Comparison of Read Mapping and Variant Calling Tools for the Analysis of Plant NGS Data




Abstract
:1. Introduction
2. Results
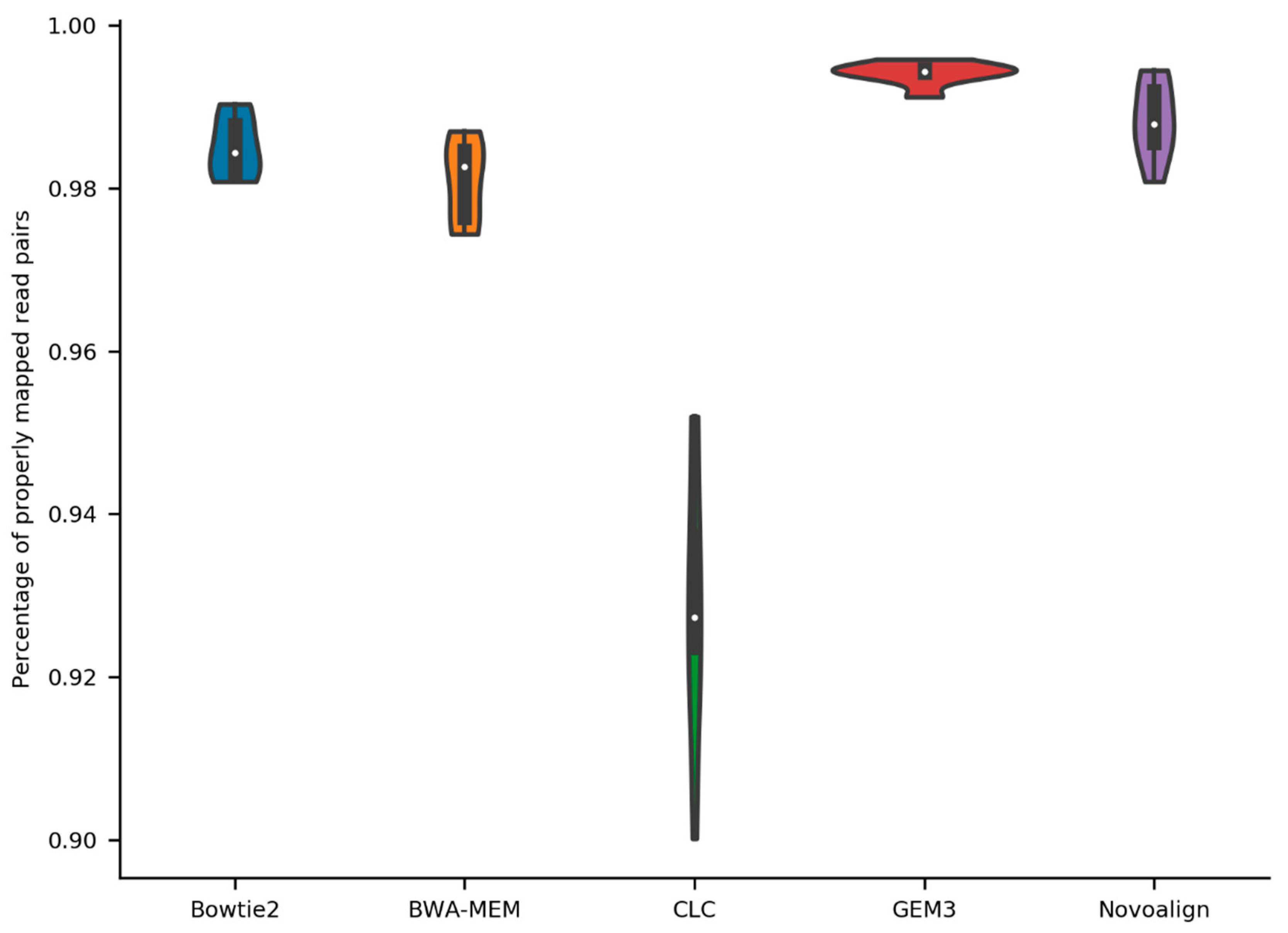
2.1. General Stats about Mapping of Reads
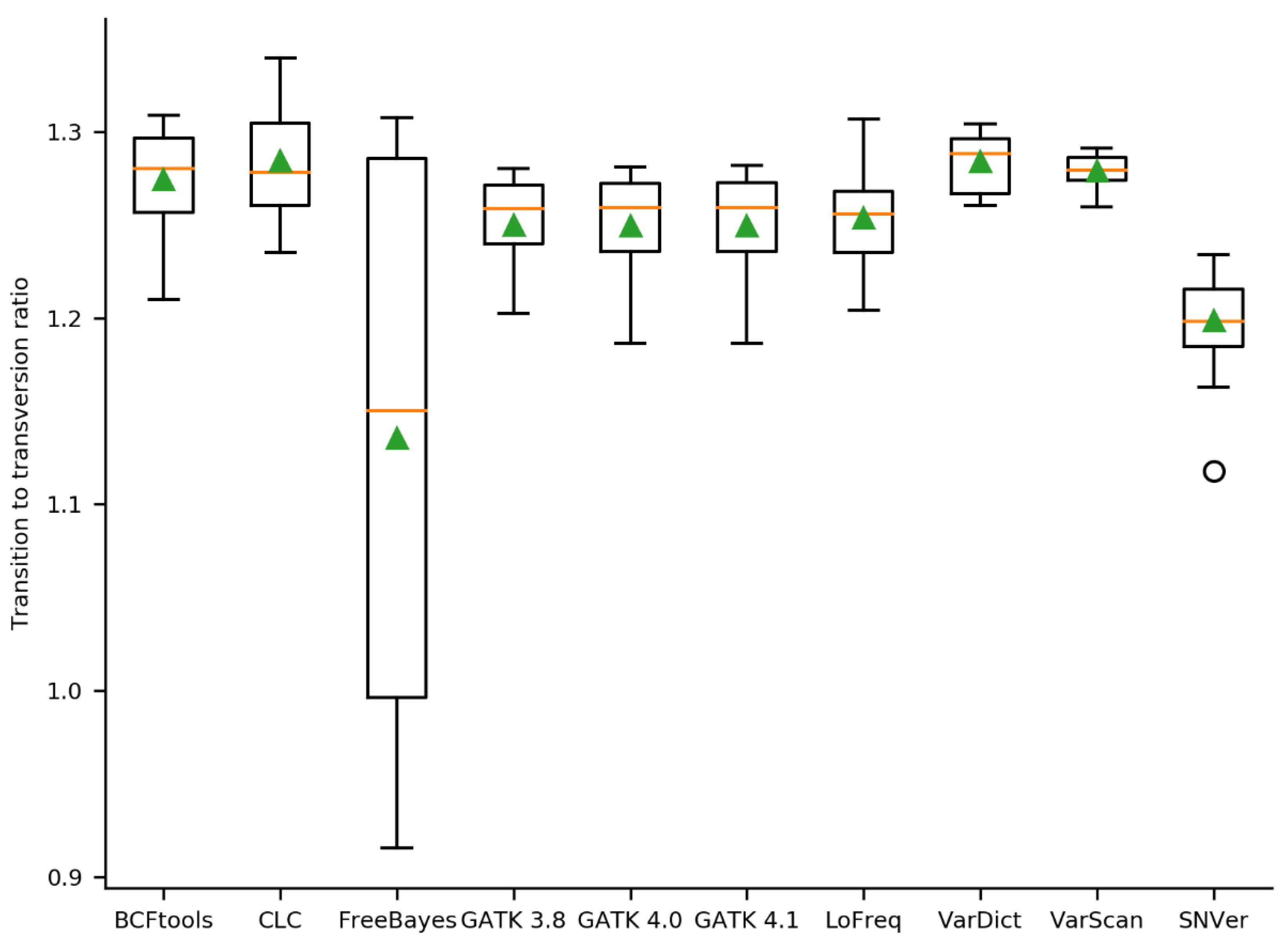
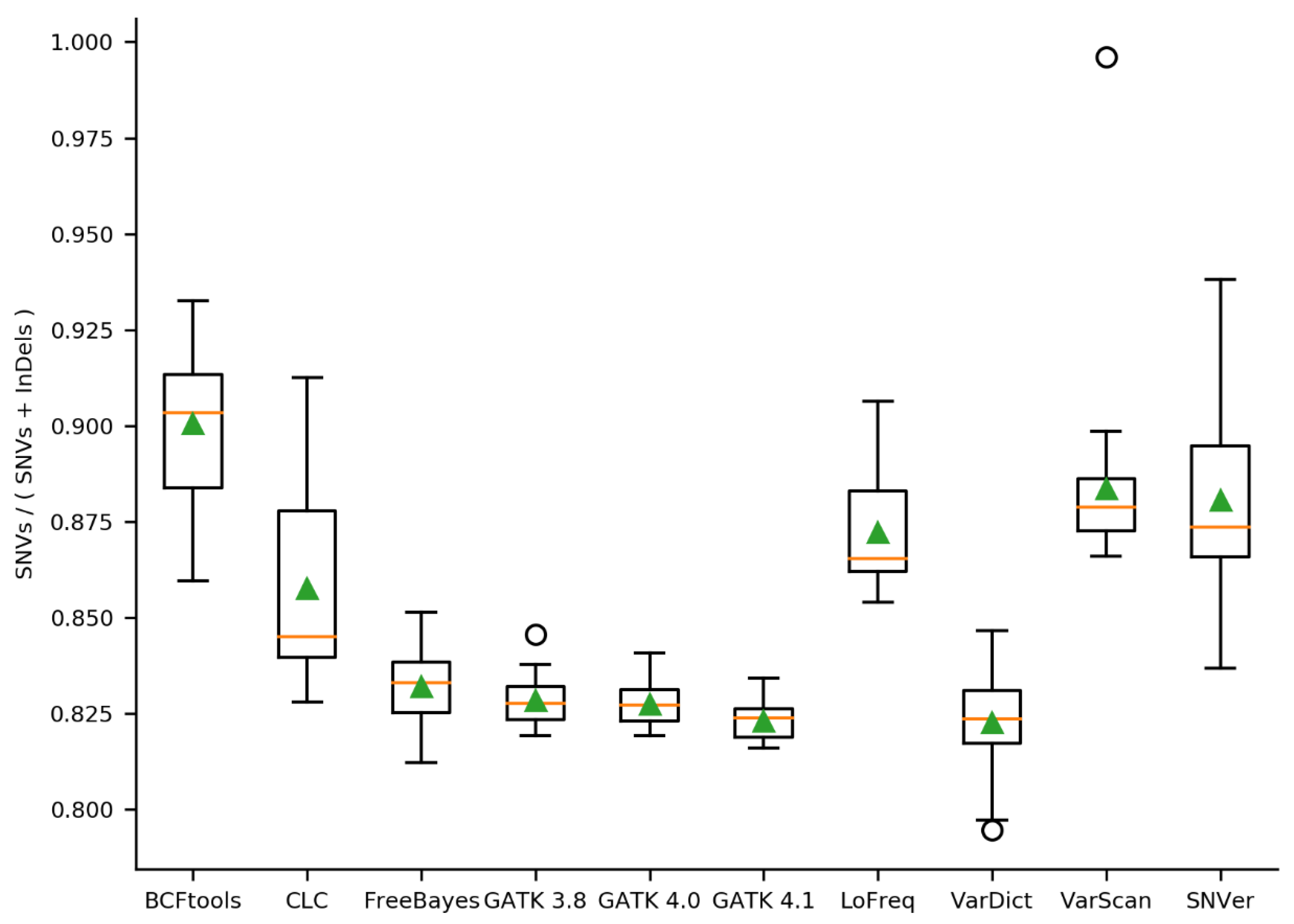
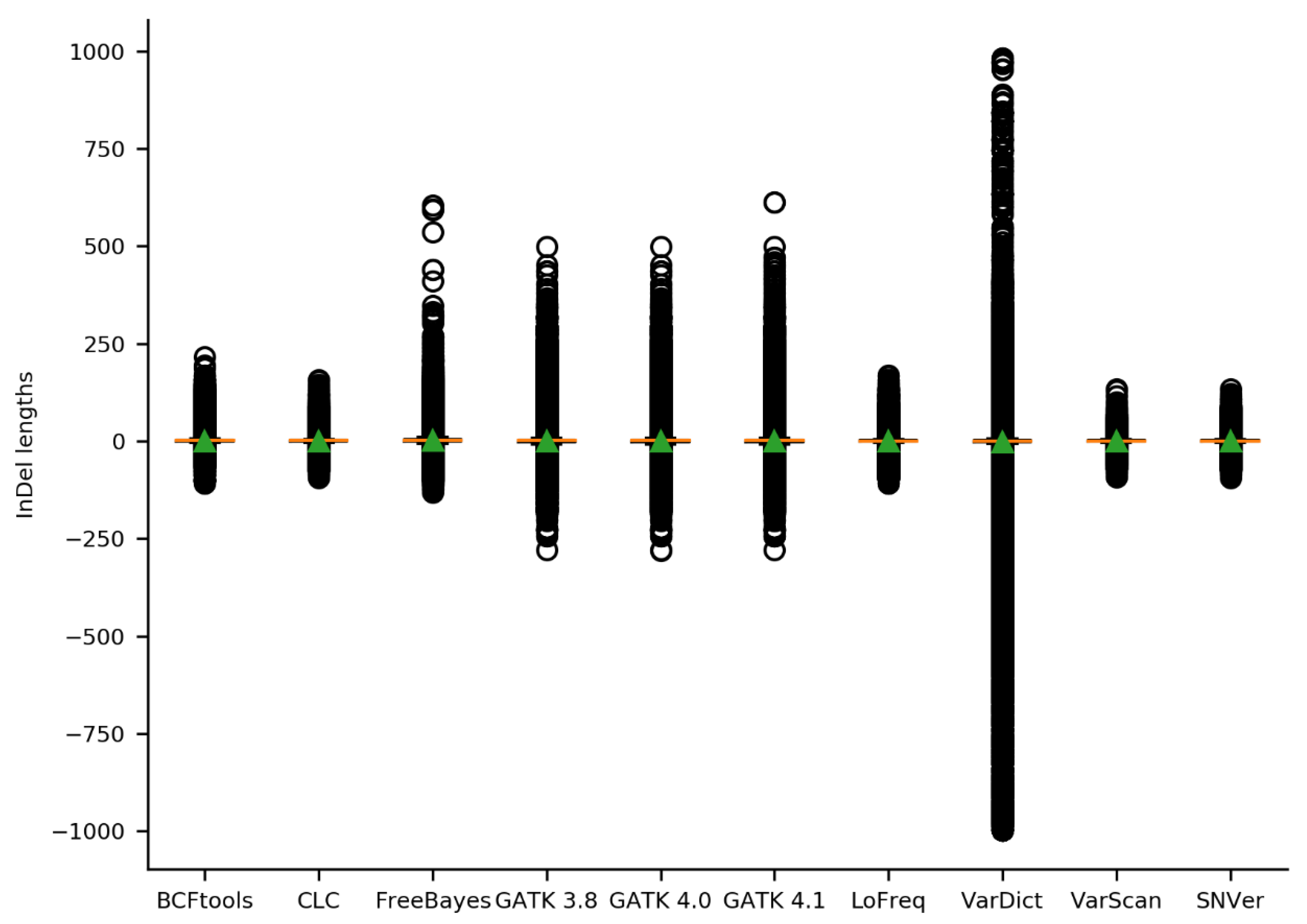
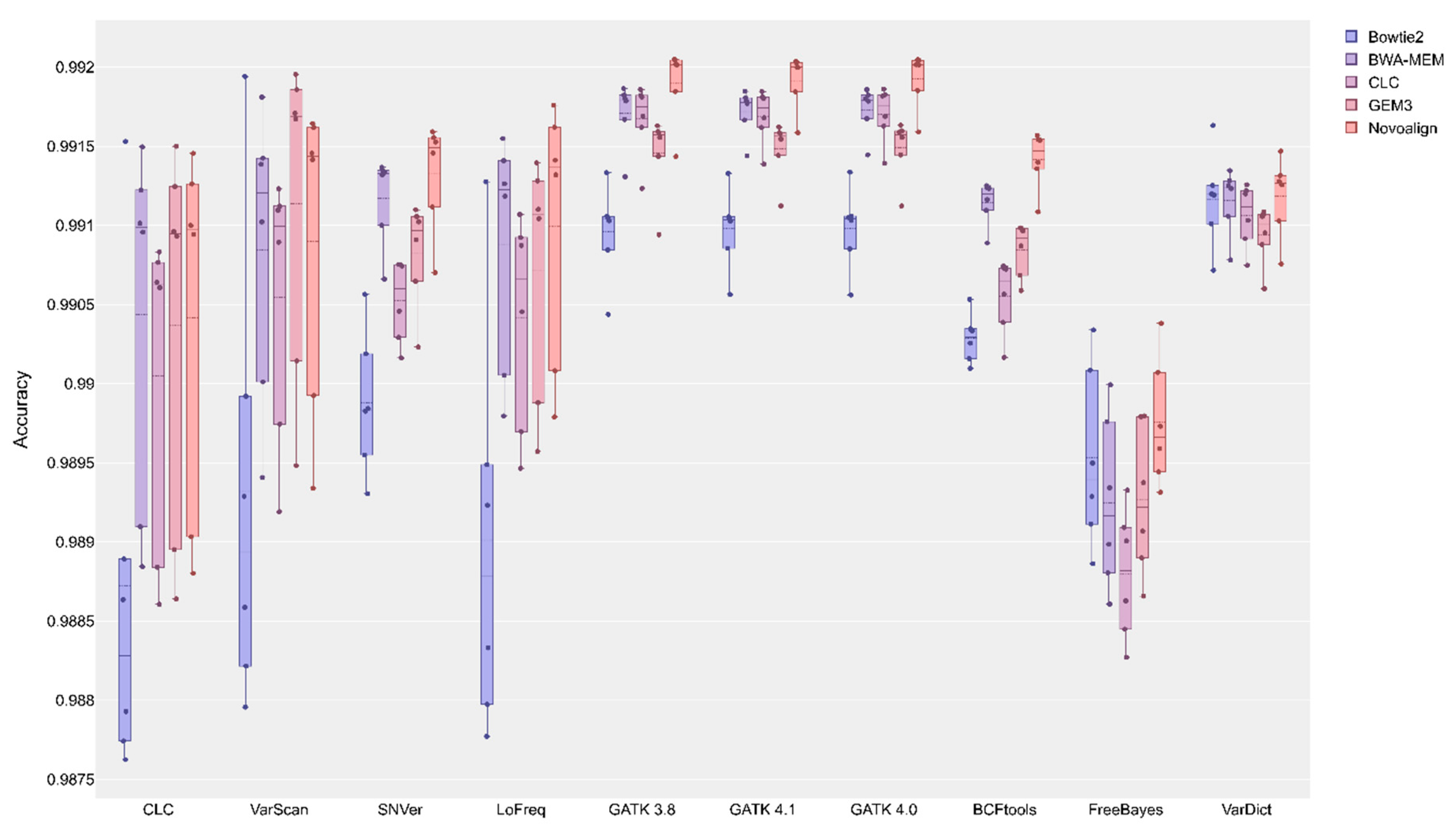
2.2. Initial Variant Calling Results & Validation Results
