
The Identification of a Yield-Related Gene Controlling Multiple Traits Using GWAS in Sorghum (Sorghum bicolor L.)
Abstract
:1. Introduction
2. Result
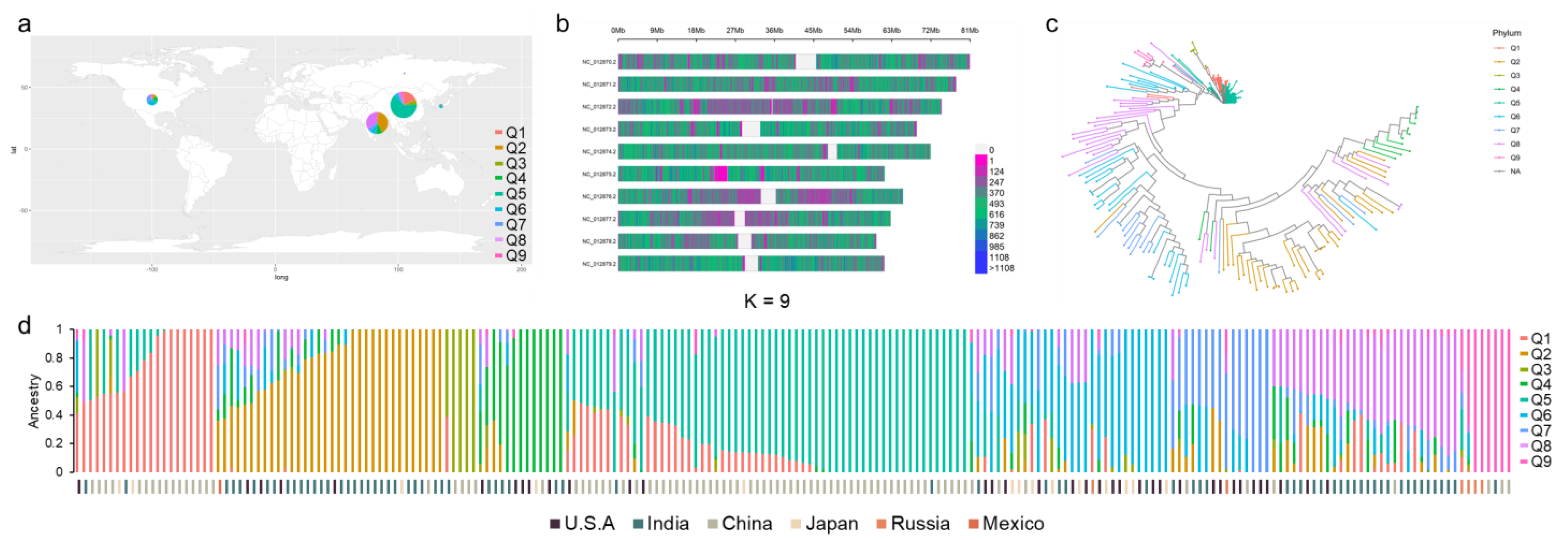
2.1. Population Collection and Genotyping
2.2. SNP Markers Selection and Population Structure Analysis
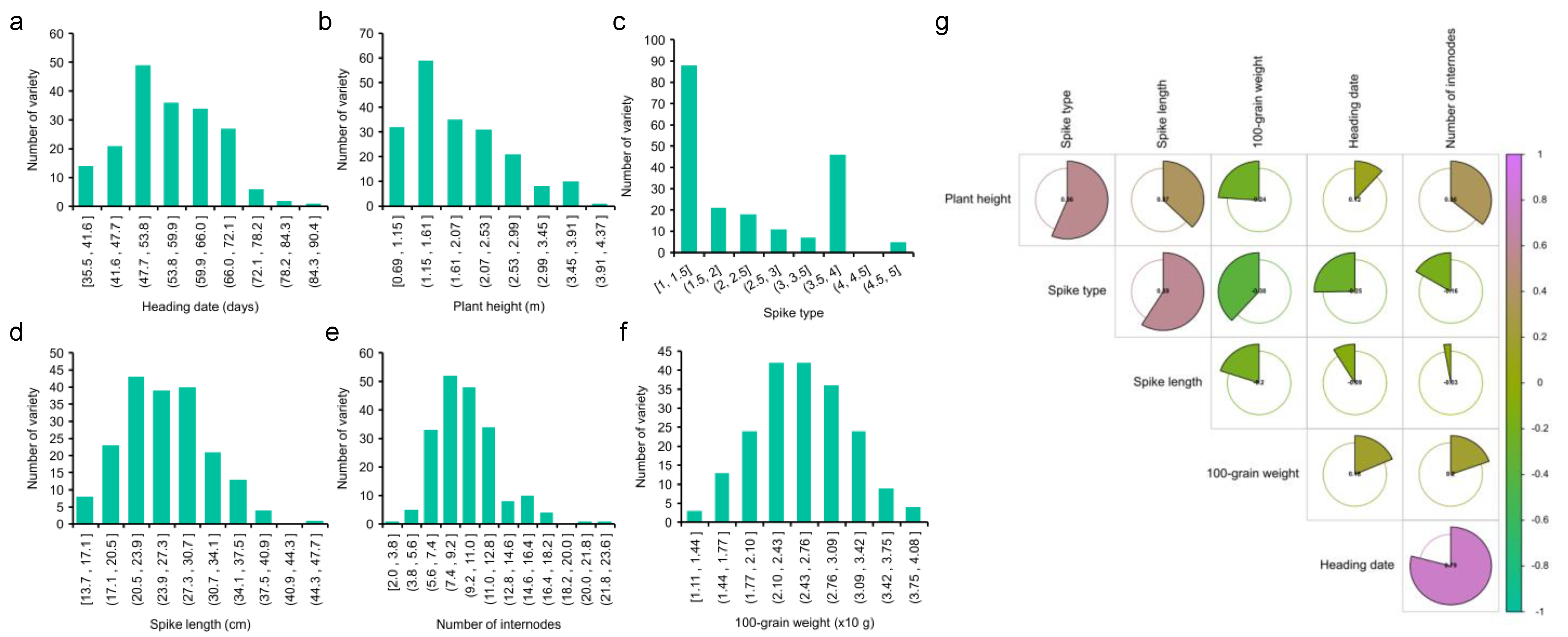
2.3. Phenotype Analysis
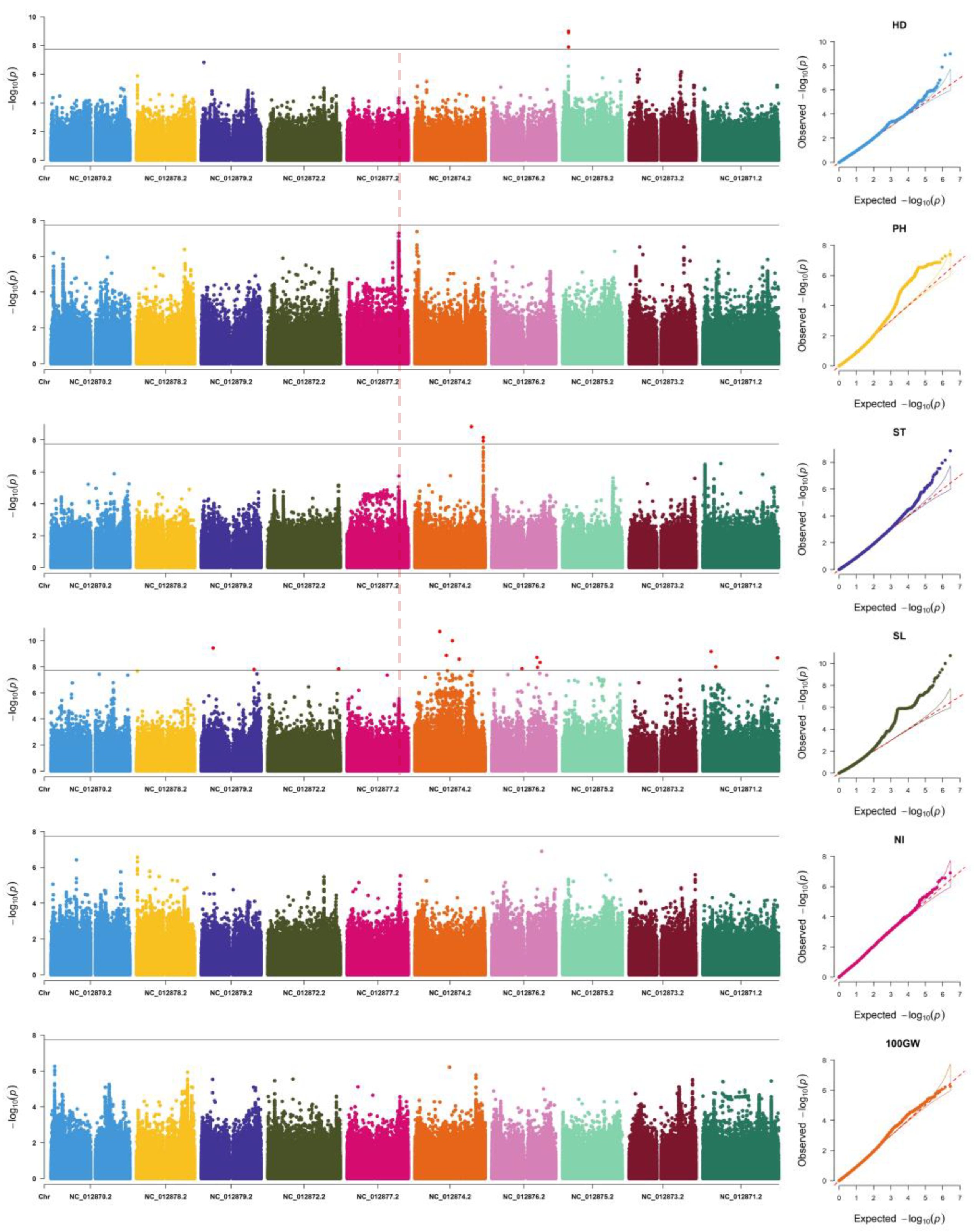
2.4. LD Decay Analysis and GWAS
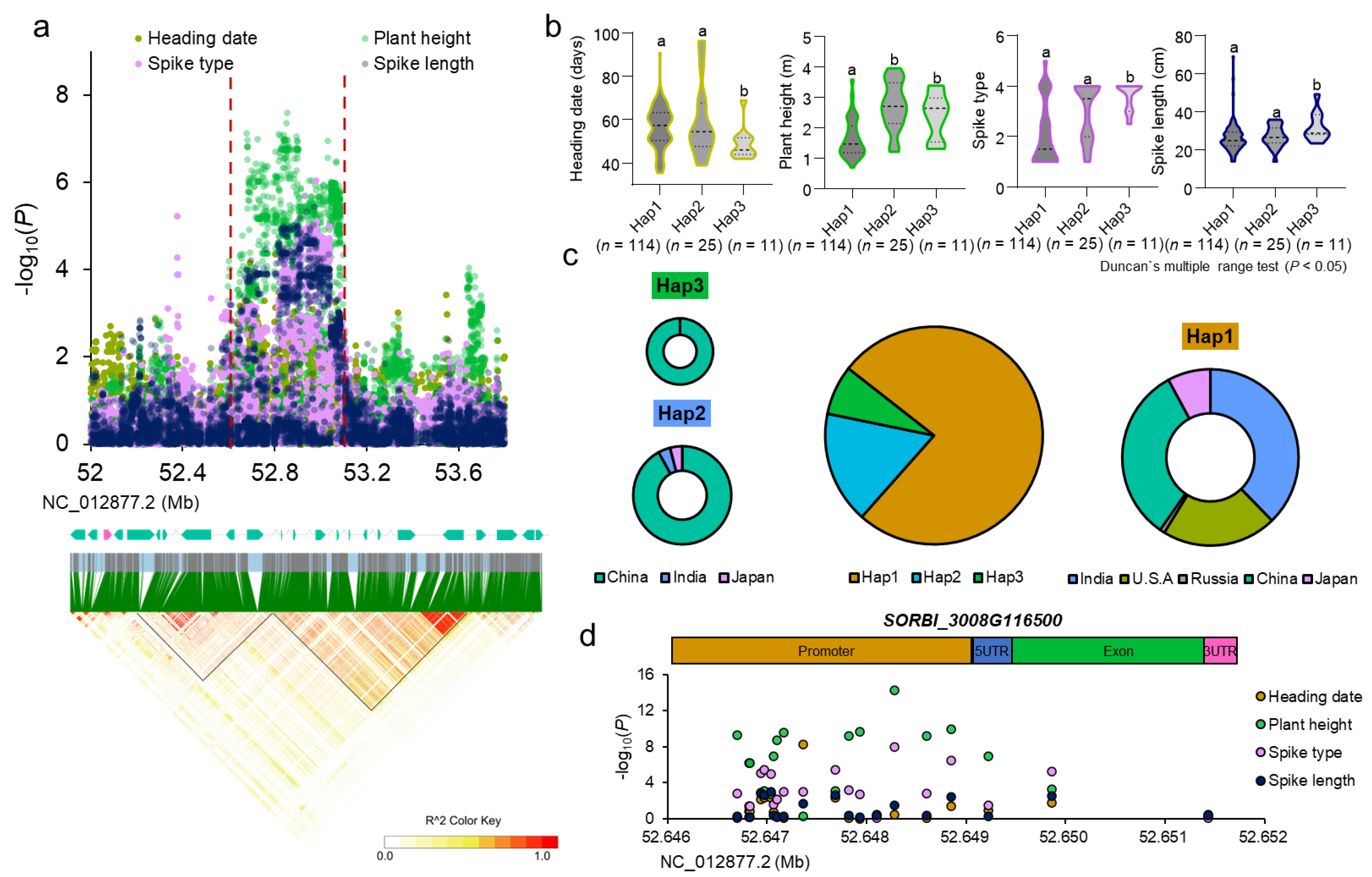
2.5. Prediction and Analysis of Yield-Related Gene
3. Discussion
3.1. GWAS Results and Observed Traits
3.2. A More Appropriate Model or Lower Threshold for Detection
3.3. SORBI_3008G116500 Breeding
4. Material and Method
4.1. Plant Materials and Environment Conditions
4.2. Genotyping
4.3. Phenotypic Trait Evaluation and Data Analysis
4.4. Population Structure, Relative Kinship, Principal Component Analysis (PCA), and Linkage Disequilibrium (LD)
4.5. Genome-Wide Association Study (GWAS) and QTL analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Ananda, G.K.; Myrans, H.; Norton, S.L.; Gleadow, R.; Furtado, A.; Henry, R.J. Wild sorghum as a promising resource for crop improvement. Front. Plant Sci. 2020, 11, 1108. [Google Scholar] [CrossRef] [PubMed]
- Ordonio, R.; Ito, Y.; Morinaka, Y.; Sazuka, T.; Matsuoka, M. Chapter Five—Molecular Breeding of Sorghum bicolor, A Novel Energy Crop. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 321, pp. 221–257. [Google Scholar]
- Calviño, M.; Messing, J. Sweet sorghum as a model system for bioenergy crops. Curr. Opin. Biotechnol. 2012, 23, 323–329. [Google Scholar] [CrossRef]
- Regassa, T.H.; Wortmann, C.S. Sweet sorghum as a bioenergy crop: Literature review. Biomass Bioenergy 2014, 64, 348–355. [Google Scholar] [CrossRef]
