
The Double-Edged Proteins in Cancer Proteomes and the Generation of Induced Tumor-Suppressing Cells (iTSCs)

Abstract
:1. Introduction
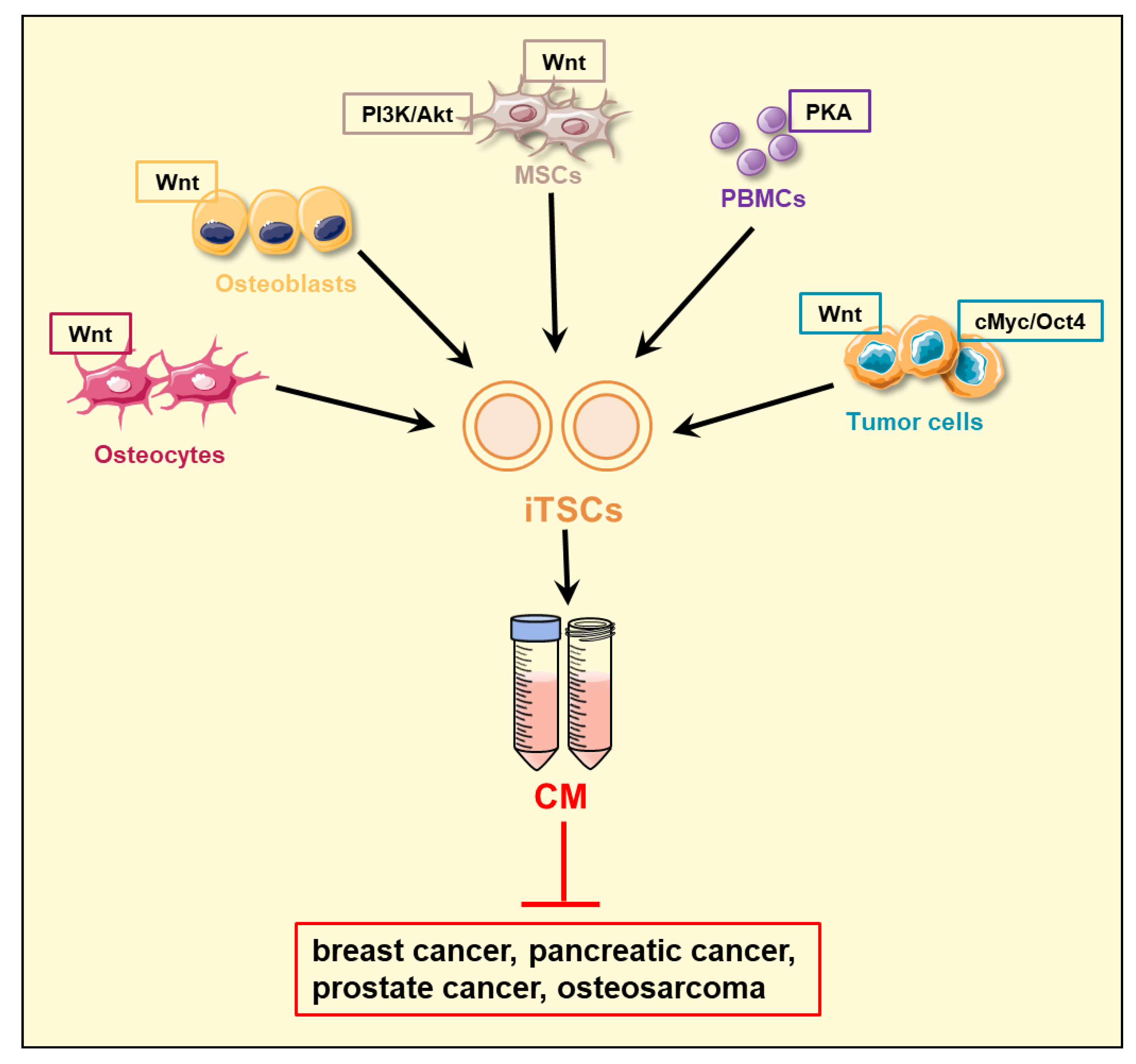
2. Induced Tumor-Suppressing Cells (iTSCs) and Their Conditioned Medium (CM)
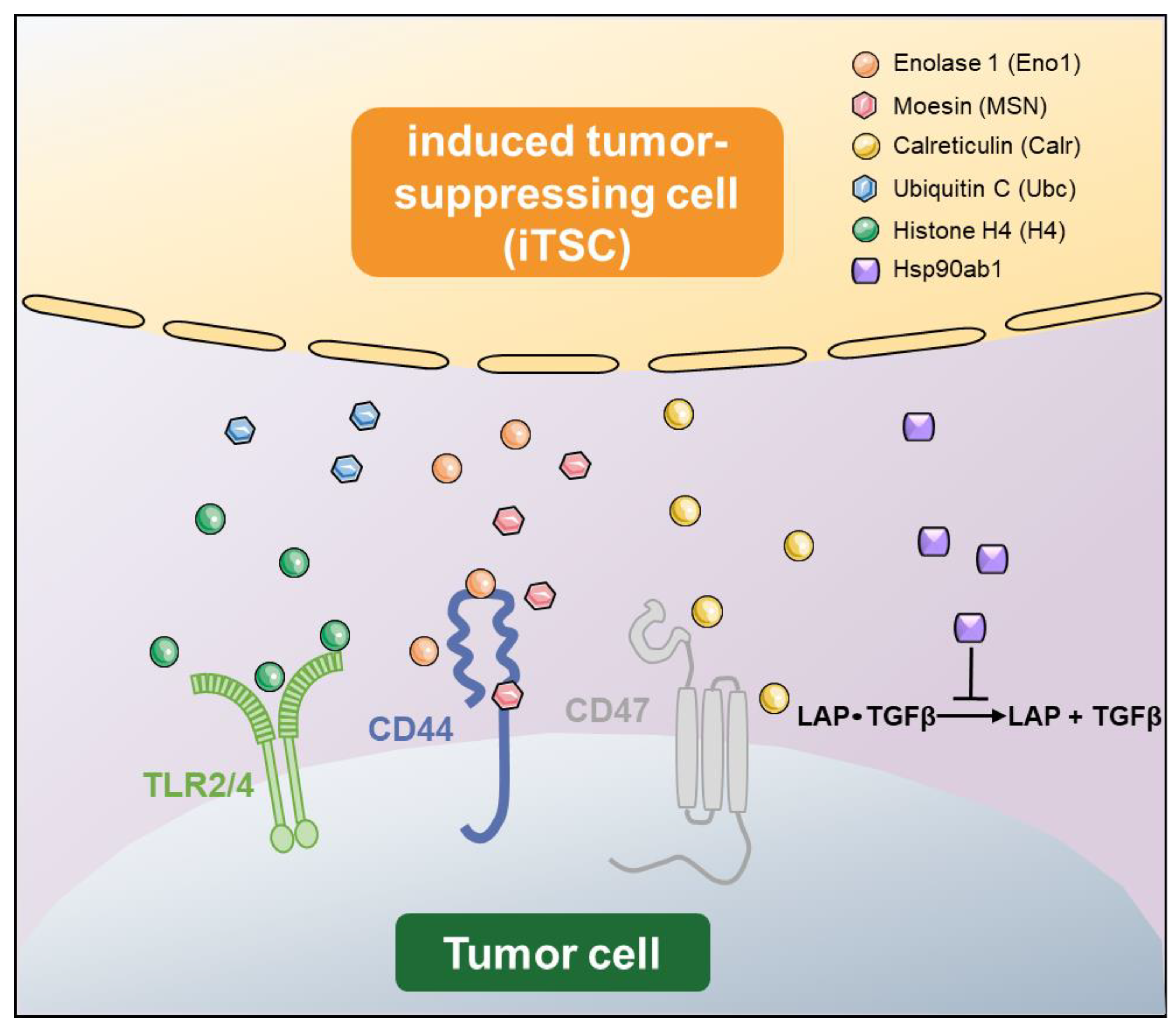
3. Double-Edged Role of Tumor-Suppressing Proteins

{kind=link}
{kind=link}
| Symbol | Name | Predicted Antitumor Action | Reference |
|---|---|---|---|
| Eno1 | Enolase 1 | Interact with CD44 | [37,39,46] |
| MSN | Moesin | Interact with CD44 and fibronectin 1 (FN1) | [37,38,39,46] |
| Calr | Calreticulin | Interact with CD47 | [44,57] |
| Ubc | Ubiquitin C | Unknown | [37] |
| H4 | Histone H4 | Interact with TLR2/4 | [47,92] |
| Hsp90ab1 | Heat shock protein 90 alpha family class B member 1 | Immunoprecipitates latent TGFβ and inactivate TGFβ | [38,39,44,45,46] |
| Ppib | Peptidylprolyl isomerase B | Unknown | [44] |
| Eef2 | Eukaryotic elongation factor 2 | Unknown | [39] |
| VCL | Vinculin | Unknown | [39] |
4. Cell Competition and Cooperation
5. Application
6. Perspective
- Generation of iTSCs: What determines the most effective procedure to generate iTSCs? This question is linked to the genes being overexpressed, signaling pathways to be regulated, and the compatibility of host cells with the genes and pathways.
