
Characterization of microRNAs and Target Genes in Musa acuminata subsp. burmannicoides, var. Calcutta 4 during Interaction with Pseudocercospora musae
 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sequence Statistics
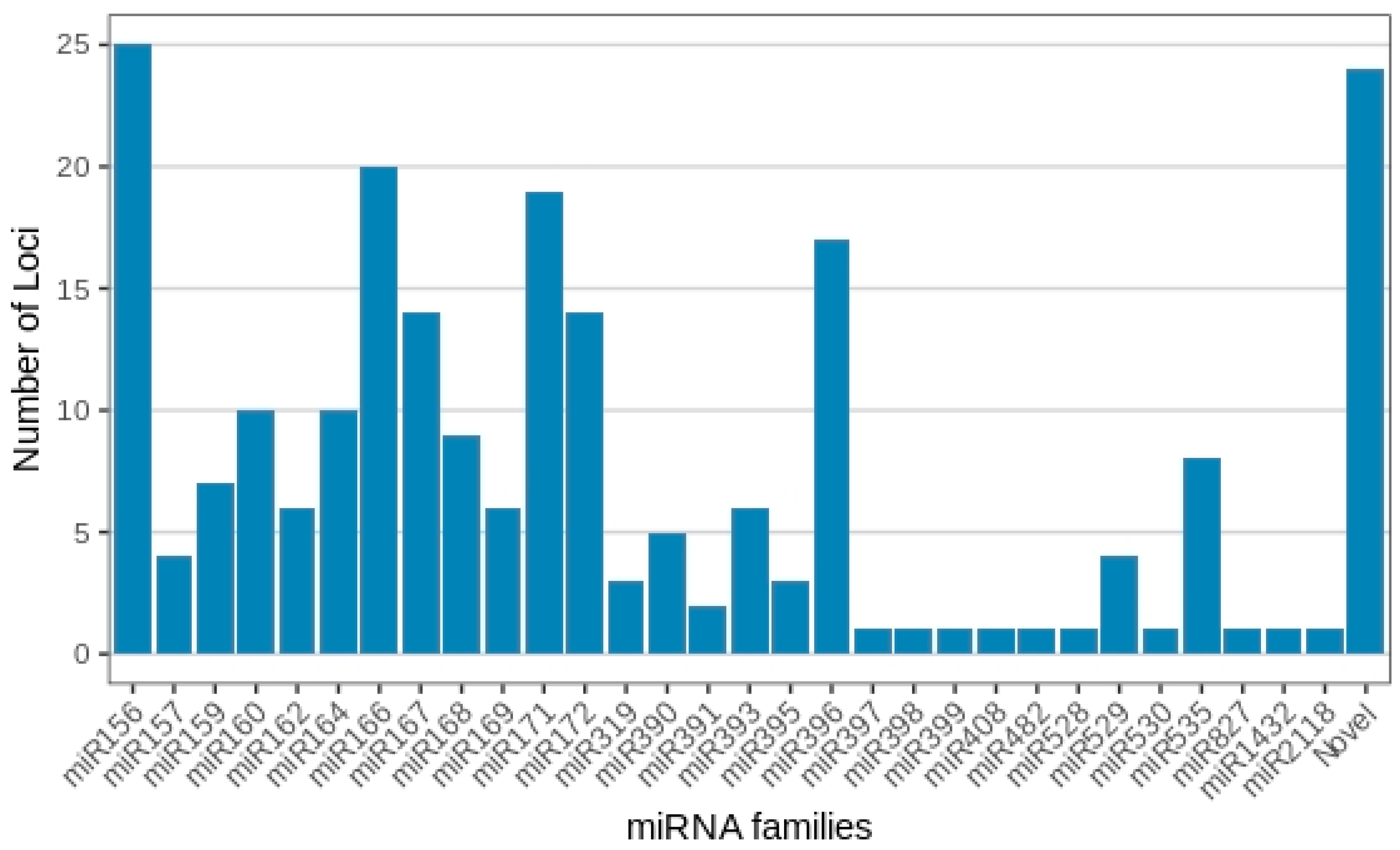
2.2. Identification of miRNAs
2.3. In Silico Expression Analysis of Musa miRNAs and Target Genes in Response to P. musae
2.4. miRNA Target Gene Prediction of Conserved and Novel M. acuminata var. Calcutta 4 miRNAs
2.5. GO Analysis of Target Genes of miRNAs
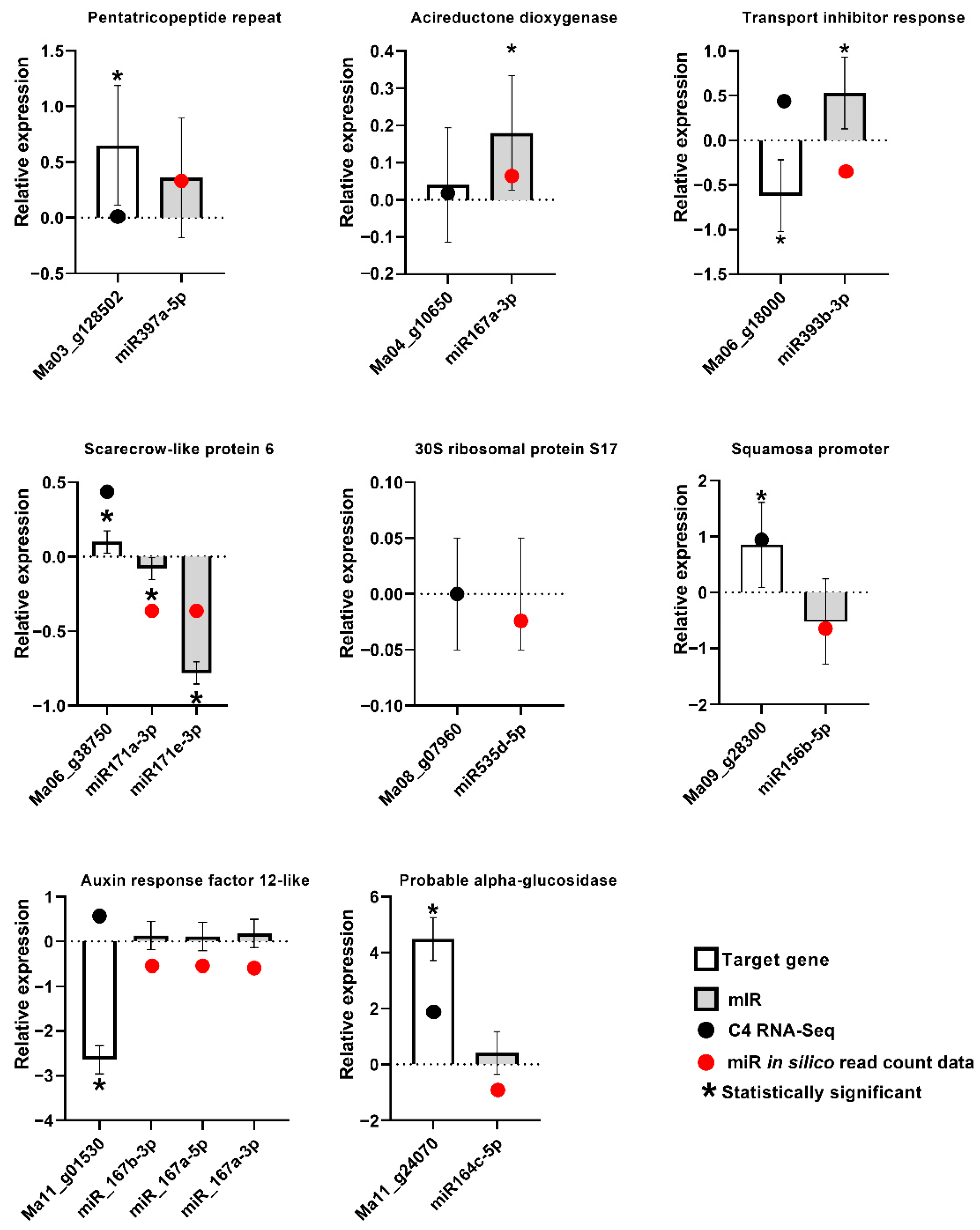
2.6. RT-qPCR Analysis of Expression of Musa miRNA and Musa Target Genes
3. Discussion
3.1. Expression Analysis
3.2. Abundant miRNAs and Target Genes Potentially Involved in the Plant Immune Response
3.3. Less Abundant miRNAs and Target Genes Potentially Involved in Plant Immune Responses
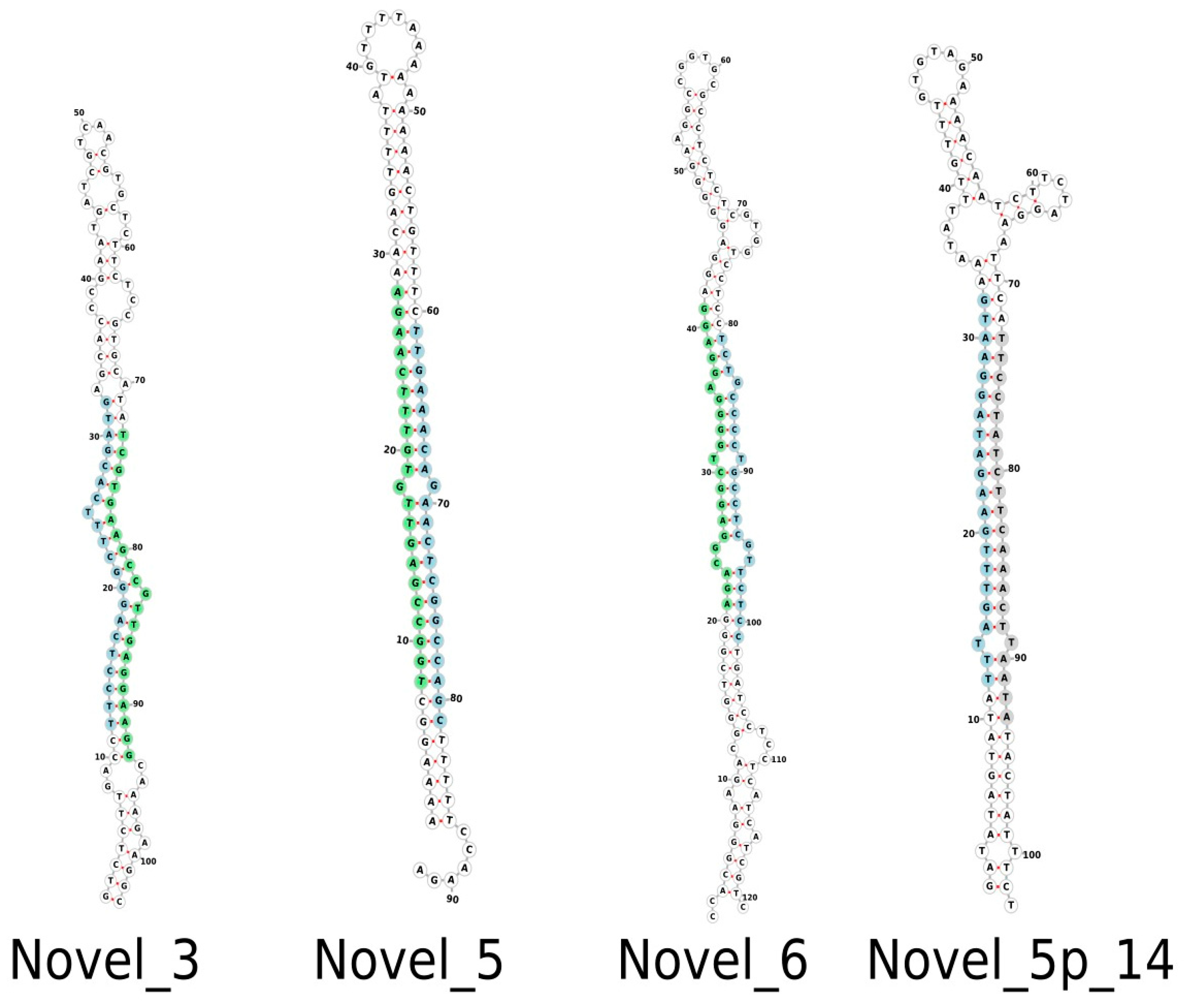
3.4. Novel Musa miRNAs
4. Materials and Methods
4.1. Plant Material and Fungal Cultures
4.2. Bioassays
4.3. Musa Total RNA Extraction and Illumina Sequencing
4.4. Gene Expression in M. acuminata var. Calcutta 4 during Interaction with P. musae
4.5. Sequence Trimming and miRNA Identification
4.6. In Silico Analysis of Differential Expression of miRNAs
4.7. miRNA Target Gene Prediction
4.8. Gene Ontology (GO) Analysis of Target Genes
4.9. In Silico Expression Analysis of Target Genes
4.10. cDNA Synthesis for Musa miRNAs
4.11. cDNA Synthesis for Musa Target Genes
4.12. RT-qPCR Expression Validation of miRNAs and Predicted Target Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayden, H.; Carlier, J.; Aitken, E. The genetic structure of Australian populations of Mycosphaerella musicola suggests restricted gene flow at the continental scale. Phytopathology 2005, 95, 489–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arzanlou, M.; Abeln, E.C.; Kema, G.H.; Waalwijk, C.; Carlier, J.; de Vries, I.; Guzmán, M.; Crous, P.W. Molecular diagnostics for the Sigatoka disease complex of banana. Phytopathology 2007, 97, 1112–1118. [Google Scholar] [CrossRef] [Green Version]
- Brito, F.S.; Santos, J.R.; Azevedo, V.C.; Peixouto, Y.S.; de Oliveira, S.A.; Ferreira, C.F.; Haddad, F.; Amorim, E.P.; Fraaije, B.; Miller, R.N. Genetic diversity and azole fungicide sensitivity in Pseudocercospora musae field populations in Brazil. Front. Microbiol. 2020, 11, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, J.; Jia, C.; Liu, J.; Li, Y.; Yin, X.; Xu, B.; Jin, Z. De novo characterization of the banana root transcriptome and analysis of gene expression under Fusarium oxysporum f. sp. cubense tropical race 4 infection. BMC Genom. 2012, 13, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-Y.; Deng, G.-M.; Yang, J.; Viljoen, A.; Jin, Y.; Kuang, R.-B.; Zuo, C.-W.; Lv, Z.-C.; Yang, Q.-S.; Sheng, O.; et al. Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genom. 2012, 13, 374. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Shao, J.; Wang, Y.; Li, W.; Guo, D.; Yan, B.; Xia, Y.; Peng, M. Analysis of banana transcriptome and global gene expression profiles in banana roots in response to infection by race 1 and tropical race 4 of Fusarium oxysporum f. sp. cubense. BMC Genom. 2013, 14, 851. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, X.; Li, C.; Sun, J.; Li, S.; Peng, M. Dual species transcript profiling during the interaction between banana (Musa acuminata) and the fungal pathogen Fusarium oxysporum f. sp. cubense. BMC Genom. 2019, 20, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.-M.; Dita, M.; Rouard, M.; Wu, W.; Roux, N.; Xie, J.-H.; Ge, X.-J. Deep RNA-seq analysis reveals key responding aspects of wild banana relative resistance to Fusarium oxysporum f. sp. cubense tropical race 4. Funct. Integr. Genom. 2020, 20, 551–562. [Google Scholar] [CrossRef]