3. Discussion
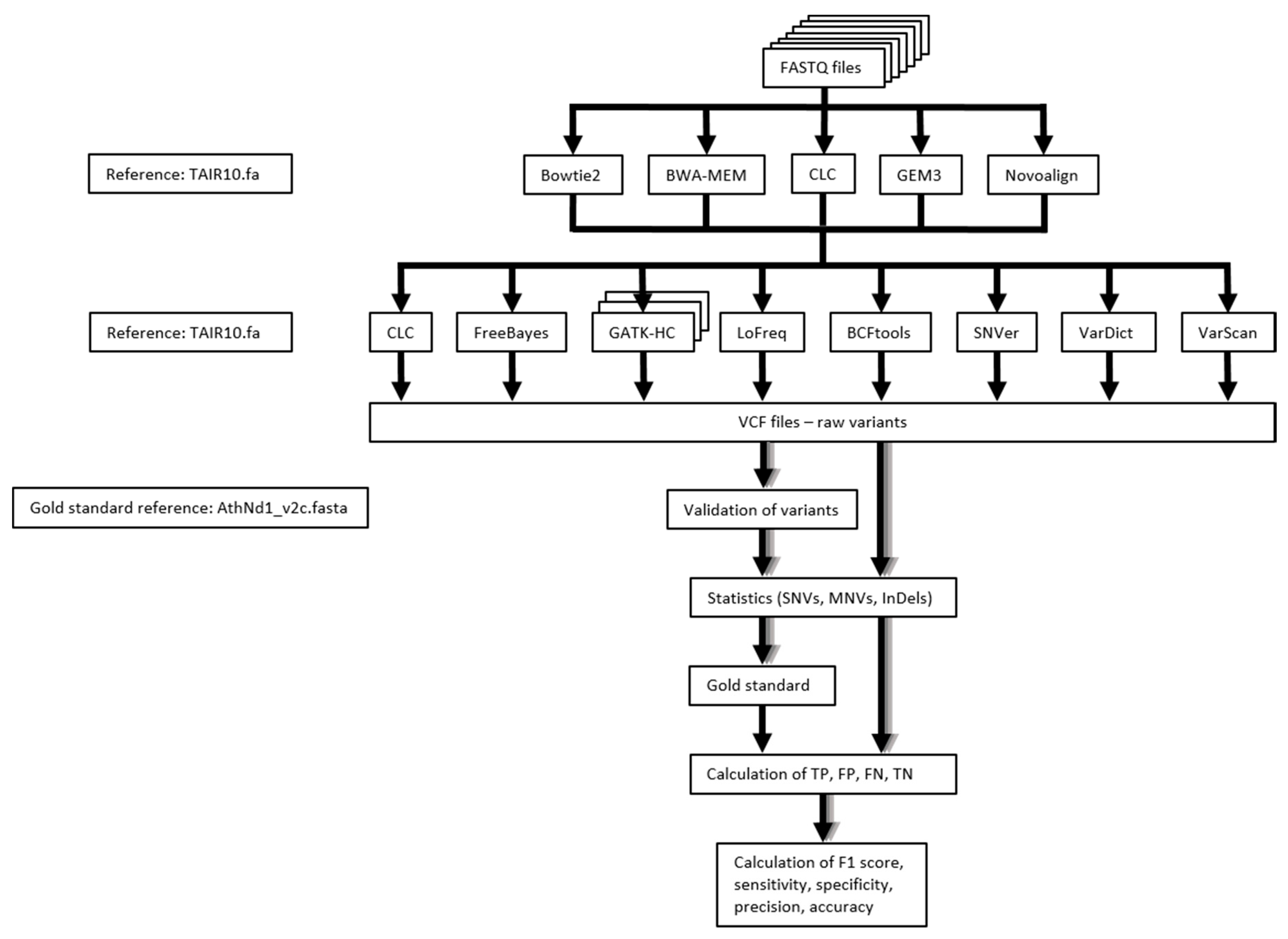
4. Materials and Methods
4.1. Sequence Read Datasets
4.2. Sequence Read Mapping
4.3. Variant Calling
4.4. Performance Measure of Variant Calling Pipelines
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weigel, D.; Mott, R. The 1001 Genomes Project for Arabidopsis thaliana. Genome Biol. 2009, 10, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Pandey, M.K.; Chitikineni, A. Plant Genetics and Molecular Biology; Springer: New York, NY, USA, 2018; ISBN 978-3-319-91313-1. [Google Scholar]
- Li, Y.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yeh, C.-T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene Mapping via Bulked Segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [Green Version]
- Mascher, M.; Jost, M.; Kuon, J.-E.; Himmelbach, A.; Aßfalg, A.; Beier, S.; Scholz, U.; Graner, A.; Stein, N. Mapping-by-sequencing accelerates forward genetics in barley. Genome Biol. 2014, 15, R78. [Google Scholar] [CrossRef] [Green Version]
- Ries, D.; Holtgräwe, D.; Viehöver, P.; Weisshaar, B. Rapid gene identification in sugar beet using deep sequencing of DNA from phenotypic pools selected from breeding panels. BMC Genom. 2016, 17, 236. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, S.P. From next-generation resequencing reads to a high-quality variant data set. Heredity 2017, 118, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Harismendy, O.; Ng, P.C.; Strausberg, R.L.; Wang, X.; Stockwell, T.B.; Beeson, K.Y.; Schork, N.J.; Murray, S.S.; Topol, E.J.; Levy, S.; et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009, 10, R32. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 14 March 2020).