- Variations among iTSCs: What is the advantage of using autologous MSCs and PBMCs as a host of iTSCs? Is there any advantage of generating iTSCs from patient-derived cancer cells?
- Target cancer types: Is iTSC-derived CM effective for all types of cancer? So far, in vitro and preclinical studies supported the efficacy for breast cancer, prostate cancer, pancreatic cancer, and osteosarcoma using cell lines, primary cells, and freshly isolated cancer tissues. Variations in efficacy were observed, however, and the question is how to enhance tumor-suppressive actions.
- Protein isoforms and modifications: Do protein isoforms and modifications such as phosphorylation alter the antitumor capability of atypical tumor-suppressing proteins? No existing studies have evaluated the role of differential splicing, post-translational modification, and DNA mutations.
- Nonprotein molecules: Do nonprotein molecules in iTSC CM contribute to tumor-suppressive capabilities? Neurotransmitters such as dopamine are shown to act as tumor suppressors, while metabolites such as cholesterol may act as a tumorigenic factor [119,120]. It is also shown that nucleic acids in exosomes affect tumor progression [121]. Further analyses are necessary to evaluate whether any nonprotein molecules significantly contribute to the antitumor action of iTSC CM.
- Mechanism of actions: Do atypical tumor-suppressing proteins exert their antitumor actions by interacting with free proteins, membrane-bound proteins, and extracellular proteins? While existing studies have been focused on interactions with plasma membrane-bound receptors, many other mechanisms can be considered, including the interaction with a tumor microenvironment. For instance, atypical tumor-suppressing proteins may interact with extracellular vesicles or the more recently described nanoparticles such as exomeres [94] and supermeres [95].
7. Limitation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johansson, H.J.; Consortia Oslo Breast Cancer Research Consortium (OSBREAC); Socciarelli, F.; Vacanti, N.M.; Haugen, M.H.; Zhu, Y.; Siavelis, I.; Fernandez, A.; Aure, M.R.; Sennblad, B.; et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat. Commun. 2019, 10, 1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183, 1436–1456. [Google Scholar] [CrossRef] [PubMed]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 2021, 11, 688377. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Huang, C.; Cui Zhou, D.; Hu, Y.; Lih, T.M.; Savage, S.R.; Krug, K.; Clark, D.J.; Schnaubelt, M.; Chen, L.; et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 2021, 184, 5031–5052. [Google Scholar] [CrossRef]
- Saha, K.; Banerjee, A.; Jash, D.; Saha, D. Osteosarcoma relapse as pleural metastasis. South Asian J. Cancer 2013, 2, 56. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, J.; Bai, J.; Shen, H.; Zhang, B.; Deng, L.; Sun, C.; Liu, Y.; Zhang, J.; Zheng, J. Risk and clinicopathological features of osteosarcoma metastasis to the lung: A population-based study. J. Bone Oncol. 2019, 16, 100230. [Google Scholar] [CrossRef]
- Gebhard, C.; Miller, I.; Hummel, K.; Ondrovics, M.N.N.; Schlosser, S.; Walter, I. Comparative proteome analysis of monolayer and spheroid culture of canine osteosarcoma cells. J. Proteom. 2018, 177, 124–136. [Google Scholar] [CrossRef]
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Torry, D.S.; Cooper, G.M. Proto-Oncogenes in Development and Cancer. Am. J. Reprod. Immunol. 1991, 25, 129–132. [Google Scholar] [CrossRef]
- Ala, M. Target c-Myc to treat pancreatic cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M. The Quest for Targets Executing MYC-Dependent Cell Transformation. Front. Oncol. 2016, 6, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riely, G.J.; Marks, J.; Pao, W. KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 201–205. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Lee, E.Y.; Muller, W.J. Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect Biol. 2010, 2, a003236. [Google Scholar] [CrossRef] [Green Version]
