
Comparative Transcriptome Analysis of Gleditsia sinensis Thorns at Different Stages of Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
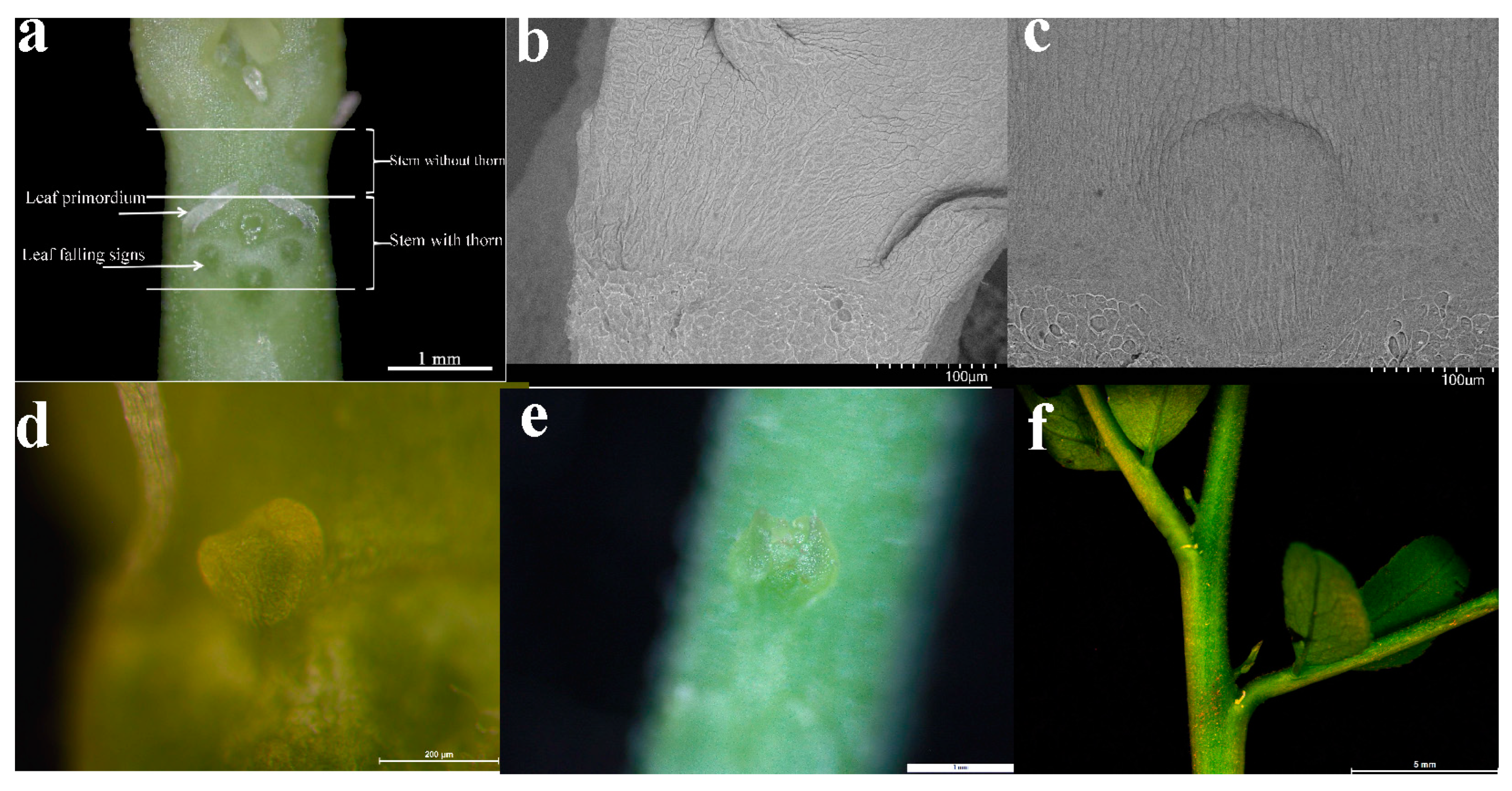
Abstract
:1. Introduction
2. Results
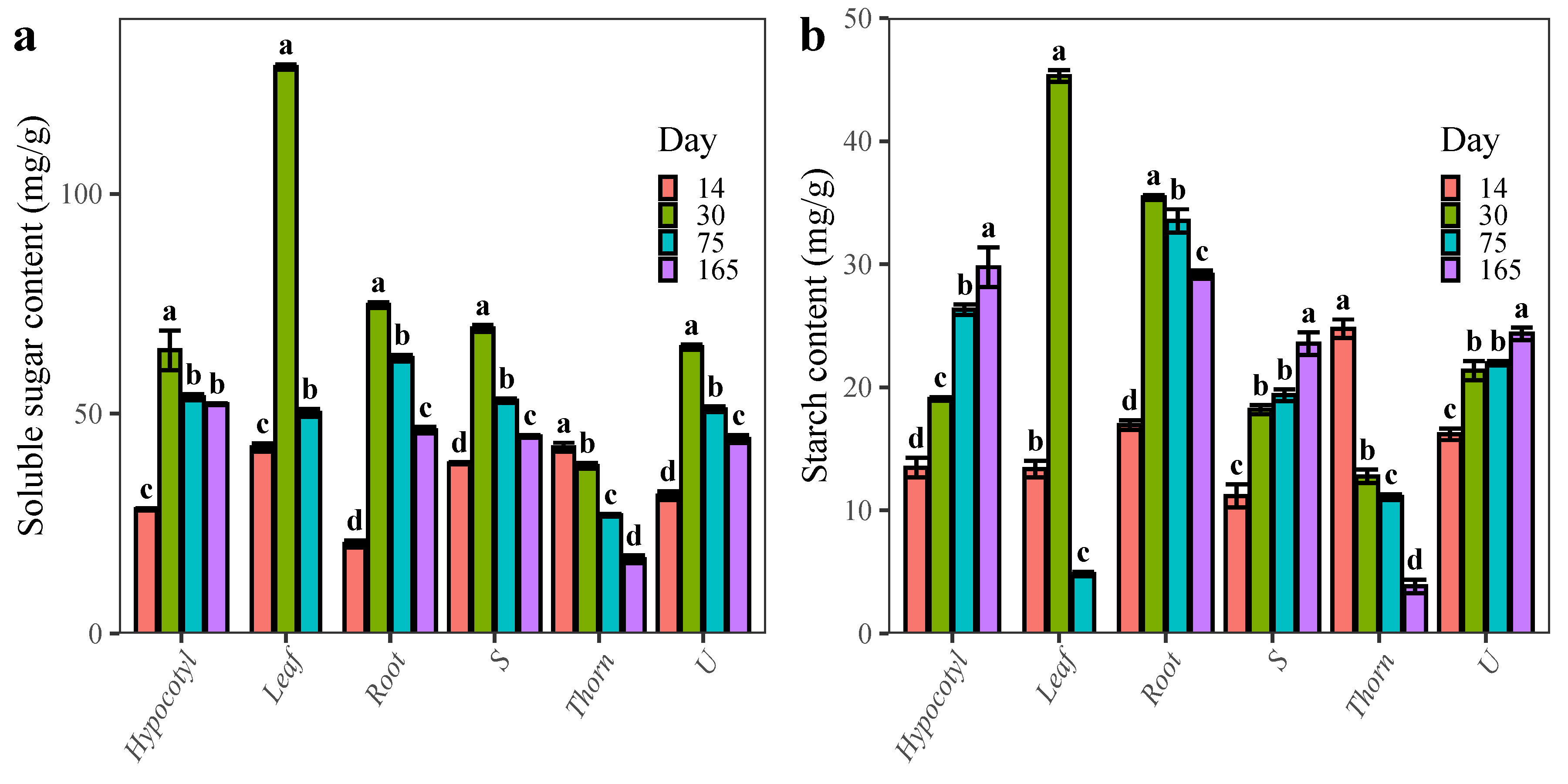
2.1. Changes in the Content of Carbohydrates during the Development of ZJC
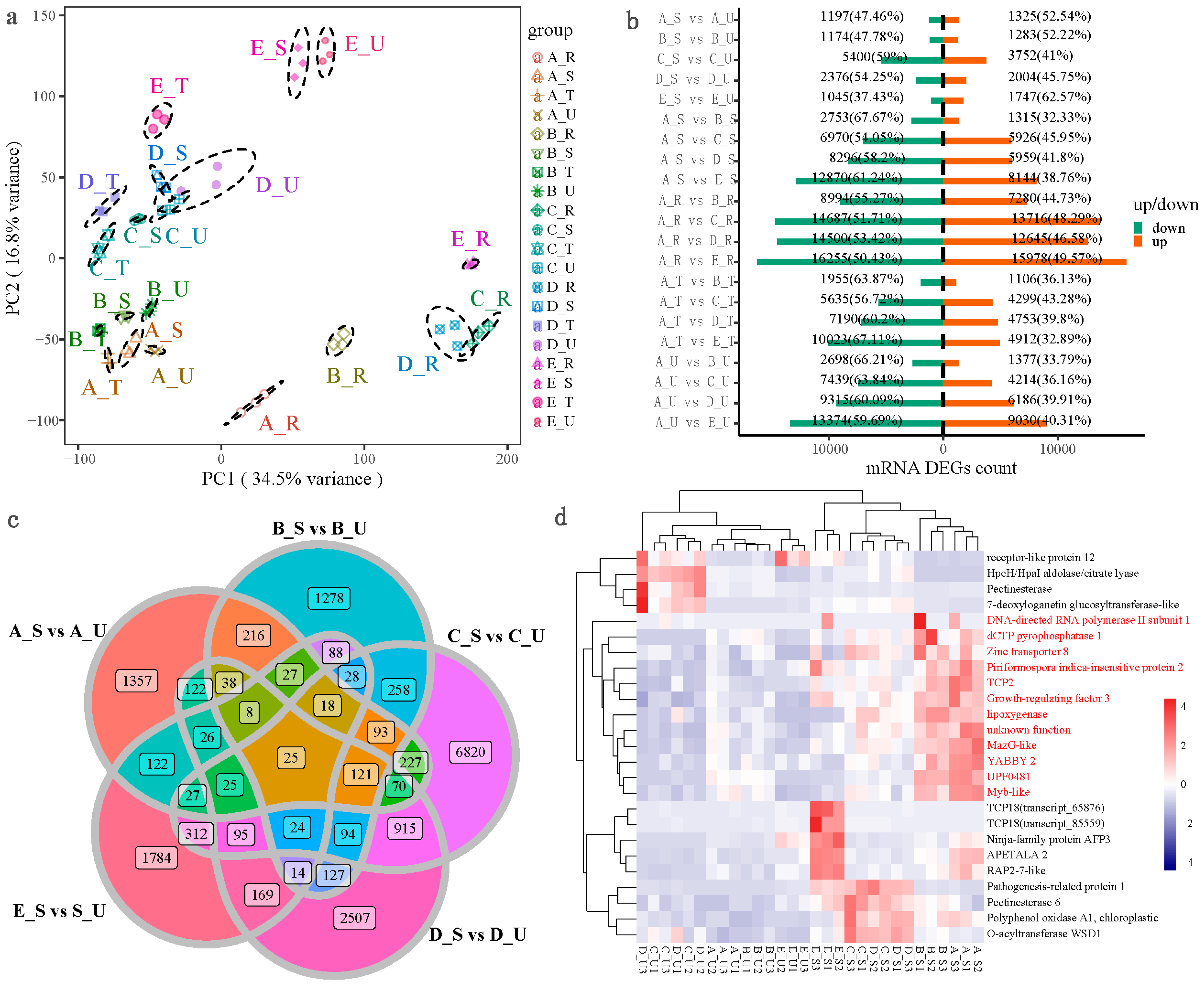
2.2. Transcriptome Principal Component Analysis (PCA) and Quantitative Analysis of DEGs
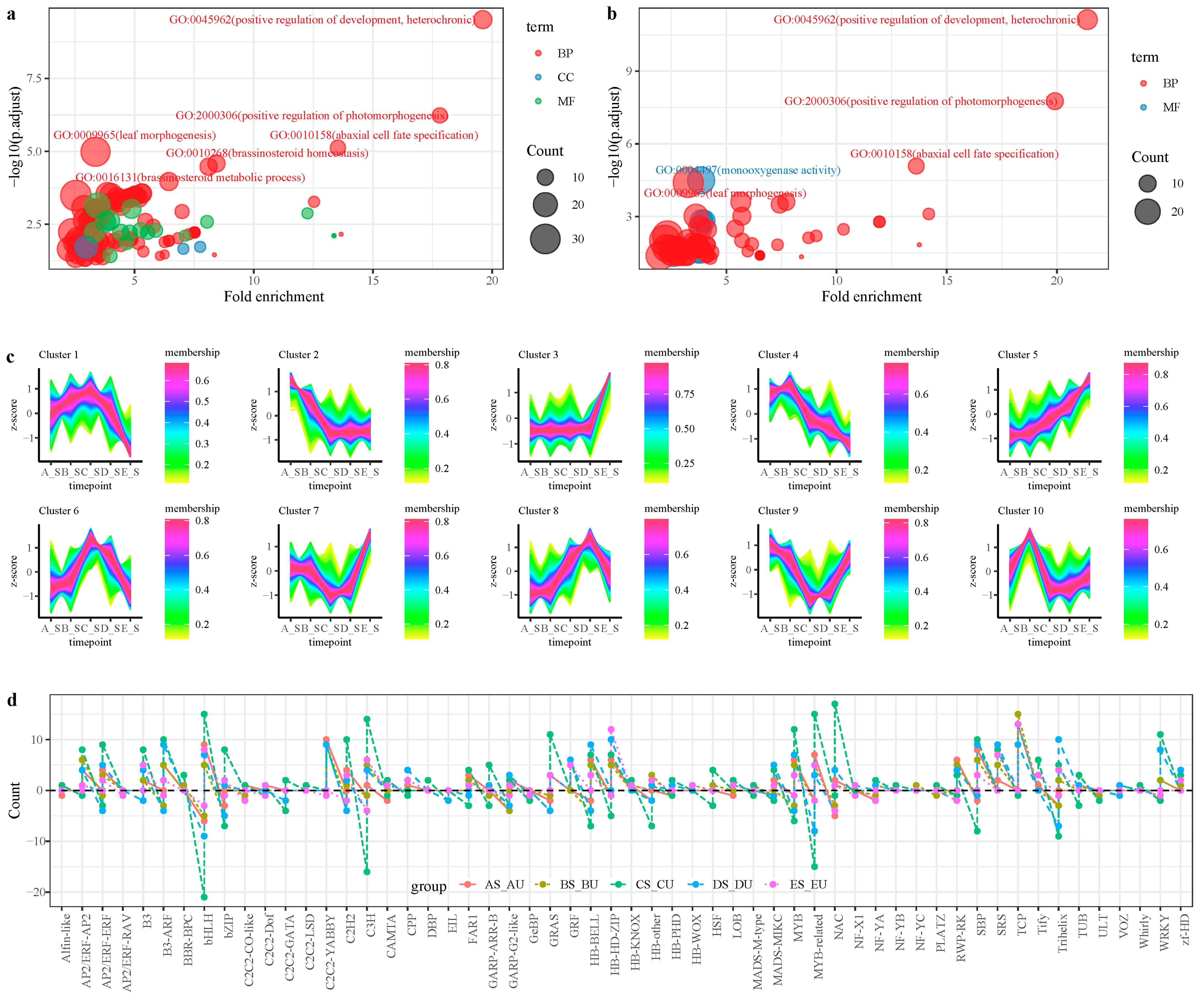
2.3. Functional Enrichment Analysis of DEGs
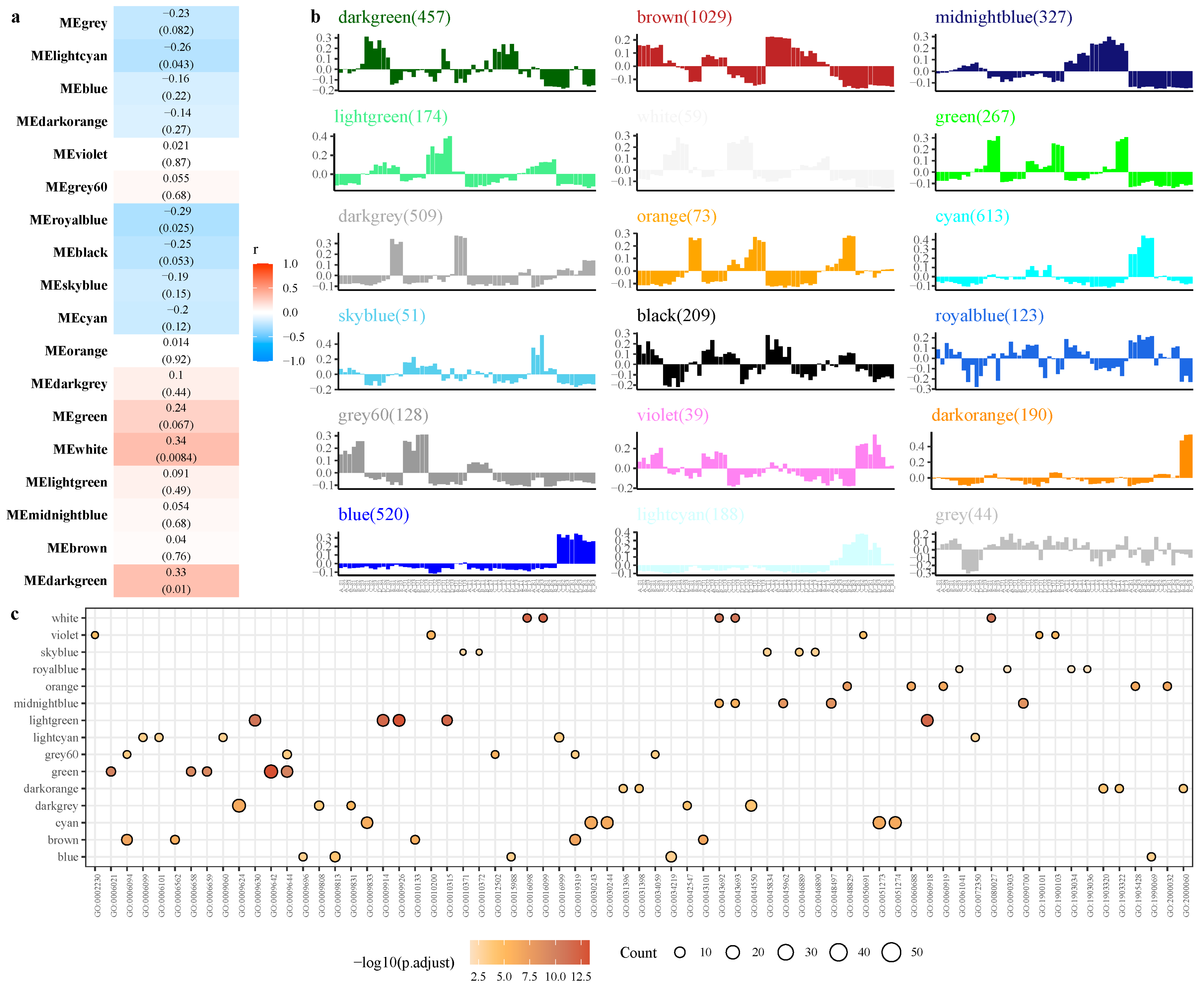
2.4. Weighted Co-Expression Network Analysis (WGCNA)
2.5. Verification of RNA-Seq Data
3. Discussion
4. Materials and Methods
4.1. Acquisition of Test Materials
4.2. RNA Extraction and Library Preparation
4.3. Data Processing and Analysis
4.4. Validation of the DEGs by Real-Time Quantitative PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, J.J.; Shang, X.C.; Ma, J.X.; Zhang, G.T. Research on morphological characteristics change of quercetin and polyphenols of accumulation dynamic of spines of Gleditsia sinensis. China J. Chin. Mater. Med. 2018, 43, 3249–3254. [Google Scholar] [CrossRef]