- Planet, E.; Attolini, C.S.-O.; Reina, O.; Flores, O.; Rossell, D. htSeqTools: High-throughput sequencing quality control, processing and visualization in R. Bioinformatics 2012, 28, 589–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, M.; Thompson, R.C.; Maher, C.; Contreras-Galindo, R.; Kaplan, M.H.; Markovitz, D.M.; Omenn, G.; Meng, F. NGSQC: Cross-platform quality analysis pipeline for deep sequencing data. BMC Genom. 2010, 11 (Suppl. S4), S7. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, T.; Hayashizaki, Y.; Daub, C.O. SAMStat: Monitoring biases in next generation sequencing data. Bioinformatics 2011, 27, 130–131. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Ezpeleta, N.; Hackenberg, M.; Aransay, A.M. Bioinformatics for High Throughput Sequencing; Springer Science & Business Media: Berlin, Germany, 2011; ISBN 978-1-4614-0782-9. [Google Scholar]
- Reinert, K.; Langmead, B.; Weese, D.; Evers, D.J. Alignment of Next-Generation Sequencing Reads. Annu. Rev. Genom. Hum. Genet. 2015, 16, 133–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Zhu, F.; Vongsangnak, W.; Tang, Y.; Zhang, W.; Shen, B. Evaluation and Comparison of Multiple Aligners for Next-Generation Sequencing Data Analysis. Available online: https://www.hindawi.com/journals/bmri/2014/309650/ (accessed on 22 January 2020).
- Pabinger, S.; Dander, A.; Fischer, M.; Snajder, R.; Sperk, M.; Efremova, M.; Krabichler, B.; Speicher, M.R.; Zschocke, J.; Trajanoski, Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014, 15, 256–278. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The GEM mapper: Fast, accurate and versatile alignment by filtration. Nat. Methods 2012, 9, 1185–1188. [Google Scholar] [CrossRef]
- Novoalign. Available online: http://novocraft.com/ (accessed on 22 January 2020).
- Li, R.; Yu, C.; Li, Y.; Lam, T.-W.; Yiu, S.-M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [Green Version]
- Ruffalo, M.; LaFramboise, T.; Koyutürk, M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics 2011, 27, 2790–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Guda, K.; Willis, J.; Veigl, M.; Wang, Z.; Markowitz, S.; Adams, M.D.; Sun, S. How do alignment programs perform on sequencing data with varying qualities and from repetitive regions? Biodata Min. 2012, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018, 201178. [Google Scholar] [CrossRef] [Green Version]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Wang, W.; Hu, P.; Lyon, G.J.; Hakonarson, H. SNVer: A statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 2011, 39, e132. [Google Scholar] [CrossRef]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucker, B.; Schilbert, H. Genomics and Transcriptomics Advances in Plant Sciences. In Molecular Approaches in Plant Biology and Environmental Challenges; Springer: Singapore, 2019; ISBN 9789811506895. [Google Scholar]
- Fumagalli, M. Assessing the effect of sequencing depth and sample size in population genetics inferences. PLoS ONE 2013, 8, e79667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krøigård, A.B.; Thomassen, M.; Lænkholm, A.-V.; Kruse, T.A.; Larsen, M.J. Evaluation of Nine Somatic Variant Callers for Detection of Somatic Mutations in Exome and Targeted Deep Sequencing Data. PLoS ONE 2016, 11, e0151664. [Google Scholar] [CrossRef] [Green Version]
- Bian, X.; Zhu, B.; Wang, M.; Hu, Y.; Chen, Q.; Nguyen, C.; Hicks, B.; Meerzaman, D. Comparing the performance of selected variant callers using synthetic data and genome segmentation. BMC Bioinform. 2018, 19, 429. [Google Scholar] [CrossRef]