- Stachelscheid, J.; Jiang, Q.; Herling, M. The Modes of Dysregulation of the Proto-Oncogene T-Cell Leukemia/Lymphoma 1A. Cancers 2021, 13, 5455. [Google Scholar] [CrossRef]
- Dai, M.; Chen, S.; Teng, X.; Chen, K.; Cheng, W. KRAS as a Key Oncogene in the Clinical Precision Diagnosis and Treatment of Pancreatic Cancer. J. Cancer 2022, 13, 3209–3220. [Google Scholar] [CrossRef]
- Eliades, P.; Flaherty, K.T.; Tsao, H. Oncogene-directed small molecule inhibitors for the treatment of cutaneous melanoma. Melanoma Manag. 2015, 2, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target Myc for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Ricarte-Filho, J.C.; Laetsch, T.W.; Bauer, A.J. Oncogene-specific inhibition in the treatment of advanced pediatric thyroid cancer. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The path to the clinic: A comprehensive review on direct KRAS(G12C) inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [Green Version]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef]
- Krisenko, M.O.; Geahlen, R.L. Calling in SYK: SYK’s dual role as a tumor promoter and tumor suppressor in cancer. Biochim. Biophys. Acta 2015. 1853, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front. Pharmacol. 2019, 10, 38. [Google Scholar] [CrossRef]
- Shortt, J.; Johnstone, R.W. Oncogenes in Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4, a009829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, E.; Poulos, R.C.; Cai, Z.; Barthorpe, S.; Manda, S.S.; Lucas, N.; Beck, A.; Bucio-Noble, D.; Dausmann, M.; Hall, C.; et al. Pan-cancer proteomic map of 949 human cell lines. Cancer Cell 2022, 40, 835–849. [Google Scholar] [CrossRef] [PubMed]
- de la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila myc regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Senoo-Matsuda, N.; Johnston, L.A. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila Myc. Proc. Natl. Acad. Sci. USA 2007, 104, 18543–18548. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Sun, X.; Li, K.; Zha, R.; Feng, Y.; Sano, T.; Dong, C.; Liu, Y.; Aryal, U.K.; Sudo, A.; et al. Generation of the tumor-suppressive secretome from tumor cells. Theranostics 2021, 11, 8517–8534. [Google Scholar] [CrossRef]
- Sun, X.; Li, K.; Hase, M.; Zha, R.; Feng, Y.; Li, B.-Y.; Yokota, H. Suppression of breast cancer-associated bone loss with osteoblast proteomes via Hsp90ab1/moesin-mediated inhibition of TGFβ/FN1/CD44 signaling. Theranostics 2022, 12, 929–943. [Google Scholar] [CrossRef]
- Li, K.; Sun, X.; Zha, R.; Liu, S.; Feng, Y.; Sano, T.; Aryal, U.K.; Sudo, A.; Li, B.-Y.; Yokota, H. Counterintuitive production of tumor-suppressive secretomes from Oct4- and c-Myc-overexpressing tumor cells and MSCs. Theranostics 2022, 12, 3084–3103. [Google Scholar] [CrossRef]
- Bosurgi, L.; Bernink, J.H.; Cuevas, V.D.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef]
- Monti, N.; Verna, R.; Piombarolo, A.; Querqui, A.; Bizzarri, M.; Fedeli, V. Paradoxical Behavior of Oncogenes Undermines the Somatic Mutation Theory. Biomolecules 2022, 12, 662. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Zhuang, Y. Paradoxical role of Id proteins in regulating tumorigenic potential of lymphoid cells. Front. Med. 2018, 12, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wu, D.; Sun, X.; Fan, Y.; Zha, R.; Jalali, A.; Feng, Y.; Li, K.; Sano, T.; Vike, N.; et al. Overexpression of Lrp5 enhanced the anti-breast cancer effects of osteocytes in bone. Bone Res. 2021, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, K.; Zha, R.; Liu, S.; Fan, Y.; Wu, D.; Hase, M.; Aryal, U.K.; Lin, C.