- Ryu, S.; Park, K.M.; Lee, S.H. Gleditsia sinensis thorn attenuates the collagen-based migration of PC3 prostate cancer cells through the suppression of α2β1 integrin expression. Int. J. Mol. Sci. 2016, 17, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.; Goel, R.; Bhardwaj, A.; Asif, M.H.; Sawant, S.V.; Misra, P. Transcriptome analysis provides insight into prickle development and its link to defense and secondary metabolism in Solanum viarum Dunal. Sci. Rep. 2018, 8, 17092. [Google Scholar] [CrossRef] [Green Version]
- Khadgi, A.; Weber, C.A. Morphological characterization of prickled and prickle-free Rubus using scanning electron microscopy. HortScience 2020, 55, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Rinehart, J.A.; Petersen, M.W.; John, M.E. Tissue-specific and developmental regulation of cotton gene FbL2A (demonstration of promoter activity in transgenic plants). Plant Physiol. 1996, 112, 1331–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber: A powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3, 104. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, Z.; Li, F. Updates on molecular mechanisms in the development of branched trichome in Arabidopsis and nonbranched in cotton. Plant Biotechnol. J. 2019, 17, 1706–1722. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, X.; Zhu, D.; Cui, S.; Li, X.; Cao, Y.; Ma, L. A single amino acid substitution in IIIf subfamily of basic helix-loop-helix transcription factor AtMYC1 leads to trichome and root hair patterning defects by abolishing its interaction with partner proteins in Arabidopsis. J. Biol. Chem. 2012, 287, 14109–14121. [Google Scholar] [CrossRef] [Green Version]
- Kellogg, A.A.; Branaman, T.J.; Jones, N.M.; Little, C.Z.; Swanson, J.-D. Morphological studies of developing Rubus prickles suggest that they are modified glandular trichomes. Botany 2011, 89, 217–226. [Google Scholar] [CrossRef]
- Yang, A.; Qi, X.; Wang, Q.-M.; Wang, H.; Wang, Y.; Li, L.; Liu, W.; Qiao, Y. The branch-thorn occurrence of Lycium ruthenicum is associated with leaf DNA hypermethylation in response to soil water content. Mol. Biol. Rep. 2022, 49, 1925–1934. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, H.; Xu, T.; Shi, T.; Li, Z.; Hou, W. Comparative transcriptome analysis reveals key genes and pathways involved in prickle development in eggplant. Genes 2021, 12, 341. [Google Scholar] [CrossRef]