- Hwang, K.-B.; Lee, I.-H.; Li, H.; Won, D.-G.; Hernandez-Ferrer, C.; Negron, J.A.; Kong, S.W. Comparative analysis of whole-genome sequencing pipelines to minimize false negative findings. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, R.R.; Chebotarov, D.; Duitama, J.; Smith, S.; De la Hoz, J.F.D.; Mohiyuddin, M.; Wing, R.A.; McNally, K.L.; Tatarinova, T.; Grigoriev, A.; et al. Structural variants in 3000 rice genomes. Genome Res. 2019, 29, 870–880. [Google Scholar] [CrossRef]
- Claros, M.G.; Bautista, R.; Guerrero-Fernández, D.; Benzerki, H.; Seoane, P.; Fernández-Pozo, N. Why Assembling Plant Genome Sequences Is So Challenging. Biology 2012, 1, 439–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Heffelfinger, C.; Zhao, H.; Dellaporta, S.L. Benchmarking variant identification tools for plant diversity discovery. BMC Genom. 2019, 20, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, J.; Tyagi, A.; Comai, L. Large-scale polymorphism of heterochromatic repeats in the DNA of Arabidopsis thaliana. BMC Plant Biol. 2007, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinboelting, N.; Huep, G.; Appelhagen, I.; Viehoever, P.; Li, Y.; Weisshaar, B. The Structural Features of Thousands of T-DNA Insertion Sites Are Consistent with a Double-Strand Break Repair-Based Insertion Mechanism. Mol. Plant. 2015, 8, 1651–1664. [Google Scholar] [CrossRef]
- Pucker, B.; Holtgräwe, D.; Rosleff Sörensen, T.; Stracke, R.; Viehöver, P.; Weisshaar, B. A De Novo Genome Sequence Assembly of the Arabidopsis thaliana Accession Niederzenz-1 Displays Presence/Absence Variation and Strong Synteny. PLoS ONE 2016, 11, e0164321. [Google Scholar] [CrossRef] [Green Version]
- Pucker, B.; Holtgräwe, D.; Stadermann, K.B.; Frey, K.; Huettel, B.; Reinhardt, R.; Weisshaar, B. A chromosome-level sequence assembly reveals the structure of the Arabidopsis thaliana Nd-1 genome and its gene set. PLoS ONE 2019, 14, e0216233. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Han, S.; Wang, Z.; Gelernter, J.; Yang, B.-Z. Variant Callers for Next-Generation Sequencing Data: A Comparison Study. PLoS ONE 2013, 8, e75619. [Google Scholar] [CrossRef] [Green Version]
- Kavak, P.; Lin, Y.-Y.; Numanagić, I.; Asghari, H.; Güngör, T.; Alkan, C.; Hach, F. Discovery and genotyping of novel sequence insertions in many sequenced individuals. Bioinformatics 2017, 33, i161–i169. [Google Scholar] [CrossRef] [Green Version]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Picard Tools. Available online: https://broadinstitute.github.io/picard/ (accessed on 22 January 2020).
- Baasner, J.-S.; Howard, D.; Pucker, B. Influence of neighboring small sequence variants on functional impact prediction. bioRxiv 2019, 596718. [Google Scholar] [CrossRef]
- Li, H. Improving SNP discovery by base alignment quality. Bioinformatics 2011, 27, 1157–1158. [Google Scholar] [CrossRef] [PubMed]
- Schilbert, H.; Rempel, A.; Pucker, B. Gold Standard of Nd1 vs. TAIR10 Sequence Variants; Bielefeld University: Bielefeld, Germany, 2020. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BCFtools | CLC | FreeBayes | GATK v3.8 | GATK v4.0 | GATK v4.1 | LoFreq | SNVer | VarDict | VarScan | |
|---|---|---|---|---|---|---|---|---|---|---|
| Bowtie2 | 26.701 sen | 7.979 sen | 43.729 sen | 33.836 sen | 33.819 sen | 33.869 sen | 12.282 sen | 21.844 sen | 37.343 sen | 13.910 sen |
| 99.928 spe | 99.987 spe | 99.566 spe | 99.937 spe | 99.937 spe | 99.936 spe | 99.985 spe | 99.969 spe | 99.884 spe | 99.979 spe | |
| 81.800 pre | 88.244 pre | 53.735 pre | 87.286 pre | 87.342 pre | 87.035 pre | 91.607 pre | 90.107 pre | 79.803 pre | 89.530 pre | |