-C.; Li, B.-Y.; et al. Preventing tumor progression to the bone by induced tumor-suppressing MSCs. Theranostics 2021, 11, 5143–5159. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, K.; Aryal, U.K.; Li, B.-Y.; Yokota, H. PI3K-activated MSC proteomes inhibit mammary tumors via Hsp90ab1 and Myh9. Mol. Ther.-Oncolytics 2022, 26, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Li, K.-X.; Sun, X.; Li, B.-Y.; Yokota, H. Conversion of Osteoclasts into Bone-Protective, Tumor-Suppressing Cells. Cancers 2021, 13, 5593. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Sun, X.; Feng, Y.; Liu, S.; Hase, M.; Fan, Y.; Zha, R.; Wu, D.; Aryal, U.K.; Li, B.-Y.; et al. Inhibition of the Growth of Breast Cancer-Associated Brain Tumors by the Osteocyte-Derived Conditioned Medium. Cancers 2021, 13, 1061. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Liu, X.; Wang, J.; Chen, X.; Zhang, H.; Kim, S.H.; Cui, J.; Li, R.; Zhang, W.; Kong, Y.; et al. Wnt signaling in bone formation and its therapeutic potential for bone diseases. Ther. Adv. Musculoskelet. Dis. 2013, 5, 13–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Sawakami, K.; Robling, A.G.; Ai, M.; Pitner, N.D.; Liu, D.; Warden, S.J.; Li, J.; Maye, P.; Rowe, D.W.; Duncan, R.L.; et al. The Wnt Co-receptor LRP5 Is Essential for Skeletal Mechanotransduction but Not for the Anabolic Bone Response to Parathyroid Hormone Treatment. J. Biol. Chem. 2006, 281, 23698–23711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, I.; Halleux, C.; Keller, H.; Pegurri, M.; Gooi, J.H.; Weber, P.B.; Feng, J.Q.; Bonewald, L.F.; Kneissel, M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell. Biol. 2010, 30, 3071–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The relation between PI3K/AKT signalling pathway and cancer. Gene 2019, 698, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef]
- London, E.; Bloyd, M.; Stratakis, C.A. PKA functions in metabolism and resistance to obesity: Lessons from mouse and human studies. J. Endocrinol. 2020, 246, R51–R64. [Google Scholar] [CrossRef]
- Sapio, L.; Di Maiolo, F.; Illiano, M.; Esposito, A.; Chiosi, E.; Spina, A.; Naviglio, S. Targeting protein kinase A in cancer therapy: An update. EXCLI J. 2014, 13, 843–855. [Google Scholar]
- Li, K.; Sun, X.; Li, H.; Ma, H.; Zhou, M.; Minami, K.; Tamari, K.; Ogawa, K.; Pandya, P.H.; Saadatzadeh, M.R.; et al. Suppression of osteosarcoma progression by engineered lymphocyte-derived proteomes. Genes Dis. in press. 2022. [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.-C.; Chou, C.-C.; Kulp, S.K.; Chen, C.-S. AMPK as a potential anticancer target—friend or foe? Curr. Pharm. Des. 2014, 20, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.-M.; Hay, N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch. Pharmacal Res. 2015, 38, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.; Wu, A.; Chen, X.; Tian, Y.; Lin, X. Enolase 1, a Moonlighting Protein, as a Potential Target for Cancer Treatment. Int. J. Biol. Sci. 2021, 17, 3981–3992. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Wang, H.; Bai, L.; Chen, Y.; Chen, S.; Gao, K.; Wang, H.; Wu, S.; Song, H.; Ma, K.; et al. Activation of TMEM16A Ca(2+)-activated Cl(-) channels by ROCK1/moesin promotes breast cancer metastasis. J. Adv. Res. 2021, 33, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Spisek, R.; Kroemer, G.; Galluzzi, L. Calreticulin and cancer. Cell Res. 2020, 31, 5–16. [Google Scholar] [CrossRef]
- Radón, V.; Czesla, M.; Reichelt, J.; Fehlert, J.; Hammel, A.; Rosendahl, A.; Knop, J.-H.; Wiech, T.; Wenzel, U.O.; Sachs, M.; et al. Ubiquitin C-Terminal Hydrolase L1 is required for regulated protein degradation through the ubiquitin proteasome system in kidney. Kidney Int. 2018, 93, 110–127. [Google Scholar] [CrossRef] [Green Version]