- Liu, X.; He, X.; Liu, Z.; Wu, P.; Tang, N.; Chen, Z.; Zhang, W.; Rao, S.; Cheng, S.; Luo, C. Transcriptome mining of genes in Zanthoxylum armatum revealed ZaMYB86 as a negative regulator of prickly development. Genomics 2022, 114, 110374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, M.; Zhu, W.; Shi, C.; Bao, M.; Zhang, W. Nonglandular prickle formation is associated with development and secondary metabolism-related genes in Rosa multiflora. Physiol. Plant. 2021, 173, 1147–1162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Rossignol, P.; Huang, T.; Wang, Y.; May, A.; Dupont, C.; Orbovic, V.; Irish, V.F. Reprogramming of stem cell activity to convert thorns into branches. Curr. Biol. 2020, 30, 2951–2961.e5. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Y.; Irish, V.F. CENTRORADIALIS maintains shoot meristem indeterminacy by antagonizing THORN IDENTITY1 in Citrus. Curr. Biol. 2021, 31, 2237–2242.e4. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Li, N.; Deng, Z.; Luo, F.; Duan, Y. Selection and evaluation of a thornless and HLB-tolerant bud-sport of pummelo citrus with an emphasis on molecular mechanisms. Front. Plant Sci. 2021, 12, 739108. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, Y.; He, K.; Lu, Y. Study on morphological and structural observations of Gleditsia sinensis thorn during development process. Acta Bot. Boreali-Occident. Sin. 2021, 41, 2080–2086. [Google Scholar]
