. Protein kinases and phosphatases: The Yin and Yang of protein phosphorylation and signaling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef]
- Varshney, N.; Schaekel, A.; Singha, R.; Chakraborty, T.; van Wijlick, L.; Ernst, J.F.; Sanyal, K. A surprising role for the Sch9 protein kinase in chromosome segregation in Candida albicans. Genetics 2015, 199, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Varshney, N.; Sanyal, K. Aurora kinase Ipl1 facilitates bilobed distribution of clustered kinetochores to ensure error-free chromosome segregation in Candida albicans. Mol. Microbiol. 2019, 112, 569–587. [Google Scholar] [CrossRef]
- Varshney, N.; Som, S.; Chatterjee, S.; Sridhar, S.; Bhattacharyya, D.; Paul, R.; Sanyal, K. Spatio-temporal regulation of nuclear division by Aurora B kinase Ipl1 in Cryptococcus neoformans. PLoS Genet. 2019, 15, e1007959. [Google Scholar] [CrossRef]
- Humphrey, S.J.; James, D.E.; Mann, M. Protein phosphorylation: A major switch mechanism for metabolic regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. [Google Scholar] [CrossRef]
- Mishra, A.K.; Sharma, V.; Mutsuddi, M.; Mukherjee, A. Signaling cross-talk during development: Context-specific networking of Notch, NF-κB and JNK signaling pathways in Drosophila. Cell. Signal. 2021, 82, 109937. [Google Scholar] [CrossRef]
- Mishra, A.K.; Sachan, N.; Mutsuddi, M.; Mukherjee, A. Kinase active Misshapen regulates Notch signaling in Drosophila melanogaster. Exp. Cell Res. 2015, 339, 51–60. [Google Scholar] [CrossRef]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Cohen, P. The role of protein phosphorylation in the hormonal control of enzyme activity. Eur. J. Biochem. 1985, 151, 439–448. [Google Scholar] [CrossRef]
- Meyerovitch, J.; Backer, J.M.; Kahn, C.R. Hepatic phosphotyrosine phosphatase activity and its alterations in diabetic rats. J. Clin. Investig. 1989, 84, 976–983. [Google Scholar] [CrossRef]
- Hijazi, M.; Smith, R.; Rajeeve, V.; Bessant, C.; Cutillas, P.R. Reconstructing kinase network topologies from phosphoproteomics data reveals cancer-associated rewiring. Nat. Biotechnol. 2020, 38, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Kettenbach, A.N.; Rush, J.; Gerber, S.A. Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat. Protoc. 2011, 6, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.E.; Zamborelli, T.; Herman, A.C.; Yeh, C.-H.; Tedeschi, N.I.; Luedke, E.S. Detection of acetylated lysine residues using sequencing by edman degradation and mass spectrometry. In Techniques in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1996; Volume 7, pp. 201–208. ISBN 9780124735569. [Google Scholar]
- Fuchs, S.M.; Strahl, B.D. Antibody recognition of histone post-translational modifications: Emerging issues and future prospects. Epigenomics 2011, 3, 247–249. [Google Scholar] [CrossRef]
- Witze, E.S.; Old, W.M.; Resing, K.A.; Ahn, N.G. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 2007, 4, 798–806. [Google Scholar] [CrossRef]
- Paul, P.; Muthu, M.; Chilukuri, Y.; Haga, S.W.; Chun, S.; Oh, J.-W. In silico tools and phosphoproteomic software exclusives. Processes 2019, 7, 869. [Google Scholar] [CrossRef]
- Nakai, K.; Kanehisa, M. Prediction of in-vivo modification sites of proteins from their primary structures. J. Biochem. 1988, 104, 693–699. [Google Scholar] [CrossRef]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013, 41, D344–D347. [Google Scholar] [CrossRef]
- Puntervoll, P.; Linding, R.; Gemünd, C.; Chabanis-Davidson, S.; Mattingsdal, M.; Cameron, S.; Martin, D.M.A.; Ausiello, G.; Brannetti, B.; Costantini, A.; et al. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 2003, 31, 3625–3630. [Google Scholar] [CrossRef] [PubMed]
- Peri, S.; Navarro, J.D.; Amanchy, R.; Kristiansen, T.Z.; Jonnalagadda, C.K.; Surendranath, V.; Niranjan, V.; Muthusamy, B.; Gandhi, T.K.B.; Gronborg, M.; et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003, 13, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.; Matsuyama, A.; Yoshida, M.; Kim, D. PostMod: Sequence based prediction of kinase-specific phosphorylation sites with indirect relationship. BMC Bioinform. 2010, 11 (Suppl. S1), S10. [Google Scholar] [CrossRef] [PubMed]
- Suo, S.-B.; Qiu, J.-D.; Shi, S.-P.; Chen, X.; Liang, R.-P. PSEA: Kinase-specific prediction and analysis of human phosphorylation substrates. Sci. Rep. 2014, 4, 4524. [Google Scholar] [CrossRef]
- Avery, C.; Patterson, J.; Grear, T.; Frater, T.; Jacobs, D.J. Protein Function Analysis through Machine Learning. Biomolecules 2022, 12, 1246. [Google Scholar] [CrossRef]
- Auslander, N.; Gussow, A.B.; Koonin, E.V. Incorporating Machine Learning into Established Bioinformatics Frameworks. Int. J. Mol. Sci. 2021, 22, 2903. [Google Scholar] [CrossRef]