- Bai, T.-T.; Xie, W.-B.; Zhou, P.-P.; Wu, Z.-L.; Xiao, W.-C.; Zhou, L.; Sun, J.; Ruan, X.-L.; Li, H.-P. Transcriptome and expression profile analysis of highly resistant and susceptible banana roots challenged with Fusarium oxysporum f. sp. cubense tropical race 4. PLoS ONE 2013, 8, e73945. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhang, J.; Fang, H.; Peng, L.; Wei, S.; Li, C.; Zheng, S.; Lu, J. Comparative transcriptome analysis reveals resistance-related genes and pathways in Musa acuminata banana ‘Guijiao 9′ in response to Fusarium wilt. Plant Physiol. Biochem. 2019, 141, 83–94. [Google Scholar] [CrossRef]
- Zhang, L.; Cenci, A.; Rouard, M.; Zhang, D.; Wang, Y.; Tang, W.; Zheng, S.-J. Transcriptomic analysis of resistant and susceptible banana corms in response to infection by Fusarium oxysporum f. sp. cubense tropical race 4. Sci. Rep. 2019, 9, 8199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, M.; Mahuku, G.; Swennen, R. Comparative transcriptome and expression profiling of resistant and susceptible banana cultivars during infection by Fusarium oxysporum. Int. J. Mol. Sci. 2021, 22, 3002. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, N.E.N.; Alves, G.S.C.; Almeida, R.M.; Amorim, E.P.; Fortes Ferreira, C.; Togawa, R.C.; Costa, M.M.D.C.; Grynberg, P.; Santos, J.R.P.; Cares, J.E.; et al. Gene expression analysis in Musa acuminata during compatible interactions with Meloidogyne incognita. Ann. Bot. 2017, 119, 915–930. [Google Scholar]
- D’hont, A.; Denoeud, F.; Aury, J.-M.; Baurens, F.-C.; Carreel, F.; Garsmeur, O.; Noel, B.; Bocs, S.; Droc, G.; Rouard, M.; et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012, 488, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, M.A.; de Cruz, V.O.; Emediato, F.L.; de Teixeira, C.C.; Azevedo, V.C.R.; Brasileiro, A.; Amorim, E.P.; Ferreira, C.F.; Martins, N.F.; Togawa, R.C.; et al. Analysis of the leaf transcriptome of Musa acuminata during interaction with Mycosphaerella musicola: Gene assembly, annotation and marker development. BMC Genom. 2013, 14, 78. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, T.D.M.; Rego, E.C.S.; Alves, G.S.C.; Fonseca, F.C.D.A.; Cotta, M.G.; Antonino, J.D.; Gomes, T.G.; Amorim, E.P.; Ferreira, C.F.; Costa, M.M.D.C.; et al. Transcriptome Profiling of the Resistance Response of Musa acuminata subsp. burmannicoides, var. Calcutta 4 to Pseudocercospora musae. Int. J. Mol. Sci. 2022, 23, 13589. [Google Scholar] [CrossRef]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Prabu, G.; Mandal, A.K.A. Computational identification of miRNAs and their target genes from expressed sequence tags of tea (Camellia sinensis). CBPD 2010, 8, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, Y.; Watanabe, Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. USA 2004, 101, 12753–12758. [Google Scholar] [CrossRef] [Green Version]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unver, T.; Namuth-Covert, D.M.; Budak, H. Review of current methodological approaches for characterizing microRNAs in plants. Int. J. Plant Genom. 2009, 2009, 262463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanpuria, P.; Yadav, S.K. Characterization of novel small RNAs from tea (Camellia sinensis L.). Mol. Biol. Rep. 2012, 39, 3977–3986. [Google Scholar] [CrossRef]