- Xu, R.; Hao, X.; Gao, H. Effect of Gibberellin of the Growthe of main branches and thorns of Gleditsia sinensis Lam. Shanxi For. Sci. Technol. 2019, 48, 36–38. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Ma, W.-T.; Lu, M.; An, H.-M.; Yi, Y. Comparative transcriptomic analysis of Rosa sterilis inflorescence branches with different trichome types reveals an R3-MYB transcription factor that negatively regulates trichome formation. J. Integr. Agric. 2022, 21, 2926–2942. [Google Scholar] [CrossRef]
- Swarnkar, M.K.; Kumar, P.; Dogra, V.; Kumar, S. Prickle morphogenesis in rose is coupled with secondary metabolite accumulation and governed by canonical MBW transcriptional complex. Plant Direct 2021, 5, e00325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-P.; Tian, X.-H.; Yang, Y.-X.; Liu, Q.-X.; Wang, Q.; Chen, L.-P.; Li, H.-L.; Zhang, W.-D. Gleditsia species: An ethnomedical, phytochemical and pharmacological review. J. Ethnopharmacol. 2016, 178, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, G.; Mu, Y.; Zhou, H.; Wang, X.; Yang, P. Anti-breast cancer triterpenoid saponins from the thorns of Gleditsia sinensis. Nat. Prod. Res. 2019, 33, 2308–2313. [Google Scholar] [CrossRef]