- Xin, Z.; Wang, M.; Cuevas, H.E.; Chen, J.; Harrison, M.; Pugh, N.A.; Morris, G. Sorghum genetic, genomic, and breeding resources. Planta 2021, 254, 114. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Lyu, Y.; Wu, X.; Li, H.; Cheng, L.; Zhang, C.; Yuan, L.; Jiang, R.; Jiang, B.; Rengel, Z.; et al. Grain production versus resource and environmental costs: Towards increasing sustainability of nutrient use in China. J. Exp. Bot. 2016, 67, 4935–4949. [Google Scholar] [CrossRef] [Green Version]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927. [Google Scholar] [CrossRef]
- Hao, H.; Li, Z.; Leng, C.; Lu, C.; Luo, H.; Liu, Y.; Wu, X.; Liu, Z.; Shang, L.; Jing, H.C. Sorghum breeding in the genomic era: Opportunities and challenges. TAG. Theor. Appl. Genetics. Theor. Und Angew. Genet. 2021, 134, 1899–1924. [Google Scholar] [CrossRef]
- Li, S.; Tian, Y.; Wu, K.; Ye, Y.; Yu, J.; Zhang, J.; Liu, Q.; Hu, M.; Li, H.; Tong, Y.; et al. Modulating plant growth-metabolism coordination for sustainable agriculture. Nature 2018, 560, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qian, Q.; Liu, Z.; Sun, H.; He, S.; Luo, D.; Xia, G.; Chu, C.; Li, J.; Fu, X. Natural variation at the DEP1 locus enhances grain yield in rice. Nat. Genet. 2009, 41, 494–497. [Google Scholar] [CrossRef]
- Wei, X.; Xu, J.; Guo, H.; Jiang, L.; Chen, S.; Yu, C.; Zhou, Z.; Hu, P.; Zhai, H.; Wan, J. DTH8 suppresses flowering in rice, influencing plant height and yield potential simultaneously. Plant Physiol. 2010, 153, 1747–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimaru, K.; Hirotsu, N.; Madoka, Y.; Murakami, N.; Hara, N.; Onodera, H.; Kashiwagi, T.; Ujiie, K.; Shimizu, B.-i.; Onishi, A.; et al. Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat. Genet. 2013, 45, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, M.; Liu, R.; Zhang, M.; Lv, Y.; Liu, Y.; Xiao, X.; Yuan, J.; Cai, H. BIOMASS YIELD 1 regulates sorghum biomass and grain yield via the shikimate pathway. J. Exp. Bot. 2020, 71, 5506–5520. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhao, Y.; Burks, P.; Pauly, M.; Brown, P.J. A sorghum NAC gene is associated with variation in biomass properties and yield potential. Plant Direct 2018, 2, e00070. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Mantilla Perez, M.B.; Hu, J.; Salas Fernandez, M.G. Genome-Wide Association Study for Nine Plant Architecture Traits in Sorghum. Plant Genome 2016, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and Prospects of Association Mapping in Plants. Plant Genome J. 2008, 1, 5. [Google Scholar] [CrossRef]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [Green Version]
- Atwell, S.; Huang, Y.S.; Vilhjalmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T.; et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Crowell, S.; Korniliev, P.; Falcao, A.; Ismail, A.; Gregorio, G.; Mezey, J.; McCouch, S. Genome-wide association and high-resolution phenotyping link Oryza sativa panicle traits to numerous trait-specific QTL clusters. Nat. Commun. 2016, 7, 10527. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef]
- Yano, K.; Morinaka, Y.; Wang, F.; Huang, P.; Takehara, S.; Hirai, T.; Ito, A.; Koketsu, E.; Kawamura, M.; Kotake, K.; et al. GWAS with principal component analysis identifies a gene comprehensively controlling rice architecture. Proc. Natl. Acad. Sci. USA 2019, 116, 21262–21267. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Casella, L.; Riccardi, P.; Vattari, A.; Orasen, G.; Perrini, R.; Tacconi, G.; Tondelli, A.; Biselli, C.; et al. Genome-Wide Association Study for Traits Related to Plant and Grain Morphology, and Root Architecture in Temperate Rice Accessions. PLoS ONE 2016, 11, e0155425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasky, J.R.; Upadhyaya, H.D.; Ramu, P.; Deshpande, S.; Hash, C.T.; Bonnette, J.; Juenger, T.E.; Hyma, K.; Acharya, C.; Mitchell, S.E. Genome-environment