- Kuan, T.; Zhai, Y.; Ma, W. Small RNAs regulate plant responses to filamentous pathogens. Semin. Cell Dev. Biol. 2016, 56, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, X.; Wolkenhauer, O.; Vera, J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res. 2016, 44, 6019–6035. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Jia, T.; Chen, X. The ‘how’ and ‘where’ of plant micro RNAs. New Phytol. 2017, 216, 1002–1017. [Google Scholar] [CrossRef] [Green Version]
- Samad, A.F.; Sajad, M.; Nazaruddin, N.; Fauzi, I.A.; Murad, A.M.; Zainal, Z.; Ismail, I. MicroRNA and transcription factor: Key players in plant regulatory network. Front. Plant Sci. 2017, 8, 565. [Google Scholar]
- Bhakta, S.; Tak, H.; Ganapathi, T.R. Exploring diverse roles of micro RNAs in banana: Current status and future prospective. Physiol. Plant. 2021, 173, 1323–1334. [Google Scholar] [CrossRef]
- Liu, W.-W.; Meng, J.; Cui, J.; Luan, Y.-S. Characterization and Function of MicroRNA∗s in Plants. Front. Plant Sci. 2017, 8, 2200. [Google Scholar] [CrossRef] [Green Version]
- Vaucheret, H.; Vazquez, F.; Crété, P.; Bartel, D.P. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004, 18, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA–target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar]
- Zamore, P.D.; Haley, B. Ribo-gnome: The big world of small RNAs. Science 2005, 309, 1519–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, L.I.; Chinnusamy, V.; Sunkar, R. The role of microRNAs and other endogenous small RNAs in plant stress responses. Biochim. Biophys. Acta 2008, 1779, 743–748. [Google Scholar] [CrossRef]
- Chen, X. Small RNAs–secrets and surprises of the genome. Plant J. 2010, 61, 941–958. [Google Scholar]
- Kulcheski, F.R.; de Oliveira, L.F.; Molina, L.G.; Almerão, M.P.; Rodrigues, F.A.; Marcolino, J.; Barbosa, J.F.; Stolf-Moreira, R.; Nepomuceno, A.L.; Marcelino-Guimarães, F.C. Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genom. 2011, 12, 307. [Google Scholar]
- Parent, J.-S.; Martinez de Alba, A.E.; Vaucheret, H. The origin and effect of small RNA signaling in plants. Front. Plant Sci. 2012, 3, 179. [Google Scholar] [CrossRef] [Green Version]
- Inal, B.; Türktaş, M.; Eren, H.; Ilhan, E.; Okay, S.; Atak, M.; Erayman, M.; Unver, T. Genome-wide fungal stress responsive miRNA expression in wheat. Planta 2014, 240, 1287–1298. [Google Scholar] [CrossRef]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. miRNA-based drought regulation in wheat. Funct. Integr. Genom. 2016, 16, 221–233. [Google Scholar]
- Song, S.; Chen, X.; Huang, D.; Xu, Y.; Zeng, H.; Hu, X.; Xu, B.; Jin, Z.; Wang, W. Identification of miRNAs differentially expressed in Fusarium wilt-resistant and susceptible banana varieties. S. Afr. J. Bot. 2016, 106, 244–249. [Google Scholar]
- Luan, Y.; Cui, J.; Zhai, J.; Li, J.; Han, L.; Meng, J. High-throughput sequencing reveals differential expression of miRNAs in tomato inoculated with Phytophthora infestans. Planta 2015, 241, 1405–1416. [Google Scholar] [PubMed]