- Yuxin, G.; Hongjiao, L.; Jianwei, Z.; Qin, Z.; Lina, Y.; Jiaqiu, L.; Baohui, L. Difference of secondary metabolites in spines of Gleditsia sinensis and Gleditsia microphylla. Sci. Silvae Sin. 2022, 58, 82–94. [Google Scholar]
- Mason, M.G.; Ross, J.J.; Babst, B.A.; Wienclaw, B.N.; Beveridge, C.A. Sugar demand, not auxin, is the initial regulator of apical dominance. Proc. Natl. Acad. Sci. USA 2014, 111, 6092–6097. [Google Scholar] [CrossRef] [Green Version]
- Hao, W. A Preliminary Study on the Mechanism of Branch Thorn Occurrence of Lycium ruthenicum. Master’s Thesis, Shenyang Agricultural University, Shenyang, China, 2020. Available online: https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C475KOm_zrgu4lQARvep2SAkHr3ADhkADnVu66WViDP_3Fh8y2srKEaumVCx-K1iyhdbXEz2t6QlqEMtlcMpLTZv&uniplatform=NZKPT (accessed on 20 January 2023).
- Hao, J.; Tu, L.; Hu, H.; Tan, J.; Deng, F.; Tang, W.; Nie, Y.; Zhang, X. GbTCP, a cotton TCP transcription factor, confers fibre elongation and root hair development by a complex regulating system. J. Exp. Bot. 2012, 63, 6267–6281. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.-H.; Luo, Y.; Han, H.; Liu, Y.