associations in sorghum landraces predict adaptive traits. Sci. Adv. 2015, 1, e1400218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chopra, R.; Hayes, C.; Morris, G.; Marla, S.; Burke, J.; Xin, Z.; Burow, G. Genome-Wide Association Study of Developing Leaves’ Heat Tolerance during Vegetative Growth Stages in a Sorghum Association Panel. Plant Genome 2017, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tang, W.; Zhang, Y.W.; Chen, K.N.; Wang, C.; Liu, Y.; Zhan, Q.; Wang, C.; Wang, S.B.; Xie, S.Q.; et al. Genome-Wide Association Studies for Five Forage Quality-Related Traits in Sorghum (Sorghum bicolor L.). Front. Plant Sci. 2018, 9, 1146. [Google Scholar] [CrossRef]
- Rhodes, D.H.; Hoffmann, L., Jr.; Rooney, W.L.; Ramu, P.; Morris, G.P.; Kresovich, S. Genome-Wide Association Study of Grain Polyphenol Concentrations in Global Sorghum [Sorghum bicolor (L.) Moench] Germplasm. J. Agric. Food Chem. 2014, 62, 10916–10927. [Google Scholar] [CrossRef] [PubMed]
- Kimani, W.; Zhang, L.M.; Wu, X.Y.; Hao, H.Q.; Jing, H.C. Genome-wide association study reveals that different pathways contribute to grain quality variation in sorghum (Sorghum bicolor). BMC Genom. 2020, 21, 112. [Google Scholar] [CrossRef] [Green Version]
- Boyles, R.E.; Cooper, E.A.; Myers, M.T.; Brenton, Z.; Rauh, B.L.; Morris, G.P.; Kresovich, S. Genome-Wide Association Studies of Grain Yield Components in Diverse Sorghum Germplasm. Plant Genome 2016, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Mace, E.S.; Singh, V.; Van Oosterom, E.J.; Hammer, G.L.; Hunt, C.H.; Jordan, D.R. QTL for nodal root angle in sorghum (Sorghum bicolor L. Moench) co-locate with QTL for traits associated with drought adaptation. Theor. Appl. Genet. 