- Jordan, M.I.; Mitchell, T.M. Machine learning: Trends, perspectives, and prospects. Science 2015, 349, 255–260. [Google Scholar] [CrossRef]
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.-M.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Plewczyński, D.; Tkacz, A.; Godzik, A.; Rychlewski, L. A support vector machine approach to the identification of phosphorylation sites. Cell. Mol. Biol. Lett. 2005, 10, 73–89. [Google Scholar]
- Miranda-Saavedra, D.; Barton, G.J. Classification and functional annotation of eukaryotic protein kinases. Proteins 2007, 68, 893–914. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.M.A.; Miranda-Saavedra, D.; Barton, G.J. Kinomer v. 1.0: A database of systematically classified eukaryotic protein kinases. Nucleic Acids Res. 2009, 37, D244–D250. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Del Vecchio, R.L.; Kannan, N.; Gergel, J.; Neuwald, A.F.; Tonks, N.K. Computational analysis of protein tyrosine phosphatases: Practical guide to bioinformatics and data resources. Methods 2005, 35, 90–114. [Google Scholar] [CrossRef] [PubMed]
- Damle, N.P.; Köhn, M. The human DEPhOsphorylation Database DEPOD: 2019 update. Database 2019, 2019, baz133. [Google Scholar] [CrossRef]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.ELM: A database of phosphorylation sites--update 2011. Nucleic Acids Res. 2011, 39, D261–D267. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The universal protein resource (uniprot). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar] [CrossRef]
- Lin, S.; Wang, C.; Zhou, J.; Shi, Y.; Ruan, C.; Tu, Y.; Yao, L.; Peng, D.; Xue, Y. EPSD: A well-annotated data resource of protein phosphorylation sites in eukaryotes. Brief. Bioinform. 2021, 22, 298–307. [Google Scholar] [CrossRef]
- Huang, K.-Y.; Wu, H.-Y.; Chen, Y.-J.; Lu, C.-T.; Su, M.-G.; Hsieh, Y.-C.; Tsai, C.-M.; Lin, K.-I.; Huang, H.-D.; Lee, T.-Y.; et al. RegPhos 2.0: An updated resource to explore protein kinase-substrate phosphorylation networks in mammals. Database 2014, 2014, bau034. [Google Scholar] [CrossRef]
- Zanzoni, A.; Carbajo, D.; Diella, F.; Gherardini, P.F.; Tramontano, A.; Helmer-Citterich, M.; Via, A. Phospho3D 2.0: An enhanced database of three-dimensional structures of phosphorylation sites. Nucleic Acids Res. 2011, 39, D268–D271. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, Y.; Lin, S.; Wang, C.; Zhou, J.; Peng, D.; Xue, Y. dbPSP 2.0, an updated database of protein phosphorylation sites in prokaryotes. Sci. Data 2020, 7, 164. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Vidal-Cortes, O.; Gallardo, O.; Abian, J.; Carrascal, M. LymPHOS 2.0: An update of a phosphosite database of primary human T cells. Database 2015, 2015, bav115. [Google Scholar] [CrossRef]
- Yao, Q.; Bollinger, C.; Gao, J.; Xu, D.; Thelen, J.J. P(3)DB: An Integrated Database for Plant Protein Phosphorylation. Front. Plant Sci. 2012, 3, 206. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; Ren, S.; Cox, J.; Olsen, J.V.; Macek, B.; Oroshi, M.; Mann, M. PHOSIDA (phosphorylation site database): Management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 2007, 8, R250. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zou, Q.; Zhao, L. VPTMdb: A viral posttranslational modification database. Brief. Bioinform. 2021, 22, bbaa251. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Liu, Z.; Wang, J.; Cui, Y.; Guo, Y.; Zhou, T.; Zhou, Z.; Guo, X.; Xue, Y.; Sha, J. Systematic analysis of the phosphoproteome and kinase-substrate networks in the mouse testis. Mol. Cell. Proteom. 2014, 13, 3626–3638. [Google Scholar] [CrossRef]
- Bodenmiller, B.; Campbell, D.; Gerrits, B.; Lam, H.; Jovanovic, M.; Picotti, P.; Schlapbach, R.; Aebersold, R. PhosphoPep—A database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 2008, 26, 1339–1340. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Chang, C.-H.; Yu, Y.-L.; Lin, T.-C.E.; Lee, S.-A.; Yen, C.-C.; Yang, J.-M.; Lai, J.-M.; Hong, Y.-R.; Tseng, T.-L.; et al. PhosphoPOINT: A comprehensive human kinase interactome and phospho-protein database. Bioinformatics 2008, 24, i14–i20. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef] [PubMed]
- Safaei, J.; Maňuch, J.; Gupta, A.; Stacho, L.; Pelech, S. Prediction of 492 human protein kinase substrate specificities. Proteome Sci. 2011, 9 (Suppl. S1), S6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, J.; Oh, B.; Kimm, K.; Koh, I. Prediction of phosphorylation sites using SVMs. Bioinformatics 2004, 20, 3179–3184. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.J.; Pasculescu, A.; Olhovsky, M.; Colwill, K.; Bork, P.; Yaffe, M.B.; Pawson, T. NetworKIN: A resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 2008, 36, D695–D699. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, Z.; Mai, G.; Luo, Y.; Zhou, F. jEcho: An Evolved weight vector to CHaracterize the protein’s posttranslational modification mOtifs. Interdiscip. Sci. 2015, 7, 194–199. [Google Scholar] [CrossRef]