-Z.; Alam, S.M.; Zhao, H.-X.; Li, Y.-T. Genome-wide analysis of citrus TCP transcription factors and their responses to abiotic stresses. BMC Plant Biol. 2022, 22, 325. [Google Scholar] [CrossRef]
- Airoldi, C.A.; Glover, B.J. Evo–Devo: Tinkering with the Stem Cell Niche to Produce Thorns. Curr. Biol. 2020, 30, R873–R875. [Google Scholar] [CrossRef]
- Wang, X. The Principle and Technology of Plant Physiology and Biochemistry Experiment; Higher Education Press: Beijing, China, 2006; pp. 280–281. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, gkw982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, F.; Zhao, Y.; Wang, X.; Sun, Y. Comparative Transcriptome Analysis of Gleditsia sinensis Thorns at Different Stages of Development. Plants 2023, 12, 1456. https://doi.org/10.3390/plants12071456
Xiao F, Zhao Y, Wang X, Sun Y. Comparative Transcriptome Analysis of Gleditsia sinensis Thorns at Different Stages of Development. Plants. 2023; 12(7):1456. https://doi.org/10.3390/plants12071456
Chicago/Turabian StyleXiao, Feng, Yang Zhao, Xiurong Wang, and Yanan Sun. 2023. "Comparative Transcriptome Analysis of Gleditsia sinensis Thorns at Different Stages of Development" Plants 12, no. 7: 1456. https://doi.org/10.3390/plants12071456