2012, 124, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Murray, S.C.; Rooney, W.L.; Hamblin, M.T.; Mitchell, S.E.; Kresovich, S. Sweet sorghum genetic diversity and association mapping for brix and height. Plant Genome 2009, 2, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Xing, Y.; Weng, X.; Zhao, Y.; Tang, W.; Wang, L.; Zhou, H.; Yu, S.; Xu, C.; Li, X.; et al. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet. 2008, 40, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Zhang, H.; Ludwig, P.; van Nocker, S. A mechanism related to the yeast transcriptional regulator Paf1c is required for expression of the Arabidopsis FLC/MAF MADS box gene family. Plant Cell 2004, 16, 2940–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Tian, Y.; Wang, S.; Su, Y.; Mao, T.; Huang, T.; Chen, Q.; Xu, Z.; Ding, Y. Phosphorylation of SPT5 by CDKD; 2 is required for VIP5 recruitment and normal flowering in Arabidopsis thaliana. Plant Cell 2017, 29, 277–291. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja Reddy, R.; Madhusudhana, R.; Murali Mohan, S.; Chakravarthi, D.; Mehtre, S.; Seetharama, N.; Patil, J. Mapping QTL for grain yield and other agronomic traits in post-rainy sorghum [Sorghum bicolor (L.) Moench]. Theor. Appl. Genet. 2013, 126, 1921–1939. [Google Scholar] [CrossRef]
- Tao, Y.; Zhao, X.; Wang, X.; Hathorn, A.; Hunt, C.; Cruickshank, A.W.; van Oosterom, E.J.; Godwin, I.D.; Mace, E.S.; Jordan, D.R. Large-scale GWAS in sorghum reveals common genetic control of grain size among cereals. Plant Biotechnol. J. 2020, 18, 1093–1105. [Google Scholar] [CrossRef] [Green Version]
- Sestili, F.; Pagliarello, R.; Zega, A.; Saletti, R.; Pucci, A.; Botticella, E.; Masci, S.; Tundo, S.; Moscetti, I.; Foti, S. Enhancing grain size in durum wheat using RNAi to knockdown GW2 genes. Theor. Appl. Genet. 2019, 132, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Pan, L.; He, J.; Zhao, T.; Xing, G.; Wang, Y.; Yu, D.; Chen, S.; Gai, J. Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two-stage multi-locus GWAS procedure. TAG. Theor. Appl. Genetics. Theor. Und Angew. Genet. 2018, 131, 2581–2599. [Google Scholar] [CrossRef]