- Li, T.; Li, F.; Zhang, X. Prediction of kinase-specific phosphorylation sites with sequence features by a log-odds ratio approach. Proteins 2008, 70, 404–414. [Google Scholar] [CrossRef]
- Gao, Y.; Hao, W.; Gu, J.; Liu, D.; Fan, C.; Chen, Z.; Deng, L. PredPhos: An ensemble framework for structure-based prediction of phosphorylation sites. J. Biol. Res. (Thessalon) 2016, 23, 12. [Google Scholar] [CrossRef]
- Ingrell, C.R.; Miller, M.L.; Jensen, O.N.; Blom, N. NetPhosYeast: Prediction of protein phosphorylation sites in yeast. Bioinformatics 2007, 23, 895–897. [Google Scholar] [CrossRef]
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteom. 2008, 7, 1598–1608. [Google Scholar] [CrossRef]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genom. Proteom. Bioinform. 2020, 18, 72–80. [Google Scholar] [CrossRef]
- Song, C.; Ye, M.; Liu, Z.; Cheng, H.; Jiang, X.; Han, G.; Songyang, Z.; Tan, Y.; Wang, H.; Ren, J.; et al. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol. Cell. Proteom. 2012, 11, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.K.; Noman, N.; Sikder, A.R. Machine learning approach to predict protein phosphorylation sites by incorporating evolutionary information. BMC Bioinform. 2010, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Durek, P.; Schudoma, C.; Weckwerth, W.; Selbig, J.; Walther, D. Detection and characterization of 3D-signature phosphorylation site motifs and their contribution towards improved phosphorylation site prediction in proteins. BMC Bioinform. 2009, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Maleki, F.; Kusalik, A.; Napper, S. DAPPLE 2: A Tool for the Homology-Based Prediction of Post-Translational Modification Sites. J. Proteome Res. 2016, 15, 2760–2767. [Google Scholar] [CrossRef] [PubMed]
- Kirchoff, K.E.; Gomez, S.M. EMBER: Multi-label prediction of kinase-substrate phosphorylation events through deep learning. Bioinformatics 2022, 38, 2119–2126. [Google Scholar] [CrossRef]
- Horn, H.; Schoof, E.M.; Kim, J.; Robin, X.; Miller, M.L.; Diella, F.; Palma, A.; Cesareni, G.; Jensen, L.J.; Linding, R. KinomeXplorer: An integrated platform for kinome biology studies. Nat. Methods 2014, 11, 603–604. [Google Scholar] [CrossRef]
- Xu, Y.; Wilson, C.; Leier, A.; Marquez-Lago, T.T.; Whisstock, J.; Song, J. PhosTransfer: A Deep Transfer Learning Framework for Kinase-Specific Phosphorylation Site Prediction in Hierarchy. In Advances in Knowledge Discovery and Data Mining: 24th Pacific-Asia Conference, PAKDD 2020, Singapore, 11–14 May 2020, Proceedings, Part II; Lauw, H.W., Wong, R.C.-W., Ntoulas, A., Lim, E.-P., Ng, S.-K., Pan, S.J., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2020; Volume 12085, pp. 384–395. ISBN 978-3-030-47435-5. [Google Scholar]
- Wang, D.; Zeng, S.; Xu, C.; Qiu, W.; Liang, Y.; Joshi, T.; Xu, D. MusiteDeep: A deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017, 33, 3909–3916. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Webb, G.I.; Baggag, A.; Bensmail, H.; Song, J. PROSPECT: A web server for predicting protein histidine phosphorylation sites. J. Bioinform. Comput. Biol. 2020, 18, 2050018. [Google Scholar] [CrossRef]
- Deznabi, I.; Arabaci, B.; Koyutürk, M.; Tastan, O. DeepKinZero: Zero-shot learning for predicting kinase-phosphosite associations involving understudied kinases. Bioinformatics 2020, 36, 3652–3661. [Google Scholar] [CrossRef]
- Ahmed, S.; Kabir, M.; Arif, M.; Khan, Z.U.; Yu, D.-J. DeepPPSite: A deep learning-based model for analysis and prediction of phosphorylation sites using efficient sequence information. Anal. Biochem. 2021, 612, 113955. [Google Scholar] [CrossRef]
- Lv, H.; Dao, F.-Y.; Zulfiqar, H.; Lin, H. DeepIPs: Comprehensive assessment and computational identification of phosphorylation sites of SARS-CoV-2 infection using a deep learning-based approach. Brief. Bioinform. 2021, 22, bbab244. [Google Scholar] [CrossRef]
- Lin, S.; Song, Q.; Tao, H.; Wang, W.; Wan, W.; Huang, J.; Xu, C.; Chebii, V.; Kitony, J.; Que, S.; et al. Rice_Phospho 1.0: A new rice-specific SVM predictor for protein phosphorylation sites. Sci. Rep. 2015, 5, 11940. [Google Scholar] [CrossRef] [PubMed]