| 99.030 acc | 98.828 acc | 98.939 acc | 99.104 acc | 99.104 acc | 99.104 acc | 98.878 acc | 98.983 acc | 99.120 acc | 98.894 acc | |
| 0.403 F1 | 0.145 F1 | 0.47 F1 | 0.488 F1 | 0.488 F1 | 0.488 F1 | 0.214 F1 | 0.352 F1 | 0.512 F1 | 0.239 F1 | |
| BWA-MEM | 43.186 sen | 34.992 sen | 43.053 sen | 47.130 sen | 47.099 sen | 47.136 sen | 38.874 sen | 41.435 sen | 40.302 sen | 37.506 sen |
| 99.806 spe | 99.908 spe | 99.548 spe | 99.842 spe | 99.842 spe | 99.839 spe | 99.892 spe | 99.868 spe | 99.852 spe | 99.907 spe | |
| 73.182 pre | 81.892 pre | 51.289 pre | 79.129 pre | 79.083 pre | 78.786 pre | 81.122 pre | 79.521 pre | 76.686 pre | 83.535 pre | |
| 99.120 acc | 99.099 acc | 98.916 acc | 99.179 acc | 99.179 acc | 99.178 acc | 99.122 acc | 99.133 acc | 99.124 acc | 99.120 acc | |
| 0.543 F1 | 0.492 F1 | 0.459 F1 | 0.591 F1 | 0.59 F1 | 0.59 F1 | 0.528 F1 | 0.546 F1 | 0.531 F1 | 0.518 F1 | |
| CLC | 47.682 sen | 36.382 sen | 41.564 sen | 49.237 sen | 49.357 sen | 49.413 sen | 41.730 sen | 45.352 sen | 39.508 sen | 41.349 sen |
| 99.695 spe | 99.851 spe | 99.517 spe | 99.817 spe | 99.815 spe | 99.812 spe | 99.821 spe | 99.759 spe | 99.857 spe | 99.847 spe | |
| 65.903 pre | 73.878 pre | 48.854 pre | 77.341 pre | 77.183 pre | 76.901 pre | 73.611 pre | 70.493 pre | 76.995 pre | 76.971 pre | |
| 99.064 acc | 99.062 acc | 98.882 acc | 99.175 acc | 99.175 acc | 99.174 acc | 99.066 acc | 99.060 acc | 99.112 acc | 99.100 acc | |
| 0.549 F1 | 0.491 F1 | 0.441 F1 | 0.601 F1 | 0.602 F1 | 0.602 F1 | 0.536 F1 | 0.549 F1 | 0.525 F1 | 0.539 F1 | |
| GEM3 | 35.488 sen | 32.826 sen | 44.104 sen | 40.895 sen | 40.890 sen | 40.963 sen | 33.867 sen | 34.710 sen | 39.131 sen | 41.502 sen |
| 99.887 spe | 99.932 spe | 99.523 spe | 99.902 spe | 99.902 spe | 99.900 spe | 99.941 spe | 99.925 spe | 99.847 spe | 99.904 spe | |
| 79.533 pre | 85.457 pre | 51.481 pre | 84.189 pre | 84.221 pre | 83.863 pre | 87.657 pre | 85.573 pre | 75.648 pre | 84.315 pre | |
| 99.092 acc | 99.095 acc | 98.922 acc | 99.157 acc | 99.157 acc | 99.156 acc | 99.107 acc | 99.097 acc | 99.100 acc | 99.169 acc | |
| 0.49 F1 | 0.475 F1 | 0.464 F1 | 0.55 F1 | 0.551 F1 | 0.55 F1 | 0.489 F1 | 0.494 F1 | 0.519 F1 | 0.557 F1 | |
| Novoalign | 44.160 sen | 33.858 sen | 42.986 sen | 48.599 sen | 48.575 sen | 48.620 sen | 38.892 sen | 41.436 sen | 39.863 sen | 38.170 sen |
| 99.820 spe | 99.922 spe | 99.610 spe | 99.846 spe | 99.845 spe | 99.842 spe | 99.905 spe | 99.885 spe | 99.860 spe | 99.922 spe | |
| 75.260 pre | 84.222 pre | 54.799 pre | 80.019 pre | 79.951 pre | 79.643 pre | 83.510 pre | 81.968 pre | 77.296 pre | 85.862 pre | |
| 99.147 acc | 99.097 acc | 98.966 acc | 99.202 acc | 99.202 acc | 99.200 acc | 99.137 acc | 99.149 acc | 99.127 acc | 99.144 acc | |
| 0.556 F1 | 0.483 F1 | 0.472 F1 | 0.605 F1 | 0.604 F1 | 0.604 F1 | 0.532 F1 | 0.551 F1 | 0.529 F1 | 0.529 F1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilbert, H.M.; Rempel, A.; Pucker, B. Comparison of Read Mapping and Variant Calling Tools for the Analysis of Plant NGS Data. Plants 2020, 9, 439. https://doi.org/10.3390/plants9040439
Schilbert HM, Rempel A, Pucker B. Comparison of Read Mapping and Variant Calling Tools for the Analysis of Plant NGS Data. Plants. 2020; 9(4):439. https://doi.org/10.3390/plants9040439
Chicago/Turabian StyleSchilbert, Hanna Marie, Andreas Rempel, and Boas Pucker. 2020. "Comparison of Read Mapping and Variant Calling Tools for the Analysis of Plant NGS Data" Plants 9, no. 4: 439. https://doi.org/10.3390/plants9040439