- Mena, H.A.; Carestia, A.; Scotti, L.; Parborell, F.; Schattner, M.; Negrotto, S. Extracellular histones reduce survival and angiogenic responses of late outgrowth progenitor and mature endothelial cells. J. Thromb. Haemost. 2016, 14, 397–410. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Xu, H.; Niu, M.; Yuan, X.; Wu, K.; Liu, A. CD44 as a tumor biomarker and therapeutic target. Exp. Hematol. Oncol. 2020, 9, 36. [Google Scholar] [CrossRef]
- Tsukita, S.; Yonemura, S. ERM (ezrin/radixin/moesin) family: From cytoskeleton to signal transduction. Curr. Opin. Cell Biol. 1997, 9, 70–75. [Google Scholar] [CrossRef]
- Ivetic, A.; Ridley, A.J. Ezrin/radixin/moesin proteins and Rho GTPase signalling in leucocytes. Immunology 2004, 112, 165–176. [Google Scholar] [CrossRef]
- Bao, H.; Huo, Q.; Yuan, Q.; Xu, C. Fibronectin 1: A Potential Biomarker for Ovarian Cancer. Dis. Markers 2021, 2021, 5561651. [Google Scholar] [CrossRef] [PubMed]
- Jun, B.; Guo, T.; Libring, S.; Chanda, M.; Paez, J.; Shinde, A.; Wendt, M.; Vlachos, P.; Solorio, L. Fibronectin-Expressing Mesenchymal Tumor Cells Promote Breast Cancer Metastasis. Cancers 2020, 12, 2553. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: One protein, one gene, many functions. Biochem. J. 1999, 344 Pt 2, 281–292. [Google Scholar] [CrossRef]
- Han, A.; Li, C.; Zahed, T.; Wong, M.; Smith, I.; Hoedel, K.; Green, D.; Boiko, A.D. Calreticulin is a Critical Cell Survival Factor in Malignant Neoplasms. PLOS Biol. 2019, 17, e3000402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPα Immune Checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.M.G.; Bianconi, V.; Pirro, M.; Jaafari, M.R.; Hatamipour, M.; Sahebkar, A. CD47: Role in the immune system and application to cancer therapy. Cell. Oncol. 2019, 43, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Silk, E.; Zhao, H.; Weng, H.; Ma, D. The role of extracellular histone in organ injury. Cell Death Dis. 2017, 8, e2812. [Google Scholar] [CrossRef] [Green Version]
- Alhamdi, Y.; Toh, C.-H. The role of extracellular histones in haematological disorders. Br. J. Haematol. 2016, 173, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Cramer, T.J.; Sinha, R.K.; Griffin, J.H. Reduction Of Histone H1 Cytotoxicity By Activated Protein C and Its Exosite Variants. Blood 2013, 122, 2334. [Google Scholar] [CrossRef]
- Mariño-Ramírez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert Rev. Proteom. 2005, 2, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, J.; Guan, L.; Mao, L.; Li, S.; Zhao, J. Histone H4 aggravates inflammatory injury through TLR4 in chlorine gas-induced acute respiratory distress syndrome. J. Occup. Med. Toxicol. 2020, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kulkarni, A.B. Extracellular heat shock protein HSP90beta secreted by MG63 osteosarcoma cells inhibits activation of latent TGF-beta1. Biochem. Biophys. Res. Commun. 2010, 398, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, T.; Pagnotti, G.M.; Guise, T.A.; Mohammad, K.S. The Role of TGF-β in Bone Metastases. Biomolecules 2021, 11, 1643. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2015, 1855, 50–60. [Google Scholar]
- Yao, Q.; Li, M.; Yang, H.; Chai, H.; Fisher, W.; Chen, C. Roles of Cyclophilins in Cancers and Other Organ Systems. World J. Surg. 2005, 29, 276–280. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, L.; Du, M.; Wei, X.; Zhang, J.; Hui, Y.; Chen, C.; Li, G.; Hou, J. Eukaryotic Elongation Factor 2 (eEF2) is a Potential Biomarker of Prostate Cancer. Pathol. Oncol. Res. 2017, 24, 885–890. [Google Scholar] [CrossRef]
- Shi, N.; Chen, X.; Liu, R.; Wang, D.; Su, M.; Wang, Q.; He, A.; Gu, H. Eukaryotic elongation factors 2 promotes tumor cell proliferation and correlates with poor prognosis in ovarian cancer. Tissue Cell 2018, 53, 53–60. [Google Scholar] [CrossRef]