- Kumar, D.; Dutta, S.; Singh, D.; Prabhu, K.V.; Kumar, M.; Mukhopadhyay, K. Uncovering leaf rust responsive miRNAs in wheat (Triticum aestivum L.) using high-throughput sequencing and prediction of their targets through degradome analysis. Planta 2017, 245, 161–182. [Google Scholar] [PubMed]
- Xin, M.; Wang, Y.; Yao, Y.; Xie, C.; Peng, H.; Ni, Z.; Sun, Q. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Jeyaraj, A.; Wang, X.; Wang, S.; Liu, S.; Zhang, R.; Wu, A.; Wei, C. Identification of regulatory networks of microRNAs and their targets in response to Colletotrichum gloeosporioides in tea plant (Camellia sinensis L.). Front. Plant Sci. 2019, 10, 1096. [Google Scholar]
- Jia, Y.; Li, C.; Li, Q.; Liu, P.; Liu, D.; Liu, Z.; Wang, Y.; Jiang, G.; Zhai, W. Characteristic dissection of Xanthomonas oryzae pv. oryzae responsive microRNAs in rice. Int. J. Mol. Sci. 2020, 21, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; He, S.; Fang, D.; Guo, L.; Zhou, X.; Guo, Y.; Gao, L.; Qiao, Y. High-throughput sequencing-based identification of Arabidopsis miRNAs induced by Phytophthora capsici infection. Front. Microbiol. 2020, 11, 1094. [Google Scholar]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J.D. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Shivaprasad, P.V.; Chen, H.-M.; Patel, K.; Bond, D.M.; Santos, B.A.; Baulcombe, D.C. A microRNA superfamily regulates nucleotide binding site–leucine-rich repeats and other mRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, S.; Park, G.; Atamian, H.S.; Han, C.S.; Stajich, J.E.; Kaloshian, I.; Borkovich, K.A. MicroRNAs suppress NB domain genes in tomato that confer resistance to Fusarium oxysporum. PLoS Pathog. 2014, 10, e1004464. [Google Scholar]
- Jiang, N.; Meng, J.; Cui, J.; Sun, G.; Luan, Y. Function identification of miR482b, a negative regulator during tomato resistance to Phytophthora infestans. Hortic. Res. 2018, 5, 9. [Google Scholar]
- de Vries, S.; Kukuk, A.; von Dahlen, J.K.; Schnake, A.; Kloesges, T.; Rose, L.E. Expression profiling across wild and cultivated tomatoes supports the relevance of early miR482/2118 suppression for Phytophthora resistance. Proc. Biol. Sci. 2018, 285, 20172560. [Google Scholar] [PubMed] [Green Version]
- Bundó, M.; Campo, S.; San Segundo, B. Role of microRNAs in Plant–Fungus Interactions. In Plant microRNAs: Shaping Development and Environmental Responses, 1st ed.; Springer Nature: New York, NY, USA, 2020; pp. 199–220. [Google Scholar]
- Davey, M.W.; Gudimella, R.; Harikrishna, J.A.; Sin, L.W.; Khalid, N.; Keulemans, J. A draft Musa balbisiana genome sequence for molecular genetics in polyploid, inter-and intra-specific Musa hybrids. BMC Genom. 2013, 14, 683. [Google Scholar]
- Liu, W.; Cheng, C.; Chen, F.; Ni, S.; Lin, Y.; Lai, Z. High-throughput sequencing of small RNAs revealed the diversified cold-responsive pathways during cold stress in the wild banana (Musa itinerans). BMC Plant Biol. 2018, 18, 308. [Google Scholar]
- Vidya, S.; Ravishankar, K.; Laxman, R. Genome wide analysis of heat responsive microRNAs in banana during acquired thermo tolerance. J. Hortic. Sci. 2018, 13, 61–71. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, Y.; Tang, R.; Qu, H.; Duan, X.; Jiang, Y. Banana sRNAome and degradome identify microRNAs functioning in differential responses to temperature stress. BMC Genom. 2019, 20, 33. [Google Scholar]