- Sharifpoor, S.; Nguyen Ba, A.N.; Youn, J.-Y.; van Dyk, D.; Friesen, H.; Douglas, A.C.; Kurat, C.F.; Chong, Y.T.; Founk, K.; Moses, A.M.; et al. A quantitative literature-curated gold standard for kinase-substrate pairs. Genome Biol. 2011, 12, R39. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Tkacz, A.; Wyrwicz, L.S.; Rychlewski, L.; Ginalski, K. AutoMotif Server for prediction of phosphorylation sites in proteins using support vector machine: 2007 update. J. Mol. Model. 2008, 14, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, M.; Liu, X.; Zhao, X.-M.; Li, A. PhosIDN: An integrated deep neural network for improving protein phosphorylation site prediction by combining sequence and protein-protein interaction information. Bioinformatics 2021, 37, 4668–4676. [Google Scholar] [CrossRef] [PubMed]
- Thapa, N.; Chaudhari, M.; Iannetta, A.A.; White, C.; Roy, K.; Newman, R.H.; Hicks, L.M.; Kc, D.B. A deep learning based approach for prediction of Chlamydomonas reinhardtii phosphorylation sites. Sci. Rep. 2021, 11, 12550. [Google Scholar] [CrossRef]
- Guo, L.; Wang, Y.; Xu, X.; Cheng, K.-K.; Long, Y.; Xu, J.; Li, S.; Dong, J. DeepPSP: A Global-Local Information-Based Deep Neural Network for the Prediction of Protein Phosphorylation Sites. J. Proteome Res. 2021, 20, 346–356. [Google Scholar] [CrossRef]
- Saunders, N.F.W.; Brinkworth, R.I.; Huber, T.; Kemp, B.E.; Kobe, B. Predikin and PredikinDB: A computational framework for the prediction of protein kinase peptide specificity and an associated database of phosphorylation sites. BMC Bioinform. 2008, 9, 245. [Google Scholar] [CrossRef]
- Wong, Y.-H.; Lee, T.-Y.; Liang, H.-K.; Huang, C.-M.; Wang, T.-Y.; Yang, Y.-H.; Chu, C.-H.; Huang, H.-D.; Ko, M.-T.; Hwang, J.-K. KinasePhos 2.0: A web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 2007, 35, W588–W594. [Google Scholar] [CrossRef]
- Ma, R.; Li, S.; Li, W.; Yao, L.; Huang, H.-D.; Lee, T.-Y. KinasePhos 3.0: Redesign and expansion of the prediction on kinase-specific phosphorylation sites. Genom. Proteom. Bioinform. 2022; in press. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef]
- Neuberger, G.; Schneider, G.; Eisenhaber, F. pkaPS: Prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biol. Direct 2007, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Marquez-Lago, T.T.; Leier, A.; Akutsu, T.; Purcell, A.W.; Ian Smith, A.; Lithgow, T.; Daly, R.J.; Song, J.; et al. Quokka: A comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics 2018, 34, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-Y.; Huang, H.-D.; Hung, J.-H.; Huang, H.-Y.; Yang, Y.-S.; Wang, T.-H. dbPTM: An information repository of protein post-translational modification. Nucleic Acids Res. 2006, 34, D622–D627. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Kawashima, S.; Ogata, H.; Kanehisa, M. Aaindex: Amino acid index database. Nucleic Acids Res. 1999, 27, 368–369. [Google Scholar] [CrossRef]
- Li, T.; Du, P.; Xu, N. Identifying human kinase-specific protein phosphorylation sites by integrating heterogeneous information from various sources. PLoS ONE 2010, 5, e15411. [Google Scholar] [CrossRef] [PubMed]
- Lins, L.; Thomas, A.; Brasseur, R. Analysis of accessible surface of residues in proteins. Protein Sci. 2003, 12, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.; Ali, W.; Nagpal, P.; Grover, A.; Grover, S. Predicting phosphorylation sites using machine learning by integrating the sequence, structure, and functional information of proteins. J. Transl. Med. 2021, 19, 218. [Google Scholar] [CrossRef]
- Scheidt, T.; Alka, O.; Gonczarowska-Jorge, H.; Gruber, W.; Rathje, F.; Dell’Aica, M.; Rurik, M.; Kohlbacher, O.; Zahedi, R.P.; Aberger, F.; et al. Phosphoproteomics of short-term hedgehog signaling in human medulloblastoma cells. Cell Commun. Signal. 2020, 18, 99. [Google Scholar] [CrossRef]
- Rubbi, L.; Titz, B.; Brown, L.; Galvan, E.; Komisopoulou, E.; Chen, S.S.; Low, T.; Tahmasian, M.; Skaggs, B.; Müschen, M.; et al. Global phosphoproteomics reveals crosstalk between Bcr-Abl and negative feedback mechanisms controlling Src signaling. Sci. Signal. 2011, 4, ra18. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guan, Y. Machine learning empowers phosphoproteome prediction in cancers. Bioinformatics 2020, 36, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.-Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. CPTAC Investigators Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Khorsandi, S.E.; Dokal, A.D.; Rajeeve, V.; Britton, D.J.; Illingworth, M.S.; Heaton, N.; Cutillas, P.R. Computational Analysis of Cholangiocarcinoma Phosphoproteomes Identifies Patient-Specific Drug Targets. Cancer Res. 2021, 81, 5765–5776. [Google Scholar] [CrossRef] [PubMed]
- Coker, E.A.; Stewart, A.; Ozer, B.; Minchom, A.; Pickard, L.; Ruddle, R.; Carreira, S.; Popat, S.; O’Brien, M.; Raynaud, F.; et al. Individualized Prediction of Drug Response and Rational Combination Therapy in NSCLC Using Artificial Intelligence-Enabled Studies of Acute Phosphoproteomic Changes. Mol. Cancer Ther. 2022, 21, 1020–1029. [Google Scholar] [CrossRef]