- Luo, F.; Pei, Z.; Zhao, X.; Liu, H.; Jiang, Y.; Sun, S. Genome-Wide Association Study for Plant Architecture and Bioenergy Traits in Diverse Sorghum and Sudangrass Germplasm. Agronomy 2020, 10, 1602. [Google Scholar] [CrossRef]
- Zhang, D.; Kong, W.; Robertson, J.; Goff, V.H.; Epps, E.; Kerr, A.; Mills, G.; Cromwell, J.; Lugin, Y.; Phillips, C.; et al. Genetic analysis of inflorescence and plant height components in sorghum (Panicoidae) and comparative genetics with rice (Oryzoidae). BMC Plant Biol. 2015, 15, bbaa227. [Google Scholar] [CrossRef] [Green Version]
- Springer, N.M.; Dong, H.; Zhao, H.; Xie, W.; Han, Z.; Li, G.; Yao, W.; Bai, X.; Hu, Y.; Guo, Z.; et al. A Novel Tiller Angle Gene, TAC3, together with TAC1 and D2 Largely Determine the Natural Variation of Tiller Angle in Rice Cultivars. PLoS Genet. 2016, 12, e1006412. [Google Scholar]
- Wang, J.; Yu, H.; Xiong, G.; Lu, Z.; Jiao, Y.; Meng, X.; Liu, G.; Chen, X.; Wang, Y.; Li, J. Tissue-Specific Ubiquitination by IPA1 Interacting PROTEIN1 Modulates IPA1 Protein Levels to Regulate Plant Architecture in Rice. Plant Cell 2017, 29, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Doyle, M.R.; Amasino, R.M. PAF1-complex-mediated histone methylation of FLOWERING LOCUS C chromatin is required for the vernalization-responsive, winter-annual habit in Arabidopsis. Genes Dev. 2004, 18, 2774–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; van Nocker, S. The Vernalization Independence 4 gene encodes a novel regulator of Flowering Locus C. Plant J. Cell Mol. Biol. 2002, 31, 663–673. [Google Scholar] [CrossRef]