- Song, S.; Xu, Y.; Huang, D.; Ashraf, M.A.; Li, J.; Hu, W.; Jin, Z.; Zeng, C.; Tang, F.; Xu, B. Identification and characterization of miRNA169 family members in banana (Musa acuminata L.) that respond to Fusarium oxysporum f. sp. cubense infection in banana cultivars. PeerJ 2018, 6, e6209. [Google Scholar]
- Fei, S.; Czislowski, E.; Fletcher, S.; Peters, J.; Batley, J.; Aitken, E.; Mitter, N. Small RNA profiling of Cavendish banana roots inoculated with Fusarium oxysporum f. sp. cubense race 1 and tropical race 4. Phytopathol. Res. 2019, 1, 22. [Google Scholar]
- Seo, J.-K.; Wu, J.; Lii, Y.; Li, Y.; Jin, H. Contribution of small RNA pathway components in plant immunity. Mol. Plant Microbe Interact. 2013, 26, 617–625. [Google Scholar]
- Li, Y.; Zhang, Q.; Zhang, J.; Wu, L.; Qi, Y.; Zhou, J.-M. Identification of microRNAs involved in pathogen-associated molecular pattern-triggered plant innate immunity. Plant Physiol. 2010, 152, 2222–2231. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lu, Y.-G.; Shi, Y.; Wu, L.; Xu, Y.-J.; Huang, F.; Guo, X.-Y.; Zhang, Y.; Fan, J.; Zhao, J.-Q. Multiple rice microRNAs are involved in immunity against the blast fungus Magnaporthe oryzae. Plant Physiol. 2014, 164, 1077–1092. [Google Scholar] [CrossRef] [Green Version]
- Baldrich, P.; Campo, S.; Wu, M.-T.; Liu, T.-T.; Hsing, Y.-I.C.; Segundo, B.S. MicroRNA-mediated regulation of gene expression in the response of rice plants to fungal elicitors. RNA Biol. 2015, 12, 847–863. [Google Scholar]
- Li, Y.; Cao, X.L.; Zhu, Y.; Yang, X.M.; Zhang, K.N.; Xiao, Z.Y.; Wang, H.; Zhao, J.H.; Zhang, L.L.; Li, G.B. Osa-miR398b boosts H2O2 production and rice blast disease-resistance via multiple superoxide dismutases. New Phytol. 2019, 222, 1507–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, F.; Girma, G.; Mengiste, T. Global mRNA and microRNA expression dynamics in response to anthracnose infection in sorghum. BMC Genom. 2020, 21, 760. [Google Scholar]
- Mahesh, H.; Shirke, M.D.; Wang, G.-L.; Gowda, M. In planta transcriptome analysis reveals tissue-specific expression of pathogenicity genes and microRNAs during rice-Magnaporthe interactions. Genomics 2021, 113, 265–275. [Google Scholar] [CrossRef]
- Muthusamy, M.; Uma, S.; Backiyarani, S.; Saraswathi, M. Computational prediction, identification, and expression profiling of microRNAs in banana (Musa spp.) during soil moisture deficit stress. J. Hortic. Sci. Biotechnol. 2014, 89, 208–214. [Google Scholar]
- Lee, W.S.; Gudimella, R.; Wong, G.R.; Tammi, M.T.; Khalid, N.; Harikrishna, J.A. Transcripts and microRNAs responding to salt stress in Musa acuminata Colla (AAA Group) cv. Berangan roots. PLoS ONE 2015, 10, e0127526. [Google Scholar]
- Ghag, S.B.; Shekhawat, U.K.; Ganapathi, T.R. Small RNA profiling of two important cultivars of banana and overexpression of miRNA156 in transgenic banana plants. PLoS ONE 2015, 10, e0127179. [Google Scholar] [CrossRef]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [Green Version]
- Chuck, G.; Cigan, A.M.; Saeteurn, K.; Hake, S. The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat. Genet. 2007, 39, 544–549. [Google Scholar]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar]