- Izard, T.; Brown, D.T. Mechanisms and Functions of Vinculin Interactions with Phospholipids at Cell Adhesion Sites. J. Biol. Chem. 2016, 291, 2548–2555. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Xu, H.; Gong, L.; Cao, D.; Jin, T.; Wang, Y.; Pi, J.; Yang, Y.; Yi, X.; Liao, D.; et al. Vinculin orchestrates prostate cancer progression by regulating tumor cell invasion, migration, and proliferation. Prostate 2021, 81, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Monestier, M.; Esmon, N.L.; Esmon, C.T. Extracellular Histones Are Mediators of Death through TLR2 and TLR4 in Mouse Fatal Liver Injury. J. Immunol. 2011, 187, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Han, X.; Bo, J.; Han, W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Zhang, Q.; Jeppesen, D.K.; Higginbotham, J.N.; Graves-Deal, R.; Trinh, V.Q.; Ramirez, M.A.; Sohn, Y.; Neininger, A.C.; Taneja, N.; McKinley, E.T.; et al. Supermeres are functional extracellular nanoparticles replete with disease biomarkers and therapeutic targets. Nature 2021, 23, 1240–1254. [Google Scholar] [CrossRef]
- Archetti, M.; Pienta, K.J. Cooperation among cancer cells: Applying game theory to cancer. Nat. Rev. Cancer 2018, 19, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowling, S.; Lawlor, K.; Rodríguez, T.A. Cell competition: The winners and losers of fitness selection. Development 2019, 146, dev167486. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.E. Emerging mechanisms of cell competition. Nat. Rev. Genet. 2020, 21, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Tamori, Y.; Deng, W.-M. Cell competition and its implications for development and cancer. J. Genet. Genom. 2011, 38, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.E.; Li, W. Cell Competition and Its Possible Relation to Cancer. Cancer Res. 2008, 68, 5505–5507. [Google Scholar] [CrossRef] [Green Version]
- Kajita, M.; Fujita, Y. EDAC: Epithelial defence against cancer--cell competition between normal and transformed epithelial cells in mammals. J. Biochem. 2015, 158, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdus-Saboor, I.; Mancuso, V.P.; Murray, J.I.; Palozola, K.; Norris, C.; Hall, D.H.; Howell, K.; Huang, K.; Sundaram, M.V. Notch and Ras promote sequential steps of excretory tube development in C. elegans. Development 2011, 138, 3545–3555. [Google Scholar] [CrossRef] [PubMed]
- Casás-Selves, M.; DeGregori, J. How Cancer Shapes Evolution and How Evolution Shapes Cancer. Evol. Educ. Outreach 2011, 4, 624–634. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Lytle, N.K.; Chen, B.; Jyotsana, N.; Novak, S.W.; Cho, C.J.; Caplan, L.; Ben-Levy, O.; Neininger, A.C.; Burnette, D.T.; et al. Single-Cell Transcriptomics Reveals a Conserved Metaplasia Program in Pancreatic Injury. Gastroenterology 2022, 162, 604–620. [Google Scholar] [CrossRef]
- Schlesinger, Y.; Yosefov-Levi, O.; Kolodkin-Gal, D.; Granit, R.Z.; Peters, L.; Kalifa, R.; Xia, L.; Nasereddin, A.; Shiff, I.; Amran, O.; et al. Single-cell transcriptomes of pancreatic preinvasive lesions and cancer reveal acinar metaplastic cells’ heterogeneity. Nat. Commun. 2020, 11, 4516. [Google Scholar] [CrossRef]
- Ling, L.; Feng, X.; Wei, T.; Wang, Y.; Wang, Y.; Wang, Z.; Tang, D.; Luo, Y.; Xiong, Z. Human amnion-derived mesenchymal stem cell (hAD-MSC) transplantation improves ovarian function in rats with premature ovarian insufficiency (POI) at least partly through a paracrine mechanism. Stem Cell Res. Ther. 2019, 10, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanate, A.S.; Majhail, N.S.; Savani, B.N.; Bredeson, C.; Champlin, R.E.; Crawford, S.; Giralt, S.A.; LeMaistre, C.F.; Marks, D.I.; Omel, J.L.; et al. Indications for Hematopoietic Cell Transplantation and Immune Effector Cell Therapy: Guidelines from the American Society for Transplantation and Cellular Therapy. Biol. Blood Marrow Transplant. 2020, 26, 1247–1256. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Fischer, J.W.; Bhattarai, N. CAR-T Cell Therapy: Mechanism, Management, and Mitigation of Inflammatory Toxicities. Front. Immunol. 2021, 12, 693016. [Google Scholar] [CrossRef]