- Qingshan, L.; Dahlberg, J.A. Chinese sorghum genetic resources. Econ. Bot. 2001, 55, 401–425. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Lippert, C.; Listgarten, J.; Liu, Y.; Kadie, C.M.; Davidson, R.I.; Heckerman, D. FaST linear mixed models for genome-wide association studies. Nat. Methods 2011, 8, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Segura, V.; Vilhjálmsson, B.J.; Platt, A.; Korte, A.; Seren, Ü.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.-S.; He, W.-M.; Ji, J.-J.; Zhang, C.; Guo, Y.; Yang, T.-L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 2021, 22, 4. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Min | Max | Mean | CV |
|---|---|---|---|---|
| Heading date (days) | 35.50 | 104.90 | 57.69 | 0.21 |
| Plant height (m) | 0.69 | 3.96 | 1.90 | 0.40 |
| Spike type | 1.00 | 5.00 | 2.34 | 0.53 |
| Spike length (cm) | 13.67 | 69.17 | 26.75 | 0.28 |
| Number of internodes | 2.00 | 22.00 | 9.79 | 0.30 |
| 100-grain weight (×10 g) | 1.11 | 4.03 | 2.56 | 0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Fan, X.; Liang, D.; Guo, Q.; Zhang, X.; Nie, M.; Li, C.; Meng, S.; Zhang, X.; Xu, P.; et al. The Identification of a Yield-Related Gene Controlling Multiple Traits Using GWAS in Sorghum (Sorghum bicolor L.). Plants 2023, 12, 1557. https://doi.org/10.3390/plants12071557
Zhang Y, Fan X, Liang D, Guo Q, Zhang X, Nie M, Li C, Meng S, Zhang X, Xu P, et al. The Identification of a Yield-Related Gene Controlling Multiple Traits Using GWAS in Sorghum (Sorghum bicolor L.). Plants. 2023; 12(7):1557. https://doi.org/10.3390/plants12071557
Chicago/Turabian StyleZhang, Yizhong, Xinqi Fan, Du Liang, Qi Guo, Xiaojuan Zhang, Mengen Nie, Chunhong Li, Shan Meng, Xianggui Zhang, Peng Xu, and et al. 2023. "The Identification of a Yield-Related Gene Controlling Multiple Traits Using GWAS in Sorghum (Sorghum bicolor L.)" Plants 12, no. 7: 1557. https://doi.org/10.3390/plants12071557