- Wang, J.-W.; Park, M.Y.; Wang, L.-J.; Koo, Y.; Chen, X.-Y.; Weigel, D.; Poethig, R.S. miRNA control of vegetative phase change in trees. PLoS Genet. 2011, 7, e1002012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K.; Shen, J.; Hou, X.; Yao, J.; Li, X.; Xiao, J.; Xiong, L. Gradual increase of miR156 regulates temporal expression changes of numerous genes during leaf development in rice. Plant Physiol. 2012, 158, 1382–1394. [Google Scholar]
- Zheng, C.; Ye, M.; Sang, M.; Wu, R. A regulatory network for miR156-SPL module in Arabidopsis thaliana. Int. J. Mol. Sci. 2019, 20, 6166. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Sun, Y.H.; Amerson, H.; Chiang, V.L. MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J. 2007, 51, 1077–1098. [Google Scholar]
- Hewezi, T.; Howe, P.; Maier, T.R.; Baum, T.J. Arabidopsis small RNAs and their targets during cyst nematode parasitism. Mol. Plant Microbe Interact. 2008, 21, 1622–1634. [Google Scholar] [CrossRef] [Green Version]
- Miyashima, S.; Koi, S.; Hashimoto, T.; Nakajima, K. Non-cell-autonomous microRNA165 acts in a dose-dependent manner to regulate multiple differentiation status in the Arabidopsis root. Development 2011, 138, 2303–2313. [Google Scholar] [PubMed] [Green Version]
- Zhang, W.; Gao, S.; Zhou, X.; Chellappan, P.; Chen, Z.; Zhou, X.; Zhang, X.; Fromuth, N.; Coutino, G.; Coffey, M. Bacteria-responsive microRNAs regulate plant innate immunity by modulating plant hormone networks. Plant Mol Biol. 2011, 75, 93–105. [Google Scholar] [PubMed] [Green Version]
- Sheeba, M.M.; Selvarajan, R.; Mustaffa, M. Prediction and Identification of MicroRNA from Banana Infected with Banana Streak Mysore Virus (BSMYV). Madras Agric. J. 2013, 100, 513–518. [Google Scholar]
- Wong, J.; Gao, L.; Yang, Y.; Zhai, J.; Arikit, S.; Yu, Y.; Duan, S.; Chan, V.; Xiong, Q.; Yan, J. Roles of small RNAs in soybean defense against Phytophthora sojae infection. Plant J. 2014, 79, 928–940. [Google Scholar]
- Balmer, A.; De Paoli, E.; Si-Ammour, A.; Mauch-Mani, B.; Balmer, D. Signs of Silence: Small RNAs and Antifungal Responses in Arabidopsis thaliana and Zea mays. In Plant Engineering; IntechOpen: London, UK, 2017. [Google Scholar]
- Liu, J.; Rice, J.H.; Chen, N.; Baum, T.J.; Hewezi, T. Synchronization of developmental processes and defense signaling by growth regulating transcription factors. PLoS ONE 2014, 9, e98477. [Google Scholar]
- Rawat, N.; Kiran, S.P.; Du, D.; Gmitter, F.G.; Deng, Z. Comprehensive meta-analysis, co-expression, and miRNA nested network analysis identifies gene candidates in citrus against Huanglongbing disease. BMC Plant Biol. 2015, 15, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Suárez, M.; Baldrich, P.; Weigel, D.; Rubio-Somoza, I.; San Segundo, B. The Arabidopsis miR396 mediates pathogen-associated molecular pattern-triggered immune responses against fungal pathogens. Sci. Rep. 2017, 7, 44898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Jue, D.; Li, W.; Zhang, R.; Chen, M.; Yang, Q. Identification of MiRNA from eggplant (Solanum melongena L.) by small RNA deep sequencing and their response to Verticillium dahliae infection. PLoS ONE 2013, 8, e72840. [Google Scholar] [CrossRef]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoades, M.W.; Reinhart, B.J.; Lim, L.P.; Burge, C.B.; Bartel, B.; Bartel, D.P. Prediction of plant microRNA targets. Cell 2002, 110, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-S.; Xie, Q.; Fei, J.-F.; Chua, N.-H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Frugis, G.; Colgan, D.; Chua, N.-H. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 2000, 14, 3024–3036. [Google Scholar] [CrossRef] [Green Version]