- Tsuboi, R.; Niiyama, S.; Irisawa, R.; Harada, K.; Nakazawa, Y.; Kishimoto, J. Autologous cell–based therapy for male and female pattern hair loss using dermal sheath cup cells: A randomized placebo-controlled double-blinded dose-finding clinical study. J. Am. Acad. Dermatol. 2020, 83, 109–116. [Google Scholar] [CrossRef]
- Madrid, M.; Sume, C.; Aivio, S.; Saklayen, N. Autologous Induced Pluripotent Stem Cell-Based Cell Therapies: Promise, Progress, and Challenges. Curr. Protoc. 2021, 1, e88. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-Y.; Hsieh, C.-H.; Huang, Y.-T.; Chu, S.-Y.; Chen, C.-M.; Lee, W.-J.; Liu, S.-J. Enhanced Paclitaxel Efficacy to Suppress Triple-Negative Breast Cancer Progression Using Metronomic Chemotherapy with a Controlled Release System of Electrospun Poly-d-l-Lactide-Co-Glycolide (PLGA) Nanofibers. Cancers 2021, 13, 3350. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.-O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of Neoadjuvant Cisplatin in Triple-Negative Breast Cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Ohnuki, M.; Takahashi, K. Present and future challenges of induced pluripotent stem cells. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140367. [Google Scholar] [CrossRef]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, P.; Kim, S.I. iPSC-Derived Natural Killer Cells for Cancer Immunotherapy. Mol. Cells 2021, 44, 541–548. [Google Scholar] [CrossRef]
- Qiao, Y.; Agboola, O.S.; Hu, X.; Wu, Y.; Lei, L. Tumorigenic and Immunogenic Properties of Induced Pluripotent Stem Cells: A Promising Cancer Vaccine. Stem Cell Rev. Rep. 2020, 16, 1049–1061. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Lan, Y.-L.; Wang, X.; Xing, J.-S.; Yu, Z.-L.; Lou, J.-C.; Ma, X.-C.; Zhang, B. Anti-cancer effects of dopamine in human glioma: Involvement of mitochondrial apoptotic and anti-inflammatory pathways. Oncotarget 2017, 8, 88488–88500. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [PubMed] [Green Version]
- Smith, L.M.; Agar, J.N.; Chamot-Rooke, J.; Danis, P.O.; Ge, Y.; Loo, J.A.; Paša-Tolić, L.; Tsybin, Y.O.; Kelleher, N.L. The Consortium for Top-Down Proteomics The Human Proteoform Project: Defining the human proteome. Sci. Adv. 2021, 7, eabk0734. [Google Scholar] [CrossRef] [PubMed]


| Signaling Regulation | Drug | iTSC-Generating Cells | Reference |
|---|---|---|---|
| PKA activation | CW008 | lymphocytes, PBMCs | [57] |
| Wnt activation | BML284 | MSCs, osteocytes osteoblasts, osteoclasts, tumor cells | [38,43,44,46] |
| PI3K/AKT activation | YS49 | MSCs | [44,45] |
| cMyc overexpression | tumor cells | [39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Huo, Q.; Li, B.-Y.; Yokota, H. The Double-Edged Proteins in Cancer Proteomes and the Generation of Induced Tumor-Suppressing Cells (iTSCs). Proteomes 2023, 11, 5. https://doi.org/10.3390/proteomes11010005
Li K, Huo Q, Li B-Y, Yokota H. The Double-Edged Proteins in Cancer Proteomes and the Generation of Induced Tumor-Suppressing Cells (iTSCs). Proteomes. 2023; 11(1):5. https://doi.org/10.3390/proteomes11010005
Chicago/Turabian StyleLi, Kexin, Qingji Huo, Bai-Yan Li, and Hiroki Yokota. 2023. "The Double-Edged Proteins in Cancer Proteomes and the Generation of Induced Tumor-Suppressing Cells (iTSCs)" Proteomes 11, no. 1: 5. https://doi.org/10.3390/proteomes11010005