- Park, Y.; Kim, M.J.; Choi, Y.; Kim, N.H.; Kim, L.; Hong, S.P.D.; Cho, H.-G.; Yu, E.; Chae, Y.K. Role of mass spectrometry-based serum proteomics signatures in predicting clinical outcomes and toxicity in patients with cancer treated with immunotherapy. J. Immunother. Cancer 2022, 10, e003566. [Google Scholar] [CrossRef]
- Ramos, E.K.; Tsai, C.-F.; Jia, Y.; Cao, Y.; Manu, M.; Taftaf, R.; Hoffmann, A.D.; El-Shennawy, L.; Gritsenko, M.A.; Adorno-Cruz, V.; et al. Machine learning-assisted elucidation of CD81-CD44 interactions in promoting cancer stemness and extracellular vesicle integrity. eLife 2022, 11, e82669. [Google Scholar] [CrossRef]
- Rodrigues-Ferreira, S.; Nahmias, C. Predictive biomarkers for personalized medicine in breast cancer. Cancer Lett. 2022, 545, 215828. [Google Scholar] [CrossRef]
- Shen, J.; Qi, L.; Zou, Z.; Du, J.; Kong, W.; Zhao, L.; Wei, J.; Lin, L.; Ren, M.; Liu, B. Identification of a novel gene signature for the prediction of recurrence in HCC patients by machine learning of genome-wide databases. Sci. Rep. 2020, 10, 4435. [Google Scholar] [CrossRef]
- Azuaje, F.; Kim, S.-Y.; Perez Hernandez, D.; Dittmar, G. Connecting Histopathology Imaging and Proteomics in Kidney Cancer through Machine Learning. J. Clin. Med. 2019, 8, 1535. [Google Scholar] [CrossRef]
- Li, H.; Siddiqui, O.; Zhang, H.; Guan, Y. Joint learning improves protein abundance prediction in cancers. BMC Biol. 2019, 17, 107. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, H.; Casado, P.; Dokal, A.; Hijazi, M.; Akhtar, N.; Osuntola, R.; Rajeeve, V.; Fitzgibbon, J.; Travers, J.; Britton, D.; et al. Drug ranking using machine learning systematically predicts the efficacy of anti-cancer drugs. Nat. Commun. 2021, 12, 1850. [Google Scholar] [CrossRef] [PubMed]
- Crowl, S.; Jordan, B.T.; Ahmed, H.; Ma, C.X.; Naegle, K.M. KSTAR: An algorithm to predict patient-specific kinase activities from phosphoproteomic data. Nat. Commun. 2022, 13, 4283. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, S.; Zhang, Q.; Lu, Q.; Su, D.; Zuo, Y.; Yang, L. Analysis and prediction of animal toxins by various Chou’s pseudo components and reduced amino acid compositions. J. Theor. Biol. 2019, 462, 221–229. [Google Scholar] [CrossRef]
- Zuo, Y.; Li, Y.; Chen, Y.; Li, G.; Yan, Z.; Yang, L. PseKRAAC: A flexible web server for generating pseudo K-tuple reduced amino acids composition. Bioinformatics 2017, 33, 122–124. [Google Scholar] [CrossRef]
- Meng, L.; Chan, W.-S.; Huang, L.; Liu, L.; Chen, X.; Zhang, W.; Wang, F.; Cheng, K.; Sun, H.; Wong, K.-C. Mini-review: Recent advances in post-translational modification site prediction based on deep learning. Comput. Struct. Biotechnol. J. 2022, 20, 3522–3532. [Google Scholar] [CrossRef]
- Schoof, E.M.; Furtwängler, B.; Üresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; Keller, U.A.d.; Dick, J.E.; Porse, B.T. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12, 3341. [Google Scholar] [CrossRef]
- Lun, X.-K.; Bodenmiller, B. Profiling Cell Signaling Networks at Single-cell Resolution. Mol. Cell. Proteom. 2020, 19, 744–756. [Google Scholar] [CrossRef]
- Wei, W.; Shin, Y.S.; Xue, M.; Matsutani, T.; Masui, K.; Yang, H.; Ikegami, S.; Gu, Y.; Herrmann, K.; Johnson, D.; et al. Single-Cell Phosphoproteomics Resolves Adaptive Signaling Dynamics and Informs Targeted Combination Therapy in Glioblastoma. Cancer Cell 2016, 29, 563–573. [Google Scholar] [CrossRef]
- Pérez-Mejías, G.; Velázquez-Cruz, A.; Guerra-Castellano, A.; Baños-Jaime, B.; Díaz-Quintana, A.; González-Arzola, K.; Ángel De la Rosa, M.; Díaz-Moreno, I. Exploring protein phosphorylation by combining computational approaches and biochemical methods. Comput. Struct. Biotechnol. J. 2020, 18, 1852–1863. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L. Consortium for Top Down Proteomics Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shiyanov, P.; Green, K.B. Top-down mass spectrometry of intact phosphorylated β-casein: Correlation between the precursor charge state and internal fragments. J. Mass Spectrom. 2019, 54, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Gafken, P.R.; Lampe, P.D. Methodologies for characterizing phosphoproteins by mass spectrometry. Cell Commun. Adhes. 2006, 13, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Zabrouskov, V.; Ge, Y.; Schwartz, J.; Walker, J.W. Unraveling molecular complexity of phosphorylated human cardiac troponin I by top down electron capture dissociation/electron transfer dissociation mass spectrometry. Mol. Cell. Proteom. 2008, 7, 1838–1849. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, S.J.; Wu, Z.; Wetzel, M.; Belongia, D.; Jin, Y.; Wenger, K.; Ong, I.M.; Ge, Y. Enhancing Top-Down Proteomics Data Analysis by Combining Deconvolution Results through a Machine Learning Strategy. J. Am. Soc. Mass Spectrom. 2020, 31, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cui, C.; Chen, H.; Liu, T. Ensemble learning-based feature selection for phosphorylation site detection. Front. Genet. 2022, 13, 984068. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name | Technique | Organisms | Description/Functionality | Ref | Website |