- Vaucheret, H.; Mallory, A.C.; Bartel, D.P. AGO1 homeostasis entails coexpression of MIR168 and AGO1 and preferential stabilization of miR168 by AGO1. Mol. Cell 2006, 22, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Zanini, S.; Šečić, E.; Busche, T.; Galli, M.; Zheng, Y.; Kalinowski, J.; Kogel, K.-H. Comparative analysis of transcriptome and sRNAs expression patterns in the Brachypodium distachyon—Magnaporthe oryzae pathosystems. Int. J. Mol. Sci. 2021, 22, 650. [Google Scholar] [CrossRef]
- Šečić, E.; Kogel, K.-H.; Ladera-Carmona, M.J. Biotic stress-associated microRNA families in plants. J. Plant Physiol. 2021, 263, 153451. [Google Scholar] [CrossRef] [PubMed]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Medina, C.; Da Rocha, M.; Magliano, M.; Ratpopoulo, A.; Revel, B.; Marteu, N.; Magnone, V.; Lebrigand, K.; Cabrera, J.; Barcala, M. Characterization of microRNAs from Arabidopsis galls highlights a role for miR159 in the plant response to the root-knot nematode Meloidogyne incognita. New Phytol. 2017, 216, 882–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Hu, X.; Zhu, M.; Xu, M.; Wang, L. Transcription factors NF-YA2 and NF-YA10 regulate leaf growth via auxin signaling in Arabidopsis. Sci. Rep. 2017, 7, 1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunkar, R.; Zhu, J.-K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [Green Version]
- Gutterson, N.; Reuber, T.L. Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr. Opin. Plant Biol. 2004, 7, 465–471. [Google Scholar] [CrossRef]
- Zhai, J.; Jeong, D.-H.; De Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; González, A.J.; Yan, Z.; Kitto, S.L.; Grusak, M.A. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.-H.; Fan, L.; Liu, Y.; Xu, H.; Llewellyn, D.; Wilson, I. miR482 regulation of NBS-LRR defense genes during fungal pathogen infection in cotton. PLoS ONE 2013, 8, e84390. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Mu, X.; Liu, C.; Cai, J.; Shi, K.; Zhu, W.; Yang, Q. Overexpression of potato miR482e enhanced plant sensitivity to Verticillium dahliae infection. J. Integr. Plant Biol. 2015, 57, 1078–1088. [Google Scholar] [CrossRef]
- Wu, X.-M.; Liu, M.-Y.; Ge, X.-X.; Xu, Q.; Guo, W.-W. Stage and tissue-specific modulation of ten conserved miRNAs and their targets during somatic embryogenesis of Valencia sweet orange. Planta 2011, 233, 495–505. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, S.; Han, S.; Wu, T.; Li, X.; Li, W.; Qi, L. Genome-wide identification of microRNAs in larch and stage-specific modulation of 11 conserved microRNAs and their targets during somatic embryogenesis. Planta 2012, 236, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Zhang, S.; Yu, Y.; Luo, Y.C.; Liu, Q.; Ju, C.; Zhang, Y.C.; Qu, L.H.; Lucas, W.J.; Wang, X. MiR397b regulates both lignin content and seed number in Arabidopsis via modulating a laccase involved in lignin biosynthesis. Plant Biotechnol. J. 2014, 12, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Quan, M.; Zhang, D. Genome-wide identification of novel long non-coding RNAs in Populus tomentosa tension wood, opposite wood and normal wood xylem by RNA-seq. Planta 2015, 241, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Raeder, U.; Broda, P. Rapid preparation of DNA from filamentous fungi. Lett. Appl. Microbiol. 1985, 1, 17–20. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z.; Pongpanich, K.; Himaman, W.; Arzanlou, M.; Wingfield, M.J. Cryptic speciation and host specificity among Mycosphaerella spp. occurring on Australian Acacia species grown as exotics in the tropics. Stud. Mycol. 2004, 50, 457–469. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.; Baurens, F.-C.; Droc, G.; Rouard, M.; Cenci, A.; Kilian, A.; Hastie, A.; Doležel, J.; Aury, J.-M.; Alberti, A. Improvement of the banana “Musa acuminata” reference sequence using NGS data and semi-automated bioinformatics methods. BMC Genom. 2016, 17, 243. [Google Scholar] [CrossRef] [Green Version]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.R.; Yeoh, J.M.; Coruh, C.; Axtell, M.J. Improved placement of multi-mapping small RNAs. G3 2016, 6, 2103–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths-Jones, S.; Saini, H.K.; Van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2007, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Bo, X.; Wang, S. TargetFinder: A software for antisense oligonucleotide target site selection based on MAST and secondary structures of target mRNA. Bioinformatics 2005, 21, 1401–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Varkonyi-Gasic, E.; Wu, R.; Wood, M.; Walton, E.F.; Hellens, R.P. Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 2007, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Rego, E.C.S.; Pinheiro, T.D.M.; Antonino, J.D.; Alves, G.S.C.; Cotta, M.G.; Fonseca, F.C.D.A.; Miller, R.N.G. Stable reference genes for RT-qPCR analysis of gene expression in the Musa acuminata-Pseudocercospora musae interaction. Sci. Rep. 2019, 9, 13589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(–delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]
- Zheng, L.L.; Qu, L.H. Application of micro RNA gene resources in the improvement of agronomic traits in rice. Plant Biotechnol. J. 2015, 13, 329–336. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rego, E.C.S.; Pinheiro, T.D.M.; Fonseca, F.C.d.A.; Gomes, T.G.; Costa, E.d.C.; Bastos, L.S.; Alves, G.S.C.; Cotta, M.G.; Amorim, E.P.; Ferreira, C.F.; et al. Characterization of microRNAs and Target Genes in Musa acuminata subsp. burmannicoides, var. Calcutta 4 during Interaction with Pseudocercospora musae. Plants 2023, 12, 1473. https://doi.org/10.3390/plants12071473
Rego ECS, Pinheiro TDM, Fonseca FCdA, Gomes TG, Costa EdC, Bastos LS, Alves GSC, Cotta MG, Amorim EP, Ferreira CF, et al. Characterization of microRNAs and Target Genes in Musa acuminata subsp. burmannicoides, var. Calcutta 4 during Interaction with Pseudocercospora musae. Plants. 2023; 12(7):1473. https://doi.org/10.3390/plants12071473
Chicago/Turabian StyleRego, Erica Cristina Silva, Tatiana David Miranda Pinheiro, Fernando Campos de Assis Fonseca, Taísa Godoy Gomes, Erica de Castro Costa, Lucas Santos Bastos, Gabriel Sergio Costa Alves, Michelle Guitton Cotta, Edson Perito Amorim, Claudia Fortes Ferreira, and et al. 2023. "Characterization of microRNAs and Target Genes in Musa acuminata subsp. burmannicoides, var. Calcutta 4 during Interaction with Pseudocercospora musae" Plants 12, no. 7: 1473. https://doi.org/10.3390/plants12071473