|---|---|---|---|---|---|
| Kinome | |||||
| Kinomer | HMMER 2.3.2 | Eukaryotes | Annotated classifications for the protein kinase complements of 43 eukaryotic genomes | [32,33] | http://www.compbio.dundee.ac.uk/kinomer/ (accessed on 24 April 2023) |
| KinaseNET | - | Human | Comprehensive source on human kinases | - | http://www.kinasenet.ca/ (accessed on 24 April 2023) |
| Phosphatome | |||||
| PTP | - | - | Integrates sequence and structure with cellular and biological functions on protein tyrosine phosphatases | [34] | http://ptp.cshl.edu/ (accessed on 24 April 2023) |
| DEPOD | BLAST | Comprehensive and informative database on human kinase-phosphatase substrate | [35] | http://depod.bioss.uni-freiburg.de/ (accessed on 24 April 2023) | |
| Kinome-Phosphatome | |||||
| Phospho.ELM | BLAST | Multiple | Database designed to store in vivo and in vitro phosphorylation | [36] | http://phospho.elm.eu.org/ (accessed on 24 April 2023) |
| PSP (PhosphositePlus) | - | Human, mouse, and rat | Resource that comprehensively curates information about the structure and regulatory interactions of phosphorylation sites | [37] | https://www.phosphosite.org (accessed on 24 April 2023) |
| UniProt | - | Multispecies | A central hub for the collection of functional information on proteins | [38] | https://www.uniprot.org (accessed on 24 April 2023) |
| EPSD (Eukaryotic Phosphorylation site Database) | - | Multiple | A data resource for the collection, curation, integration, and annotation of p-sites in eukaryotic proteins | [39] | http://epsd.biocuckoo.cn (accessed on 24 April 2023) |
| RegPhos 2.0 (regulatory network in protein phosphorylation) | - | Human, mouse, and rat | A comprehensive tool to view intracellular signaling networks by integrating the information of metabolic pathways and protein–protein interactions | [40] | http://140.138.144.141/~RegPhos/ (accessed on 24 April 2023) |
| Phospho3D 2.0 | - | Multiple | A database for the collection of information on the residues surrounding the p-site in space (3D zones) | [41] | http://www.phospho3d.org/ (accessed on 24 April 2023) |
| dbPSP | - | Prokaryotes | Collection of p-sites in prokaryotic phosphoproteins | [42] | http://dbpsp.biocuckoo.cn (accessed on 24 April 2023) |
| LymPHOS | - | Human, mouse | A database for storage, sharing, and visualization of data related with the human T-lymphocyte phosphoproteome | [43] | http://www.lymphos.org (accessed on 24 April 2023) |
| P3DB | - | Plant species | Displays data in a relational, hierarchical manner that integrates proteins, peptides, phosphosites, and spectra for each phosphorylation event | [44] | http://www.p3db.org/ (accessed on 24 April 2023) |
| PHOSIDA (Phosphorylation site database) | - | Multiple | Structural and evolutionary investigation and prediction of phosphosites | [45] | http://phosida.de/ (accessed on 24 April 2023) |
| HPRD (human protein reference database) | BLAST | Human | A database of curated proteomic information including PTMs, kinase/phosphatase motifs, and binding motifs pertaining to human proteins | [46] | http://www.hprd.org (accessed on 24 April 2023) |
| VPTMdb | SVM, NB, RF | Virus | Predicts viral p-Ser | [47] | http://vptmdb.com:8787/VPTMdb/ (accessed on 24 April 2023) |
| pTestis | - | Mouse | Testis phosphorylation sites from various studies were analyzed, integrated with the iGPS prediction results, which present the potential kinase–substrate regulatory relationships | [48] | http://ptestis.biocuckoo.org/ (accessed on 24 April 2023) |
| PhosphoPep | BLAST | Multiple | Database of protein phosphorylation sites for systems level research in model organisms | [49] | http://www.unipep.org/phosphopep/index.php (accessed on 24 April 2023) |
| PhosphoPOINT | PPI, BLASTP | Human | Annotates interactions among kinases, with their downstream substrates and interacting phosphoproteins | [50] | http://kinase.bioinformatics.tw/ (accessed on 24 April 2023), https://bioregistry.io/registry/phosphopoint.protein (accessed on 24 April 2023) |
| Name | Technique | Organisms | Description/Functionality | Ref | Website |
|---|---|---|---|---|---|
| NetPhos 3.1 | ANN | Multiple | Generates NN predictions for serine, threonine, and tyrosine phosphorylation sites in eukaryotic proteins. Utilizes sequence composition features, both generic and kinase specific predictions | [30] | https://mybiosoftware.com/tag/netphos (accessed on 24 April 2023) |
| NetPhosK | ANN | Eukaryotes | Kinase-specific phosphorylation sites prediction | [51] | https://www.hsls.pitt.edu/obrc/index.php?page=URL1117048165 (accessed on 24 April 2023) |
| Scansite 2.0 | PSSM | Human | A tool built on experimental binding and/or substrate information from oriented peptide library screening and phage display experiments, together with detailed biochemical characterization to derive a weight matrix-based scoring algorithm that predicts protein–protein interactions and sites of phosphorylation | [52] | https://scansite4.mit.edu/#home (accessed on 24 April 2023) |
| PhosphoNet | PSSM | Human | An open source of putative phosphosites predicted after improvisation of kinase substrate prediction algorithm to the primary structure of proteins | [53] | http://www.phosphonet.ca (accessed on 24 April 2023) |
| Predphospho | SVM | Human | Predicts the changes in phosphorylation sites caused by amino acid variations at intra- and interspecies levels | [54] | http://www.ngri.re.kr/proteo/PredPhospho.htm (accessed on 24 April 2023) |
| NetworkKIN | ANN, PSSM | Human | Uses probabilistic protein association network (string) to model the context of kinases and substrates, combined with consensus sequence motifs | [55] | https://networkin.info/ (accessed on 24 April 2023) |
| jEcho | Weight vector | Human | Phosphorylation sites of kinases | [56] | http://www.healthinformaticslab.org/supp/resources.php (accessed on 24 April 2023) |
| PhoScan | Scoring function | Human | Predicts kinase-specific phosphorylation sites with sequence features by a log-odds ratio approach | [57] | http://bioinfo.au.tsinghua.edu.cn/phoscan/ (accessed on 24 April 2023) |
| Predphos | SVM | Multiple | Structural-based prediction of phosphorylation sites, hybrid approach, which incorporates bootstrap resampling technique, SVM-based fusion classifiers and majority voting strategy | [58] | No tool link |
| NetPhosYeast | ANN | Yeast | Prediction of protein phosphorylation sites in yeast | [59] | https://services.healthtech.dtu.dk/service.php?NetPhosYeast-1.0 (accessed on 24 April 2023) |
| GPS 6.0 (group-based prediction system) | MLS, PSSM, GA | Mammalian | Protein phosphorylation sites and their cognate kinases (addresses false positive rates in prediction) | [60,61] | http://gps.biocuckoo.org/ (accessed on 24 April 2023) |
| iGPS | GPS with PPI | Human | It is a GPS algorithm with the interaction filter, or in vivo GPS mainly for the prediction of in vivo site-specific kinase-substrate relation (ssKSRs) | [62] | http://igps.biocuckoo.org/links.php (accessed on 24 April 2023) |
| PPRED | PSSM, SVM | - | Incorporates only evolutionary information of PSSM profile of the proteins in predicting phosphorylation sites | [63] | http://www.cse.univdhaka.edu/~ashis/ppred/index.php (accessed on 24 April 2023) |
| Phos3D | SVM | Human | Prediction of phosphorylation sites (p-sites) in proteins, originally designed to investigate the advantages of including spatial information in p-site prediction | [64] | https://phos3d.mpimp-golm.mpg.de/cgi-bin/index.py (accessed on 24 April 2023) |
| DAPPLE 2 | BLAST | Human | Homology-based prediction of phosphorylation sites | [65] | http://saphire.usask.ca/saphire/dapple2 (accessed on 24 April 2023) |
| EMBER | CNN + RNN | Multiple | Embedding-based multilabel prediction of phosphorylation events (EMBER), a DL method that integrates kinase phylogenetic information and motif-dissimilarity information into a multilabel classification model for the prediction of kinase motif phosphorylation events | [66] | https://github.com/gomezlab/EMBER (accessed on 24 April 2023) |
| KinomeXplorer | NetworKIN algorithm, a novel Bayesian scoring scheme | Human and major eukaryotes | Analyze phosphorylation-dependent protein interaction networks | [67] | http://kinomexplorer.info/ (accessed on 24 April 2023) |
| PhosTransfer | CNN | Info not available | Hierarchical kinase-specific phosphorylation site (KPS) prediction | [68] | https://github.com/yxu132/PhosTransfer (accessed on 24 April 2023) |
| MusiteDeep | CNN/CapsNet | Human | Prediction and visualization for multiple PTMs and simultaneously potential PTM cross-talks | [69] | https://www.musite.net (accessed on 24 April 2023) |
| PROSPECT | CNN | E. coli | Predicts histidine phosphorylation sites from sequence information | [70] | https://bio.tools/prospect-web (accessed on 24 April 2023) |
| DeepKinZero | ZSL | Human | Predicts the kinase acting on a phosphosite for kinases with no known phosphosite information | [71] | https://github.com/Tastanlab/DeepKinZero (accessed on 24 April 2023) |
| DeepPPSite | LSTM | Mammals and Arabidopsis thaliana | Long short-term memory (LSTM) recurrent network for predicting phosphorylation sites | [72] | https://github.com/saeed344/DeepPPSite (accessed on 24 April 2023) |
| DeepIPs | CNN + LSTM | Human | Identification of phosphorylation sites using deep learning method | [73] | https://github.com/linDing-group/DeepIPs (accessed on 24 April 2023) |
| Rice_Phospho 1.0 | SVM | Rice | Predicts protein phosphorylation sites in rice | [74] | http://bioinformatics.fafu.edu.cn/rice_phospho1.0 (accessed on 24 April 2023) |
| Yeast KID | - | Yeast | The first literature-curated database for kinases that integrates a series of HTP and LTP, genetic, physical, and biochemical experimental evidence with the goal of establishing known kinase–substrate relationships. | [75] | http://www.moseslab.csb.utoronto.ca/KID/ (accessed on 24 April 2023) |
| AutoMotif | SVM | The service uses a supervised support vector machine approach to predict various types of phosphorylation sites in proteins | [76] | http://ams2.bioinfo.pl/ (accessed on 24 April 2023) | |
| PhosIDN | Multilayer DNN | Human | An integrated DNN approach for improving protein phosphorylation site prediction by combining sequence and protein–protein interaction information | [77] | https://github.com/ustchangyuanyang/PhosIDN (accessed on 24 April 2023) |
| DeepPhos | CNN | Human | Uses densely connected CNN for kinase-specific phosphorylation site prediction | [29] | https://github.com/USTC-HIlab/DeepPhos (accessed on 24 April 2023) |
| Chlamy-EnPhosSite | CNN + LSTM | Chlamydomonas reinhardtii | Can predict novel sites of phosphorylation within the entire C. reinhardtii proteome | [78] | https://github.com/dukkakc/Chlamy-EnPhosSite (accessed on 24 April 2023) |
| DeepPSP | DNN, SENet, Bi-LSTM | ? | Uses both local and global sequence information to improve phosphorylation site prediction performance | [79] | https://github.com/DeepPSP (accessed on 24 April 2023) |
| Predikin 2.0 | PSSM | Human | Utilizes the kinase sequence to build scoring matrices based on key residues in the kinase catalytic domain that are known from structural analysis to interact with the substrate phosphorylation site. | [80] | http://predikin.biosci.uq.edu.au (accessed on 24 April 2023) |
| KinasePhos2.0 | SVM | Human? | Predicts phosphorylation sites based on protein sequence profile and protein coupling pattern and the type of kinase that acts at each site | [81] | http://kinasephos2.mbc.nctu.edu.tw/document.html, https://bio.tools/kinasephos_2.0 (accessed on 24 April 2023) |
| KinasePhos 3.0 | SVM, XGBoost | Human and others | Provides comprehensive annotations of kinase-specific phosphorylation sites on multiple proteins. Shapley additive explanations (SHAP) was integrated to increase the feature interpretability | [82] | https://awi.cuhk.edu.cn/KinasePhos/index.html, https://github.com/tom-209/KinasePhos-3.0-executable-file (accessed on 24 April 2023) |
| DISPHOS (disorder-enhanced phosphorylation predictor) | BLR | Human | Position-specific amino acid frequencies and disorder information is used to improve the discrimination between phosphorylation and non-phosphorylation sites | [83] | http://www.ist.temple.edu/DISPHOS (accessed on 24 April 2023) |
| pkaPS | Scoring function | Human | Phosphorylation sites of PKA | [84] | http://mendel.imp.univie.ac.at/sat/pkaPS (accessed on 24 April 2023) |
| Quokka | Seqeunce scoring function + LR | Human | Predicts kinase-specific phosphorylation sites | [85] | http://quokka.erc.monash.edu/ (accessed on 24 April 2023) |
| PHOSIDA (phosphorylation site database) | SVM | Multiple | Structural and evolutionary investigation and prediction of phosphosites | [45] | http://phosida.de/ (accessed on 24 April 2023) |
| VPTMdb | SVM, NB, RF | Virus | Predicts viral p-Ser | [47] | http://vptmdb.com:8787/VPTMdb/ (accessed on 24 April 2023) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varshney, N.; Mishra, A.K. Deep Learning in Phosphoproteomics: Methods and Application in Cancer Drug Discovery. Proteomes 2023, 11, 16. https://doi.org/10.3390/proteomes11020016
Varshney N, Mishra AK. Deep Learning in Phosphoproteomics: Methods and Application in Cancer Drug Discovery. Proteomes. 2023; 11(2):16. https://doi.org/10.3390/proteomes11020016
Chicago/Turabian StyleVarshney, Neha, and Abhinava K. Mishra. 2023. "Deep Learning in Phosphoproteomics: Methods and Application in Cancer Drug Discovery" Proteomes 11, no. 2: 16. https://doi.org/10.3390/proteomes11020016